
Land Use Effects on Airborne Bacterial Communities Are Evident in Both Near-Surface and Higher-Altitude Air

 ,
,
Abstract
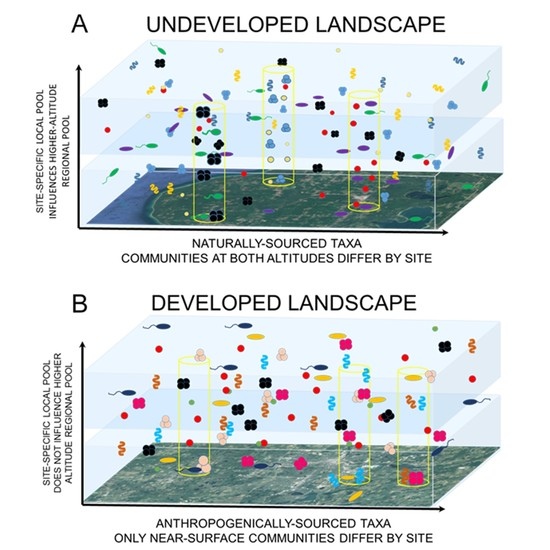
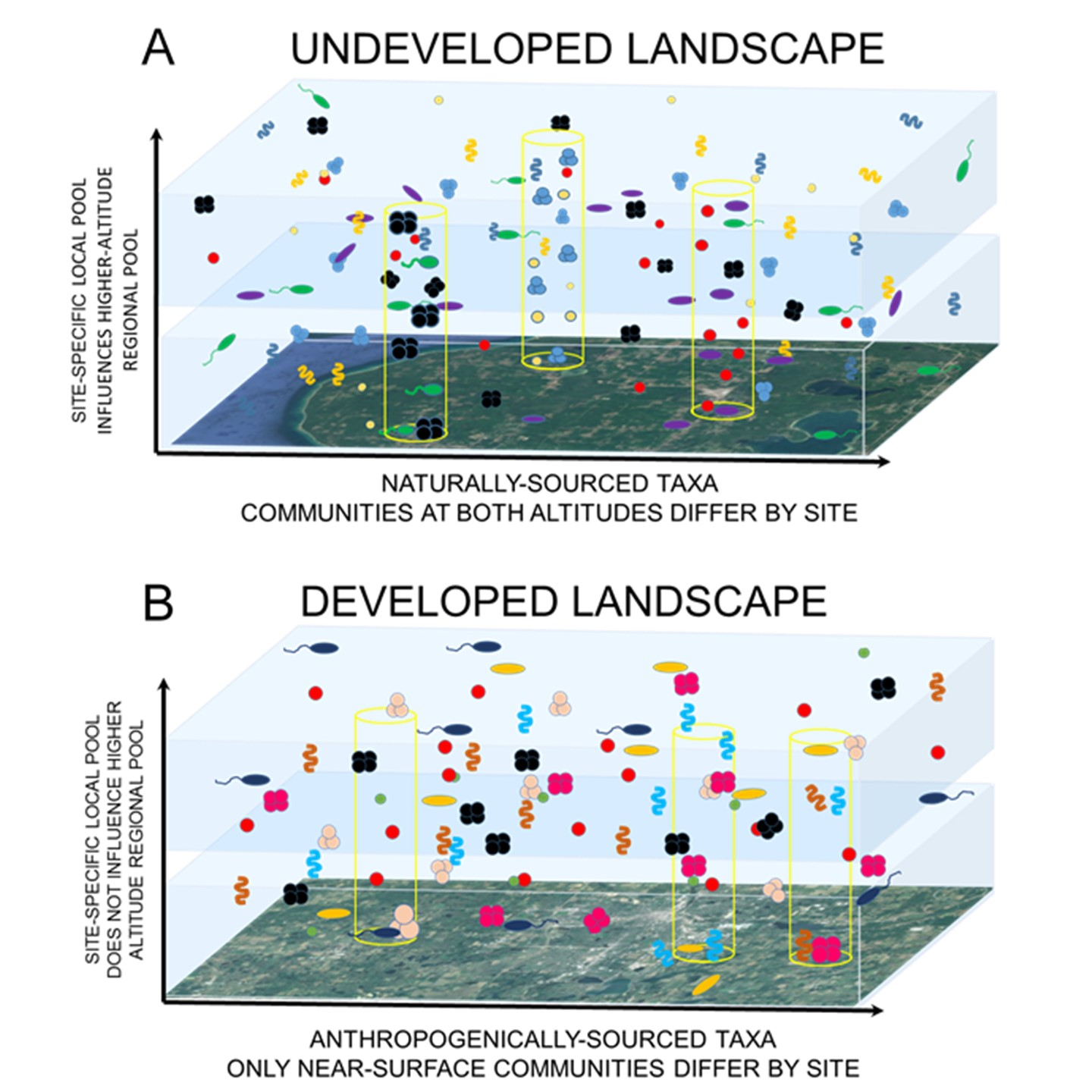
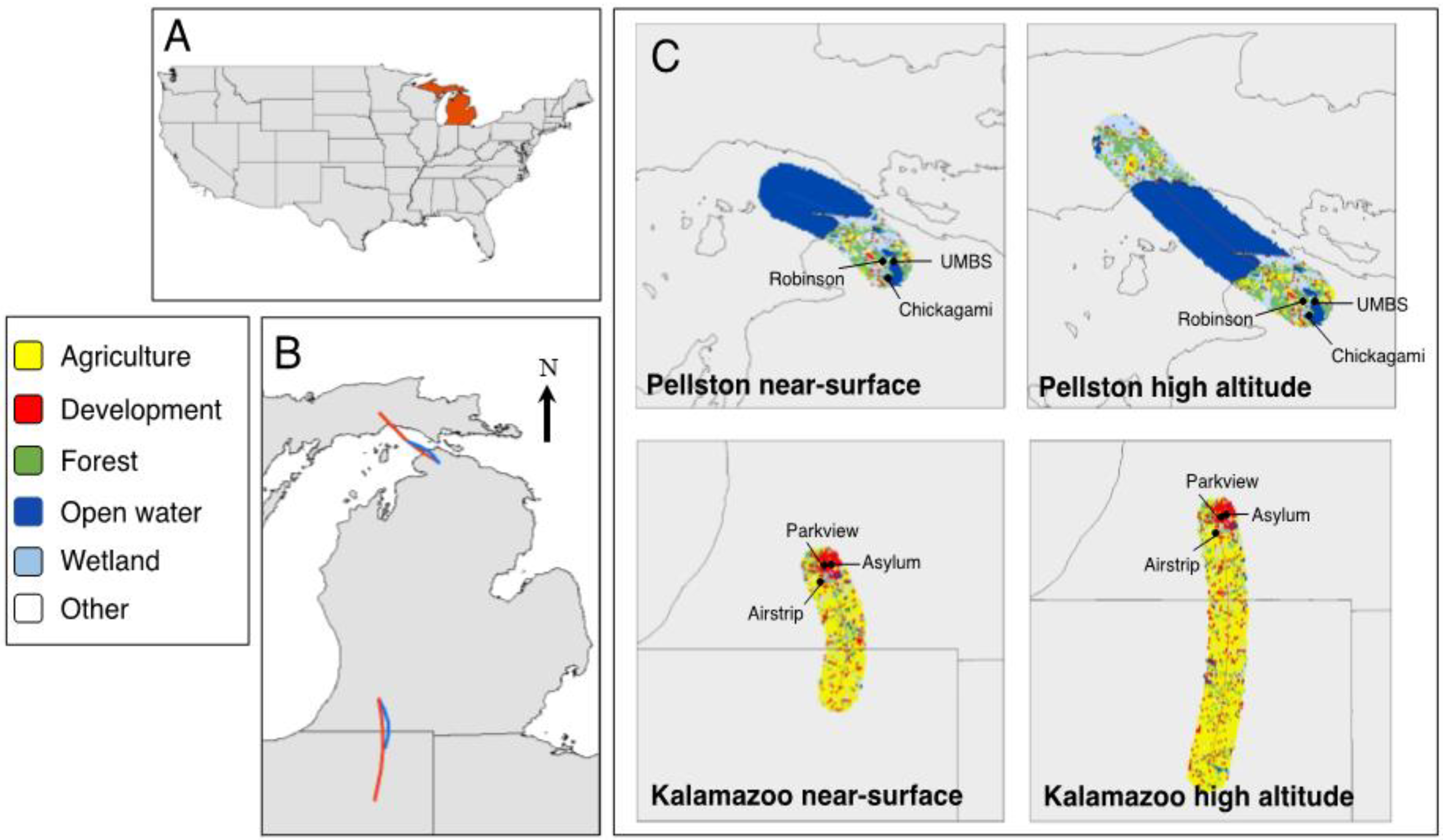
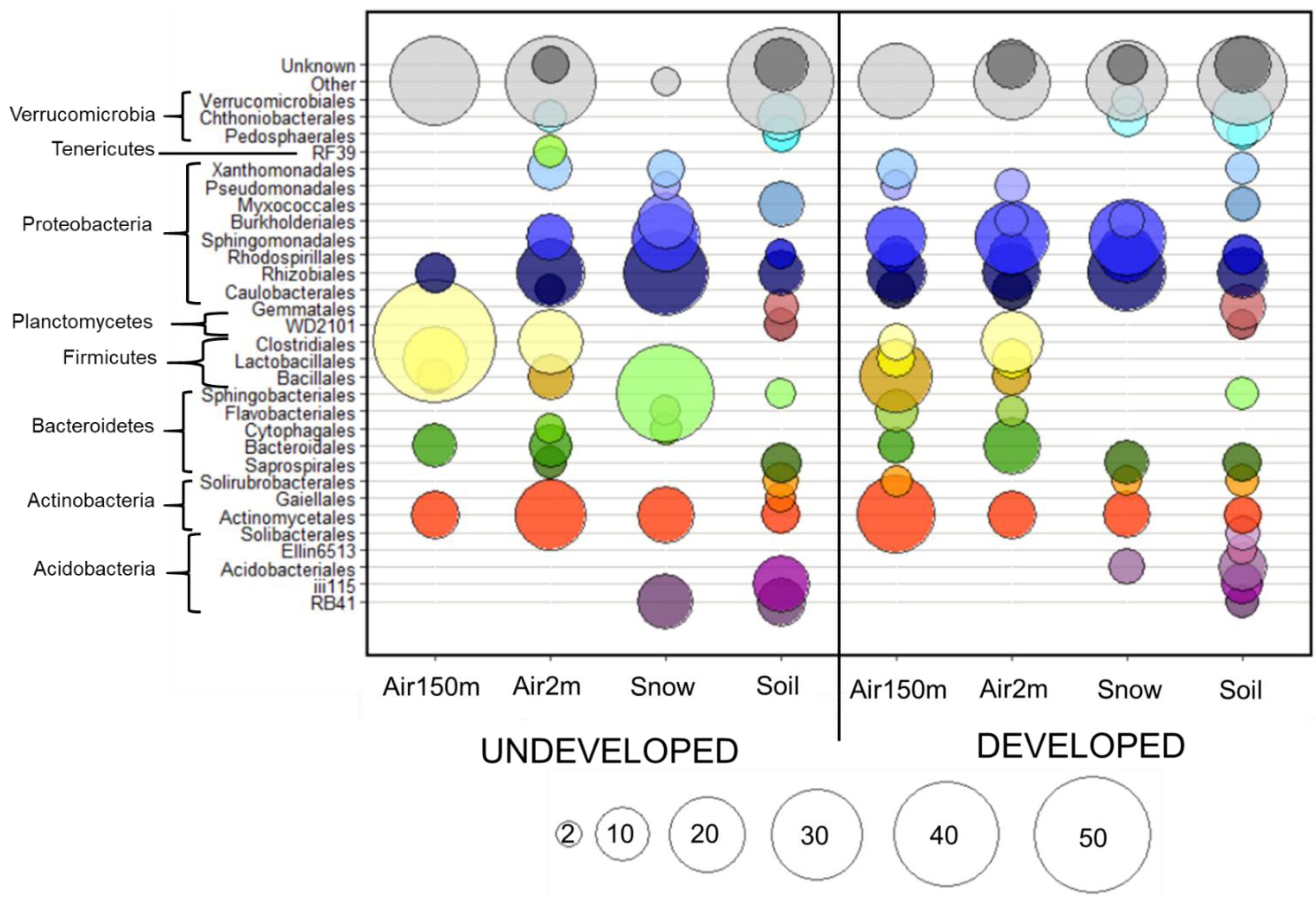
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Airborne Bacterial Community Sampling
2.2. DNA Extraction
2.3. DNA Sequencing and Bioinformatics
2.4. Land Cover and Statistical Analyses
2.5. Considerations of the Experimental Approach
3. Results
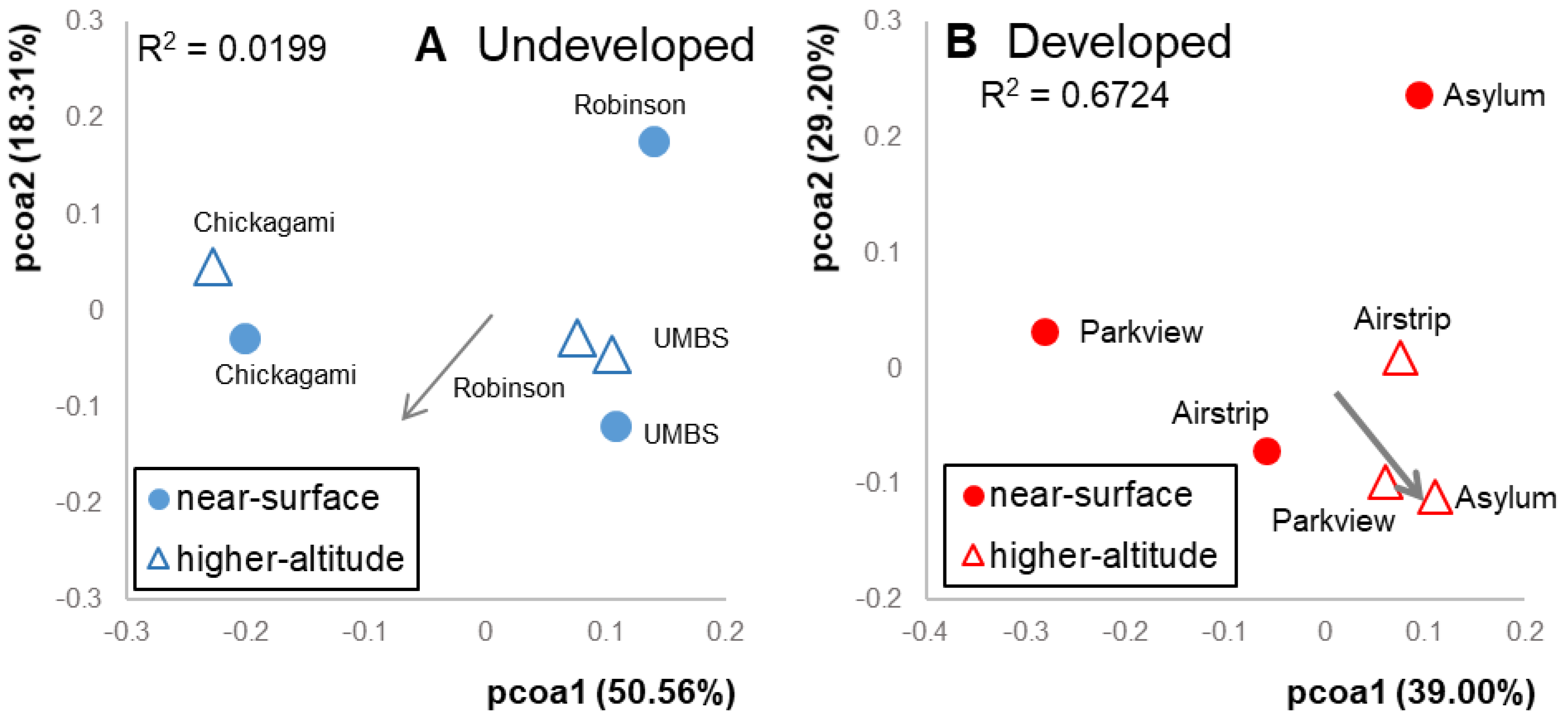
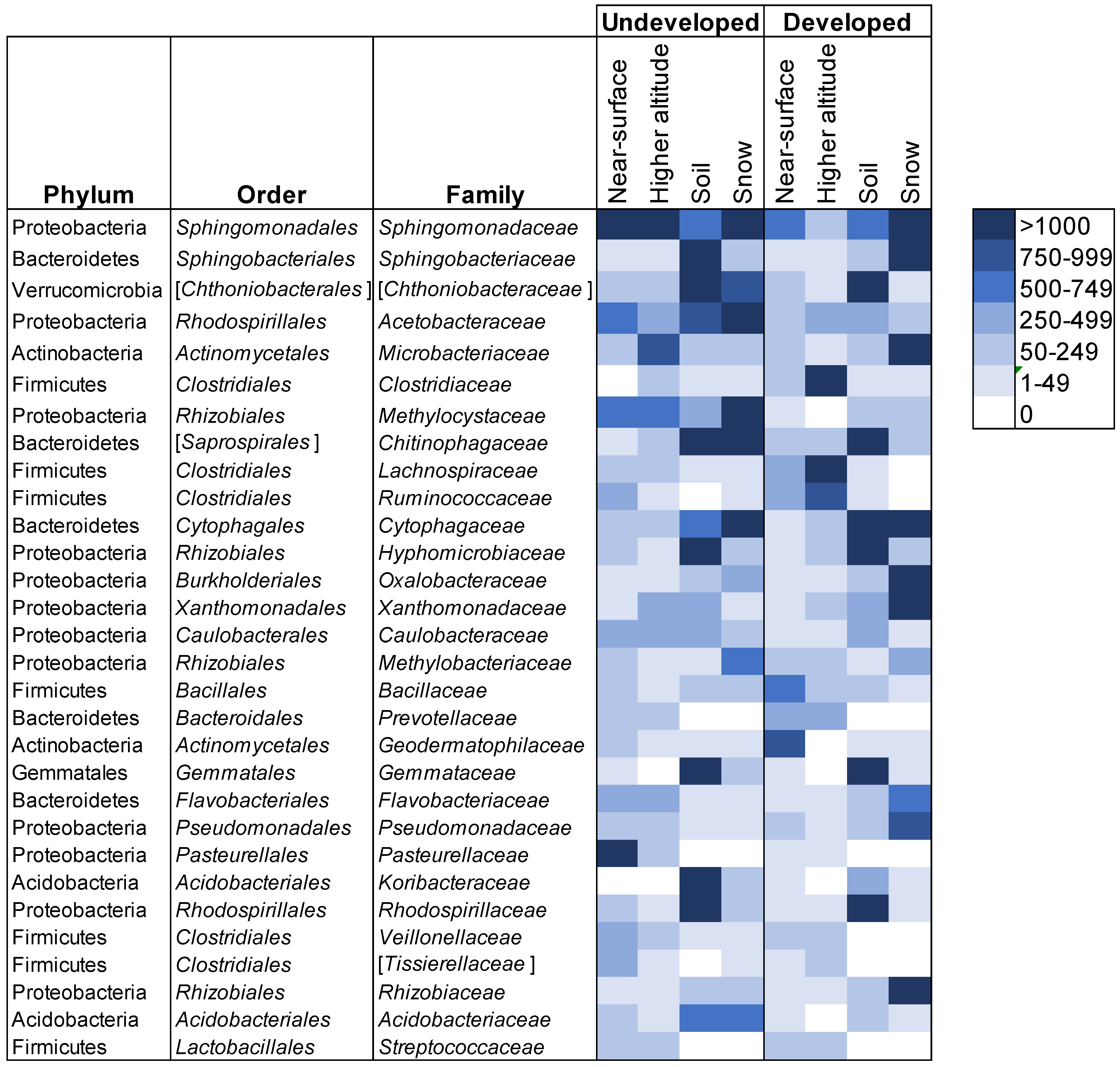
3.1. Airborne Community Results by Location
3.2. Effects of Altitude
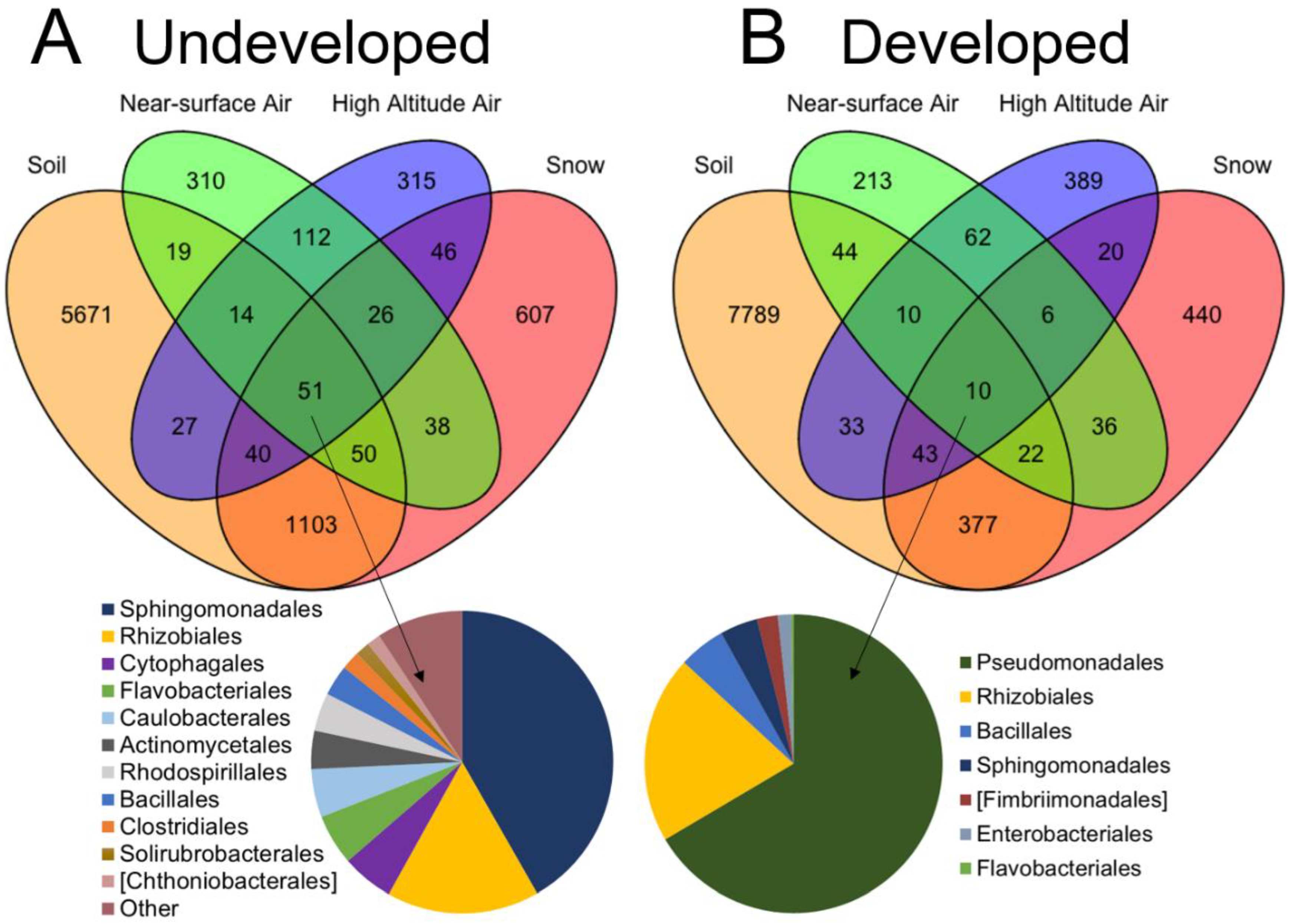
3.3. Connectivity between Sample Sources
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bowers, R.M.; Lauber, C.L.; Wiedinmyer, C.; Hamady, M.; Hallar, A.G.; Fall, R.; Knight, R.; Fierer, N. Characterization of Airborne Microbial Communities at a High-Elevation Site and Their Potential To Act as Atmospheric Ice Nuclei. Appl. Environ. Microbiol. 2009, 75, 5121–5130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmale, D.G.; Ross, S.D. Highways in the Sky: Scales of Atmospheric Transport of Plant Pathogens. Annu. Rev. Phytopathol. 2015, 53, 591–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, S.M.; Elbert, W.; Lawrence, M.G. Bacteria in the global atmosphere. Atmos. Chem. Phys. 2009, 9, 9263–9280. [Google Scholar] [CrossRef] [Green Version]
- Choudoir, M.J.; Barberán, A.; Menninger, H.L.; Dunn, R.R.; Fierer, N. Variation in range size and dispersal capabilities of microbial taxa. Ecology 2018, 99, 322–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.S.; Cohan, F. Recombination and migration rates in natural populations of Bacillus subtilis and Bacillus mojavensis. Evolution 1995, 49, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Seifried, J.S.; Wichels, A.; Gerdts, G. Spatial distribution of marine airborne bacterial communities. Microbiology 2015, 4, 475–490. [Google Scholar] [CrossRef]
- Smith, D.J.; Timonen, H.J.; Jaffe, D.A.; Griffin, D.W.; Birmele, M.N.; Perry, K.D.; Ward, P.D.; Roberts, M.S. Intercontinental Dispersal of Bacteria and Archaea by Transpacific Winds. Appl. Environ. Microbiol. 2013, 79, 1134–1139. [Google Scholar] [CrossRef] [Green Version]
- Meola, M.; Lazzaro, A.; Zeyer, J. Bacterial Composition and Survival on Sahara Dust Particles Transported to the European Alps. Front. Microbiol. 2015, 6, 1454. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.M.; Bohannan, B.J.M.; Jaffe, D.A.; Levin, D.A.; Green, J.L. Molecular Evidence for Metabolically Active Bacteria in the Atmosphere. Front. Microbiol. 2016, 7, 772. [Google Scholar] [CrossRef]
- Amato, P.; Joly, M.; Besaury, L.; Oudart, A.; Taib, N.; Moné, A.I.; Deguillaume, L.; Delort, A.-M.; Debroas, D. Active microorganisms thrive among extremely diverse communities in cloud water. PLoS ONE 2017, 12, e0182869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Qi, J.; Beibei, E.; Yin, Y.; Gao, D. Concentration, viability and size distribution of bacteria in atmospheric bioaerosols under different types of pollution. Environ. Pollut. 2020, 257, 113485. [Google Scholar] [CrossRef]
- Möhler, O.; DeMott, P.J.; Vali, G.; Levin, Z. Microbiology and atmospheric processes: The role of biological particles in cloud physics. Biogeosciences 2007, 4, 2559–2591. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.E.; Dueker, M.E.; Logan, J.R.; Weathers, K.C. The biology of fog: Results from coastal Maine and Namib Desert reveal common drivers of fog microbial composition. Sci. Total. Environ. 2019, 647, 1547–1556. [Google Scholar] [CrossRef]
- Jones, S.E.; Newton, R.J.; McMahon, K.D. Potential for atmospheric deposition of bacteria to influence bacterioplankton communities. FEMS Microbiol. Ecol. 2008, 64, 388–394. [Google Scholar] [CrossRef]
- Kaushik, R.; Balasubramanian, R.; Dunstan, H. Microbial Quality and Phylogenetic Diversity of Fresh Rainwater and Tropical Freshwater Reservoir. PLoS ONE 2014, 9, e100737. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.E.; Bell-Dereske, L.P.; Dougherty, K.M.; Kittredge, H.A. Dispersal alters soil microbial community response to drought. Environ. Microbiol. 2020, 22, 905–916. [Google Scholar] [CrossRef]
- Fröhlich-Nowoisky, J.; Kampf, C.J.; Weber, B.; Huffman, J.A.; Pöhlker, C.; Andreae, M.O.; Lang-Yona, N.; Burrows, S.M.; Gunthe, S.S.; Elbert, W.; et al. Bioaerosols in the Earth system: Climate, health, and ecosystem interactions. Atmos. Res. 2016, 182, 346–376. [Google Scholar] [CrossRef] [Green Version]
- Oie, S.; Kamiya, A. Bacterial contamination of aerosol solutions containing antibiotics. Microbios 1995, 82, 109–113. [Google Scholar]
- Solans, X.; Alonso, R.M.; Constans, A.; Alfonso, M. Exposición laboral a hongos y bacterias ambientales en una planta de selección de residuos de envases. Rev. Iberoam. Micol. 2007, 24, 131–135. [Google Scholar] [CrossRef]
- Thomas, M.; Jarobe, G.; Frazer, R. Infection Control in the Dental Office. Dent. Clin. N. Am. 2008, 52, 609–628. [Google Scholar] [CrossRef]
- Brodie, E.L.; DeSantis, T.Z.; Parker, J.P.M.; Zubietta, I.X.; Piceno, Y.M.; Andersen, G.L. Urban aerosols harbor diverse and dynamic bacterial populations. Proc. Natl. Acad. Sci. USA 2006, 104, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, V.; Gandolfi, I.; Ambrosini, R.; Bestetti, G.; Innocente, E.; Rampazzo, G.; Franzetti, A. Temporal variability and effect of environmental variables on airborne bacterial communities in an urban area of Northern Italy. Appl. Microbiol. Biotechnol. 2012, 97, 6561–6570. [Google Scholar] [CrossRef] [Green Version]
- Bowers, R.M.; Clements, N.; Emerson, J.B.; Wiedinmyer, C.; Hannigan, M.P.; Fierer, N. Seasonal Variability in Bacterial and Fungal Diversity of the Near-Surface Atmosphere. Environ. Sci. Technol. 2013, 47, 12097–12106. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Ladau, J.; Leff, J.W.; Pollard, K.S.; Menninger, H.L.; Dunn, R.R.; Fierer, N. Continental-scale distributions of dust-associated bacteria and fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 5756–5761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhou, L.; Zhang, X.; Xu, C.; Dong, L.; Yao, M. Bioaerosol emissions and detection of airborne antibiotic resistance genes from a wastewater treatment plant. Atmos. Environ. 2016, 124, 404–412. [Google Scholar] [CrossRef]
- Lymperopoulou, D.S.; Adams, R.I.; Lindow, S.E. Contribution of Vegetation to the Microbial Composition of Nearby Outdoor Air. Appl. Environ. Microbiol. 2016, 82, 3822–3833. [Google Scholar] [CrossRef] [Green Version]
- Docherty, K.M.; Pearce, D.S.; Lemmer, K.M.; Hale, R.L. Distributing regionally, distinguishing locally: Examining the underlying effects of local land use on airborne bacterial biodiversity. Environ. Microbiol. 2018, 20, 3529–3542. [Google Scholar] [CrossRef]
- Mhuireach, G.Á.; Betancourt-Román, C.M.; Green, J.L.; Johnson, B.R. Spatiotemporal Controls on the Urban Aerobiome. Front. Ecol. Evol. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Bowers, R.M.; McLetchie, S.; Knight, R.; Fierer, N. Spatial variability in airborne bacterial communities across land-use types and their relationship to the bacterial communities of potential source environments. ISME J. 2010, 5, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mhuireach, G.; Johnson, B.R.; Altrichter, A.E.; Ladau, J.; Meadow, J.F.; Pollard, K.S.; Green, J.L. Urban greenness influences airborne bacterial community composition. Sci. Total. Environ. 2016, 571, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Groffman, P.M.; Cavender-Bares, J.; Bettez, N.D.; Grove, J.M.; Hall, S.J.; Heffernan, J.B.; Hobbie, S.E.; Larson, K.L.; Morse, J.L.; Neill, C.; et al. Ecological homogenization of urban USA. Front. Ecol. Environ. 2014, 12, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, E.; Johansson, A.-M.; Ahlinder, J.; Lundkvist, M.J.; Singh, N.J.; Brodin, T.; Forsman, M.; Stenberg, P. Airborne microbial biodiversity and seasonality in Northern and Southern Sweden. PeerJ 2020, 8, e8424. [Google Scholar] [CrossRef] [Green Version]
- DeLeon-Rodriguez, N.; Lathem, T.L.; Rodriguez, R.L.M.; Barazesh, J.M.; Anderson, B.E.; Beyersdorf, A.J.; Ziemba, L.D.; Bergin, M.; Nenes, A.; Konstantinidis, K.T. Microbiome of the upper troposphere: Species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc. Natl. Acad. Sci. USA 2013, 110, 2575–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.J.; Ravichandar, J.D.; Jain, S.; Griffin, D.W.; Yu, H.; Tan, Q.; Thissen, J.; Lusby, T.; Nicoll, P.; Shedler, S.; et al. Airborne Bacteria in Earth’s Lower Stratosphere Resemble Taxa Detected in the Troposphere: Results From a New NASA Aircraft Bioaerosol Collector (ABC). Front. Microbiol. 2018, 9, 1752. [Google Scholar] [CrossRef]
- Smith, D.J.; Griffin, D.W.; McPeters, R.D.; Ward, P.D.; Schuerger, A.C. Microbial survival in the stratosphere and implications for global dispersal. Aerobiology 2011, 27, 319–332. [Google Scholar] [CrossRef]
- Bowers, R.M.; Sullivan, A.P.; Costello, E.K.; Collett, J.L.; Knight, R.; Fierer, N. Sources of Bacteria in Outdoor Air across Cities in the Midwestern United States. Appl. Environ. Microbiol. 2011, 77, 6350–6356. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.; Reponen, T.; Grinshpun, S.A.; Martuzevicius, D.; Lemasters, G. Correlation of ambient inhalable bioaerosols with particulate matter and ozone: A two-year study. Environ. Pollut. 2006, 140, 16–28. [Google Scholar] [CrossRef]
- Park, J.; Ichijo, T.; Nasu, M.; Yamaguchi, N. Investigation of bacterial effects of Asian dust events through comparison with seasonal variability in outdoor airborne bacterial community. Sci. Rep. 2016, 6, 35706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tipton, L.; Zahn, G.; Datlof, E.; Kivlin, S.N.; Sheridan, P.; Amend, A.S.; Hynson, N.A. Fungal aerobiota are not affected by time nor environment over a 13-y time series at the Mauna Loa Observatory. Proc. Natl. Acad. Sci. USA 2019, 116, 25728–25733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federal Aviation Adminstration of the United States of America. Code of Federal Regulations: Moored Balloons, Kites, Amateur Rockets, Unmanned Free Balloons and Certain Model Aircraft 1963-2016, Section 101.1–101.7. Available online: https://rgl.faa.gov/regulatory_and_guidance_library/rgfar.nsf/farsbysect?openview&count=-1&restricttocategory=part+101 (accessed on 25 January 2021).
- Homer, C.; Dewitz, J.; Yang, L.; Jin, S.; Danielson, P.; Xian, G.; Coulston, J.; Herold, N.; Wickham, J.; Megown, K. Completion of the 2011 National Land Cover Database for the Conterminous United States—Representing a Decade of Land Cover Change Information. Photogramm. Eng. Remote Sens. 2015, 81, 346–354. [Google Scholar] [CrossRef]
- Spring, A.M.; Docherty, K.M.; Domingue, K.D.; Kerber, T.V.; Mooney, M.M.; Lemmer, K.M. A Method for Collecting Atmospheric Microbial Samples from Set Altitudes for Use with Next-Generation Sequencing Techniques to Characterize Communities. Air Soil Water Res. 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.S.; Hoover, B.A.; Jennings, S.; Nevitt, G.A.; Docherty, K.M. Morphological and genetic factors shape the microbiome of a seabird species (Oceanodroma leucorhoa) more than environmental and social factors. Microbiome 2017, 5, 146. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Laurence, M.; Hatzis, C.; Brash, D.E. Common Contaminants in Next-Generation Sequencing That Hinder Discovery of Low-Abundance Microbes. PLoS ONE 2014, 9, e97876. [Google Scholar] [CrossRef]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M. Community Ecology Package: “Vegan”, version 2.5-6; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaeser, S.P.; Kämpfer, P. The Family Sphingomonadaceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 641–707. [Google Scholar]
- Average Weather at Pellston Regional Airport of Emmet County. Available online: https://weatherspark.com/y/146680/Average-Weather-at-Pellston-Regional-Airport-of-Emmet-County-Michigan-United-States-Year-Round (accessed on 2 May 2020).
- Average Weather at Kalamazoo-Battle Creek International Airport. Available online: https://weatherspark.com/y/146615/Average-Weather-at-Kalamazoo-Battle-Creek-International-Airport-Michigan-United-States-Year-Round (accessed on 2 May 2020).
- Fierer, N.; Liu, Z.; Rodríguez-Hernández, M.; Knight, R.; Henn, M.; Hernandez, M.T. Short-Term Temporal Variability in Airborne Bacterial and Fungal Populations. Appl. Environ. Microbiol. 2007, 74, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-S.; Huang, T.-Y. Fluorochrome in Monitoring Indoor Bioaerosols. Aerosol Sci. Technol. 2006, 40, 237–241. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Behzad, H.; Gojobori, T.; Mineta, K. Challenges and Opportunities of Airborne Metagenomics. Genome Biol. Evol. 2015, 7, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Fahlgren, C.; Hagström, A.; Nilsson, D.; Zweifel, U.L. Annual Variations in the Diversity, Viability, and Origin of Airborne Bacteria. Appl. Environ. Microbiol. 2010, 76, 3015–3025. [Google Scholar] [CrossRef] [Green Version]
- Kembel, S.W.; Meadow, J.F.; O’Connor, T.K.; Mhuireach, G.; Northcutt, D.; Kline, J.; Moriyama, M.; Brown, G.Z.; Bohannan, B.J.M.; Green, J.L. Architectural Design Drives the Biogeography of Indoor Bacterial Communities. PLoS ONE 2014, 9, e87093. [Google Scholar] [CrossRef]
- Jones, S.E.; Lennon, J.T. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 5881–5886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evtushenko, L.I.; Takeuchi, M. The Family Microbacteriaceae. In The Prokaryotes: Volume 3: Archaea. Bacteria: Firmicutes, Actinomycetes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 1020–1098. [Google Scholar]
- Webb, H.K.; Ng, H.J.; Ivanova, E.P. The Family Methylocystaceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 341–347. [Google Scholar]
- Chudobova, D.; Cihalova, K.; Jelinkova, P.; Zitka, J.; Nejdl, L.; Guran, R.; Klimanek, M.; Adam, V.; Kizek, R. Effects of Stratospheric Conditions on the Viability, Metabolism and Proteome of Prokaryotic Cells. Atmosphere 2015, 6, 1290–1306. [Google Scholar] [CrossRef] [Green Version]
- Waters, S.M.; Zeigler, D.R.; Nicholson, W.L. Experimental Evolution of Enhanced Growth by Bacillus subtilis at Low Atmospheric Pressure: Genomic Changes Revealed by Whole-Genome Sequencing. Appl. Environ. Microbiol. 2015, 81, 7525–7532. [Google Scholar] [CrossRef] [Green Version]
- Khodadad, C.L.; Wong, G.M.; James, L.M.; Thakrar, P.J.; Lane, M.A.; Catechis, J.A.; Smith, D.J. Stratosphere Conditions Inactivate Bacterial Endospores from a Mars Spacecraft Assembly Facility. Astrobiology 2017, 17, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares-Filho, B.S.; Nepstad, D.C.; Curran, L.M.; Cerqueira, G.C.; Garcia, R.A.; Ramos, C.A.; Voll, E.; McDonald, A.; Lefebvre, P.; Schlesinger, P. Modelling conservation in the Amazon basin. Nat. Cell Biol. 2006, 440, 520–523. [Google Scholar] [CrossRef]
- Rodrigues, J.L.M.; Pellizari, V.H.; Mueller, R.; Baek, K.; Jesus, E.D.C.; Paula, F.S.; Mirza, B.; Hamaoui, G.S.; Tsai, S.M.; Feigl, B.; et al. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2012, 110, 988–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.T.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spring, A.M.; Domingue, K.D.; Kerber, T.V.; Mooney, M.M.; Hale, R.L.; Lemmer, K.M.; Docherty, K.M. Land Use Effects on Airborne Bacterial Communities Are Evident in Both Near-Surface and Higher-Altitude Air. Diversity 2021, 13, 85. https://0-doi-org.brum.beds.ac.uk/10.3390/d13020085
Spring AM, Domingue KD, Kerber TV, Mooney MM, Hale RL, Lemmer KM, Docherty KM. Land Use Effects on Airborne Bacterial Communities Are Evident in Both Near-Surface and Higher-Altitude Air. Diversity. 2021; 13(2):85. https://0-doi-org.brum.beds.ac.uk/10.3390/d13020085
Chicago/Turabian StyleSpring, Allison M., Kenneth D. Domingue, Thomas V. Kerber, Margaret M. Mooney, Rebecca L. Hale, Kristina M. Lemmer, and Kathryn M. Docherty. 2021. "Land Use Effects on Airborne Bacterial Communities Are Evident in Both Near-Surface and Higher-Altitude Air" Diversity 13, no. 2: 85. https://0-doi-org.brum.beds.ac.uk/10.3390/d13020085