
Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities †


Instituto de Química del Sur (INQUISUR), Universidad Nacional del Sur-CONICET, 8000 Bahía Blanca, Argentina
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–5 December 2020; Available online: https://ecsoc-24.sciforum.net/ .
Chem. Proc. 2021, 3(1), 122; https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08393
Published: 14 December 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:Polyphenols are attracting increasing attention in the discovery of useful agents for the treatment of neurodegenerative diseases. Thelesperma megapotamicum (Spreng.) Kuntze belongs to the family Asteraceae, which is known to have a high antioxidant capacity. The phytochemical investigation of T. megapotamicum revealed the presence of 1′-S-isobutyroxyeugenol isobutyrate, (1), lupeol, (2), 1′-S-acetoxyeugenol isobutyrate, (3), stigmasterol, (4), β-sitosterol, (5), eriodictyol, (6), luteolin, (7), and marein, (8), as major secondary metabolites. The neuroprotective activity of this species was studied by evaluating the inhibition in vitro of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), and the antioxidant capacity of the sub-extracts and of the major metabolites isolated from them. The AChE and BChE inhibition were determined by Ellman’s method and the antioxidant activity by DPPH assay. The inhibitory activity against BChE and the antioxidant capacity of the polyphenols present in T. megapotamicum highlight this species as a promising source of active metabolites for the development of agents for the treatment of neurodegenerative diseases.
1. Introduction
In the last three decades, the role of free radicals and their participation in oxidative stress has been extensively studied in the pathophysiology of various age-related diseases, such as cancer, diabetes, cardiovascular and neurodegenerative diseases [1,2,3,4]. Numerous evidence suggest that oxidative damage caused by excessive production of free radicals, both from reactive oxygen species (ROS) and reactive nitrogen species (RNS), plays an important role in the pathogenesis of these diseases [5,6]. The role of oxidative stress and the potential therapeutic use of antioxidants has been extensively studied in several neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD) [7,8,9]. Polyphenolic compounds obtained from natural sources exhibit powerful antioxidant properties [10]. Polyphenols, such as catechins from green tea, curcumin from turmeric, and resveratrol from grape have shown significant antioxidant, anti-inflammatory, and neuroprotective effects [11,12,13,14,15,16,17,18,19].
Flavonoids and phenylpropanoids represent two large groups of polyphenols that are widely distributed in the plant kingdom and have a wide range of medicinal properties. Furthermore, flavonoids and phenylpropanoids have also been reported to show various effects on AD treatment and are gaining increasing attention. Natural flavonoids have shown neuroprotective effect and acetylcholinesterase (AChE) inhibitory activity [20,21,22], and recently it has been reported that phenylpropanoids from Prunus tomentosa are efficient β-amyloid (Aβ) aggregation inhibitors [23].
Thelesperma megapotamicum (Spreng.) Kuntze (Asteraceae) is a species native to Argentina, widely distributed from the center of the country to Patagonia. It is commonly known as Indian tea or pampas tea, and has been used in folk medicine as a digestive and antispasmodic agent [24].
Recent studies show its high antioxidant capacity, which is attributed to the total content of phenols present in the plant. Due to the especially low toxicity of these molecules, this plant species constitutes a source of natural antioxidants that could be useful in the food industry [25]. Previous to our phytochemical investigation, a single study of material from Northern Arizona, United States, has been reported, where three flavonoids present in the ethyl acetate sub-extract were identified [26].
In the present study, the phytochemical investigation from the fractionation of the ethanol extract of the aerial parts of T. megapotamicum, collected from the dunes of the Atlantic coast of Buenos Aires province, revealed the presence of two phenylpropanoids (1 and 3), a triterpene (2), two phytosterols (4 and 5), and three flavonoids (6–8), as major secondary metabolites. Isolation and purification of the phytochemicals were conducted using Silica gel, Sephadex LH-20, and Polyamide columns. All the structures were identified from spectroscopic methods and consequently compared with literature data. This work was completed by evaluating the inhibition in vitro of AChE and butyrylcholinesterase (BChE), and the antioxidant capacity of the sub-extracts and of the major metabolites isolated from them. The AChE and BChE inhibition was determined by Ellman’s method and the antioxidant activity by 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay.
2. Materials and Methods
2.1. General
AChE from electric eel (type VI-S), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), acetylthiocholine iodide (ATCI), butyrylthiocholine iodide (BTCI), tacrine, eserine and 2,2-diphenyl-1-picrylhydrazyl (DPPH), 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) were purchased from Sigma-Aldrich. BChE (horse serum) was purchased from MP Biomedicals. All the solvents used for the extractions and chromatographies were previously distilled. For column chromatographies, silica gel 60 (70–230 mesh, Merck and 200–425 mesh, Aldrich), and polyamide (0.05–0.16 mm, Macherey Nagel) were used. For the preparative chromatographies were used high-performance thin-layer chromatography plates (HPTLC) of 10 cm × 10 cm with fluorescence indicator at 254 nm with glass support (150 microns, Merck). The chromatographies were monitored by thin-layer chromatography (TLC) on silica gel plates (60F-254), visualized under UV light and/or using a p-anisaldehyde solution (5 mL p-anisaldehyde, 5 mL H2SO4 concentered, 1mL acetic acid and 90 mL ethanol). The flavonoids were identified using a buffer solution (20 mL ethyl acetate, 5.4 mL H2O, 2.2 mL formic acid and 2.2 mL acetic acid). All 1H and 13C NMR spectra were recorded at room temperature in CDCl3 or MeOD-d4 on a Bruker Avance ARX-300 spectrophotometer. Chemical shifts (δ) are reported in parts per million (ppm) from tetramethylsilane (TMS) using the residual solvent resonance. Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m= multiplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, dt = doublet of triplets, brs = broad signal. Melting points were determined using a Büchi 510 apparatus, whereas the optical rotation was measured through a Polar IBZ Messtechnik polarimeter.
Mass spectra (EI) were obtained at 70 eV on an Agilent CG-78903 instrument equipped with an MS-5977A MSD selective mass detector. The purity of volatile compounds and the chromatographic analyses (GC) were determined with a GC Shimadzu (GC-14B) with a flame ionization detector equipped with an HP-5MS column (30 m × 0.25 mm × 0.25 μm) using nitrogen as carrier gas.
UV spectra, and enzymatic and antioxidant activities, were recorded in a spectrophotometer JASCO V-630BIO, with temperature module Peltier. To calculate the pendent of absorbance curves vs time was used the program GBC Spectral 1.50 Release 1.1 1995.
2.2. Plant Material
Thelesperma megapotamicum (Spreng.) Kuntze (Asteraceae) plant specimens were collected from the dunes of the Atlantic coast of Buenos Aires province, in the town of Pehuen-Có. The taxonomic authentication of the plant was carried out by Dr María Gabriela Murray. A voucher specimen (voucher reference number: MGM575) was deposited in the Herbarium of the Department of Biology, Biochemistry and Pharmacy, Universidad Nacional del Sur, Argentina (BBB).
2.3. Extraction and Isolation
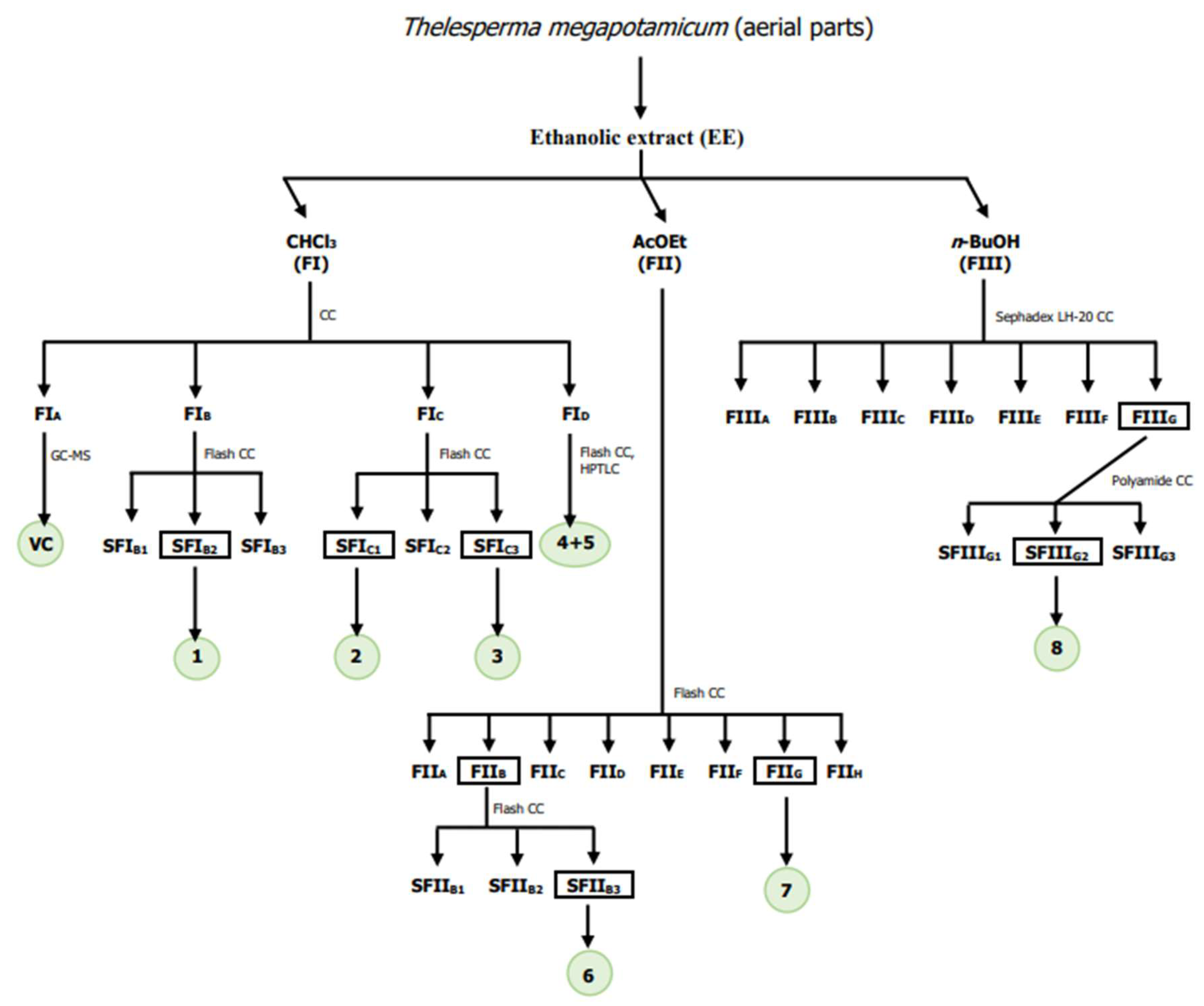
The starting plant material was from aerial parts of T. megapotamicum (365 g), which was extracted twice with 96% ethanol at room temperature for 7 days. The ethanolic extract (EE) was concentrated under reduced pressure, giving 8.0 g (2.2%); 4 g of this residue was suspended in H2O and successively partitioned with chloroform, ethyl acetate, and n-butanol. The extracted solutions were evaporated under reduced pressure to yield 3.1 g (77.5%) of chloroform sub-extract (FI), 0.7 g (17.5%) of ethyl acetate sub-extract (FII), and 0.08 g (2.0%) n-butanol sub-extract (FIII).
Chromatography on silica gel (70–230 mesh) of chloroform fraction (FI) (0.3 g), eluted with chloroform, yielded four fractions: FIA (26.4 mg), FIB (68.8 mg), FIC (128.2 mg), and FID (69.0 mg). Chromatographic separation was monitored by TLC and the fractions were pooled according to their chromatographic profile.
FIA fraction was analysed by GC-MS, and the identification of each volatile compound was achieved by comparing its mass spectrum with those of the database, and its retention time with authentic samples, when available: eugenol (tR = 9.37 min, 15.1%), 6,10,14-trimethyl-2-pentadecanone (tR = 10.33 min, 29.0%), methyl hexadecanoate (tR = 10.72 min, 6.6%), octyl acetate (tR = 11.06 min, 10.1%) and methyl linoleate (tR = 11.13 min, 38.7%).
FIB fraction was analysed by GC-MS in the same analytical conditions used for the FIA analysis. The chromatogram revealed the presence of a major component (1, tR = 11.33 min, 94.5%), and its mass spectrum did not match with those stored in the MS database. FIB fraction (34.4 mg) was subjected to flash chromatography, eluted with dichloromethane, yielding three sub-fractions (SFIB1–SFIB3), of which the sub-fraction SFIB2 led to obtaining of 1 in pure form as an oily liquid (22.0 mg, 64%), and was identified as 1′-S-isobutyroxyeugenol isobutyrate (1). [α]25D = −64.5º (0.44; CHCl3). The structure was confirmed by NMR and GC-MS spectrometry. 1H-NMR and 13C-NMR data were in agreement with previously reported findings [27].
1′-S-isobutyroxyeugenol isobutyrate (1): 1H-NMR (300 MHz, CDCl3): δ (ppm) 6.99 (d, 1H, J = 8.5 Hz, H-3), 6.93 (d, 1H, J = 2 Hz, H-2), 6.93 (dd, 1H, J = 8.5 Hz, 2Hz, H-6), 6.24 (dd, 1H, J = 5.8 Hz, 2 Hz, H-1′), 5.98 (ddd, 1H, J = 17 Hz, 10.5 Hz, 5.8 Hz, H-2′), 5.30 (dt, 1H, J = 17 Hz, 1.2 Hz, Ha-3′), 5.27 (dt, 1H, J = 10.5 Hz, 1.2 Hz, Hb-3′), 3.81 (s, 1H, OMe-H), 2.83 (m, 1H, H-2″), 2.62 (m, 1H, J = 7.0 Hz, H-2′″), 1.32 (d, 6H, J = 7.0 Hz, H-3″, H-4″), 1.20 (d, 6H, J = 7.0 Hz, H-3′″, H-4′″); 13C-NMR (75 MHz, CDCl3): δ (ppm) 175.33 (C-1″), 175.05 (C-1′″), 151.28 (C-5), 139.84 (C-4), 137.86 (C-1), 136.35 (C-2′), 122.86 (C-3), 119.54 (C-2), 117.00 (C-3′), 111.40 (C-6), 75.46 (C-1′), 56.05 (OCH3), 34.31 (C-2″), 34.12 (C-2′″), 19.17 (C-3″), 19.17 (C-4″), 19.06 (C-3′″), 19.06 (C4′″); MS (EI, 70 eV) m/z at 320 ([M]+, 4), 250 (37), 180 (100), 162 (60), 131 (60), 43 (86).
A portion of FIc fraction (42.0 mg) was subjected to flash chromatography, eluted with chloroform, yielding three sub-fractions (SFIC1–SFIC3), of which the sub-fraction SFIC1 led to obtaining of 2 in pure form as a white solid (10.6 mg, 25.2%). Compound 2 was identified as lupeol by NMR and GC-MS spectrometry, by comparison of their spectroscopic data with the literature [28].
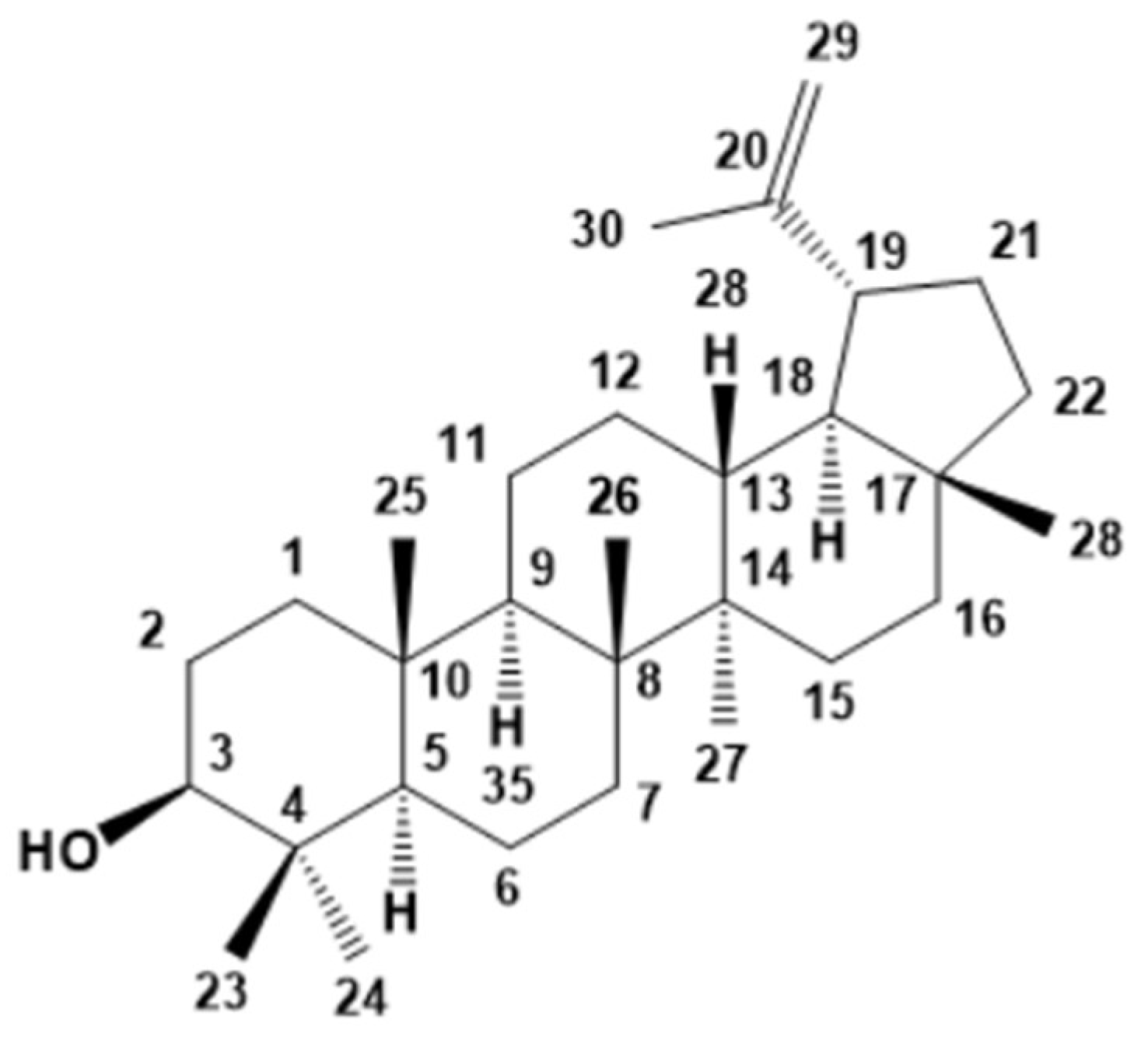
Lupeol (2): 1H-NMR (300 MHz, CDCl3): δ (ppm) 4.68 (brs, 1 H, H-29a), 4.56 (br s, 1 H, H-29b), 3.18 (dd, 1 H, J = 10.6 Hz, 5.6 Hz, H-3), 2.38 (ddd, 1 H, J = 11.2 Hz, 11.0 Hz, 5.7 Hz, H-19), 1.68 (s, 3H, H-30), 1.63 (m, 8 H), 1.38 (m, 9 H), 1.25 (m, 8 H), 1.03 (3H, s, H-26), 0.98 (3H, s, H-23), 0.95 (3H, s, H-27), 0.83 (3H, s, H-25), 0.79 (3H, s, H-28), 0.76 (3H, s, H-24); 13C-NMR (75 MHz, CDCl3): δ (ppm) 150.11 (C-20), 109.47 (C-29), 79.15 (C-3), 55.45 (C-5), 50.59 (C-9), 48.45 (C-18), 48.13 (C-19), 43.15 (C-17), 42.98 (C-14), 40.98 (C-8), 40.15 (C-22), 39.00 (C-1), 38.86 (C-4), 38.20 (C-13), 37.31 (C-10), 35.73 (C-16), 34.43 (C-7), 29.85 (C-21), 28.13 (C-23), 27.59 (C-2), 27.56 (C-15), 25.29 (C12), 21.08 (C-11), 19.45 (C-30), 18.47 (C-6), 18.15 (C-28), 16.27 (C-25), 16.12 (C-26), 15.52 (C-24), 14.69 (C-27); MS (EI, 70 eV) m/z at 426 ([M]+, 25), 411 (10), 207 (71), 189 (60), 135 (64), 121 (65), 107 (95), 93 (92), 68 (100), 55 (88), 41 (10).
Compound 3, 1′-S-acetoxyeugenol isobutyrate, (4.5 mg, 11%) was isolated from SFIC3 fraction, [α]25D = -55.7º (0.44; CHCl3). Its structure was confirmed by NMR and GC-MS spectrometry and compared with spectroscopic data of 1, and bibliographic data [27].
1′-S-acetoxyeugenol isobutyrate (3): 1H-NMR (300 MHz, CDCl3): δ (ppm) 6.99 (d, 1H, J = 8.5 Hz, H-3), 6.93 (d, 1H, J = 2 Hz, H-2), 6.93 (dd, 1H, J = 8.5 Hz, 2 Hz, H-6), 6.25 (dd, 1H, J = 5.8 Hz, 2 Hz, H-1′), 5.99 (ddd, 1H, J =17 Hz, 10.5 Hz, 5.8 Hz, H-2′), 5.31 (d, 1H, J = 17 Hz, Ha-3′), 5.25 (d, 1H, J = 10.5 Hz, Hb-3′), 2.83 (m, 1H, J = 7,0 Hz, H-2″), 2.11 (s, 1H, J = 7.0 Hz, H-2′″), 1.32 (d, 6H, J = 7.0 Hz, H-3″ y H-4″); 13C-NMR (75 MHz, CDCl3): δ (ppm) 175.32 (C-1″), 170.05 (C-1′″), 151.31 (C-5), 139.97 (C-4), 137.57 (C-1), 136.15 (C-2′), 122.88 (C-3), 119.75 (C-2), 117.00 (C-3′), 111.65 (C-6), 75.87 (C-1′), 56.07 (OCH3), 34.11 (C-2″), 21.40 (C-2′″), 19.16 (C-3″), 19.16 (C-4″); MS (EI, 70 eV) m/z at 292 ([M]+, 4), 222 (45), 180 (98), 162 (70), 131 (42), 43 (100).
FID fraction was analysed by GC-MS in the same analytical conditions used for the FIA analysis. The chromatogram revealed the presence of two major compounds: 4 (tR = 20.64 min, 61.1%) and 5 (tR = 21.66 min, 38.8%). They were identified as stigmasterol (4) and β-sitosterol (5) by comparing its mass spectrum with those of the database. Chromatography on silica gel (200–425) of the fraction FID, eluted with n-hexane/ethyl acetate (100:0 to 0:100, step-gradient system), allowed to purify the phytosterols from the other minority compounds present in this fraction; however, it was not possible to separate them. Fraction FID was subjected to a repurification on HPTLC plates, using hexane: ethyl acetate (90:10) as the mobile phase, but 1H and 13C- NMR spectra confirmed the mixture of both.
Stigmasterol (4): MS (EI, 70 eV) m/z at 412 ([M]+, 31), 300 (16), 271 (18), 255 (24), 213 (17), 159 (30), 145 (30), 135 (28), 119 (24), 105 (40), 91 (50), 79 (49), 77 (44), 69 (48), 67 (33), 55 (100), 43 (74), 41 (61).
β-sitosterol (5): MS (EI, 70 eV) m/z at 414 ([M]+, 25), 396 (15), 381 (8), 329 (13), 303 (10), 273 (8), 255 (9), 213 (16), 159 (15), 145 (23), 119 (19), 105 (27), 91 (28), 75 (23), 69 (21), 57 (42), 55 (35), 43 (100), 41 (41).
Chromatography on silica gel (200–425 mesh) of a portion of the ethyl acetate sub-extract (FII) (24.0 mg), eluted with chloroform/methanol (100:0 to 0:100, step-gradient system), yielded eight fractions: FIIA–FIIH. Chromatographic separation was monitored by TLC using the flavonoid buffer as mobile phase, and the fractions were pooled according to their chromatographic profile. Two major compounds 6 and 7, were observed in FIIB and FIIG fractions, respectively, easily seen for their stain colour in the plate. FIIB fraction (5.2 mg) was subjected to flash chromatography, eluted with chloroform/methanol mixtures, yielding three sub-fractions (SFIIB1–SFIIB3), of which the sub-fraction SFIIB3 led to obtaining of 6 in pure form (3.5 mg, 67.0%). Compound 6 was identified as eriodictyol by NMR spectrometry, by comparison of their spectroscopic data with the literature [29].
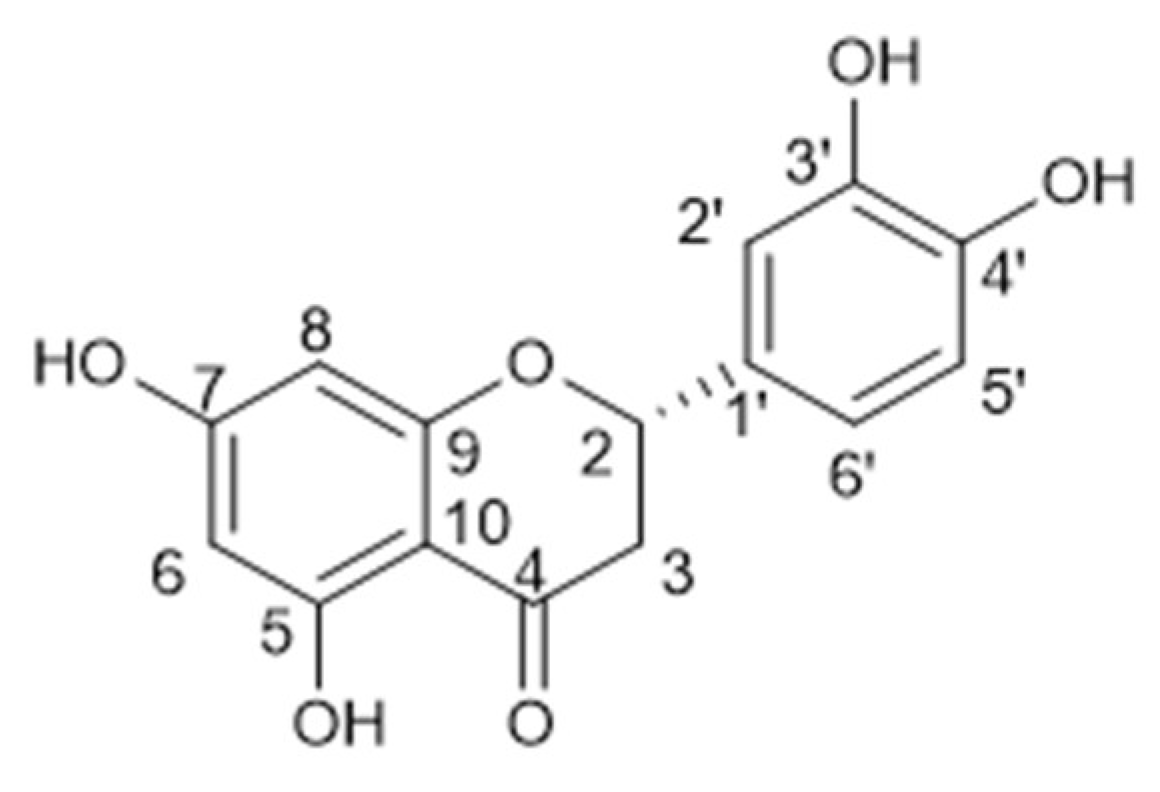
Eriodictyol (6): 1H-NMR (300 MHz, MeOD-d4): δ (ppm) 7.01 (s, 1H, H-2′), 6.88 (s, 1H, H-5′), 6.75 (s, 1H, H-6′), 5.87 (d, 1H, J = 2.1 Hz, H-8), 5.85 (d, 1 H, J = 2.1 Hz, H-6), 5.25 (dd, 1H, J = 12.6 Hz, 3.0 Hz, H-2), 2.66 (dd, 2H, J = 17.4 Hz, 3.0 Hz H-3). 13C-NMR (75 MHz, MeOD-d4): δ (ppm) 196.86 (C-4), 165.40 (C-7), 165.40 (C-5), 164.84 (C-9), 146.88 (C-4′), 146.48 (C-3′), 131.74 (C-1′), 119.30 (C-6′), 116.31 (C-5′), 114.74 (C-2′), 103.35 (C-10), 97.07 (C-6), 96.21 (C-8), 80.41 (C-2), 44.04 (C-3).
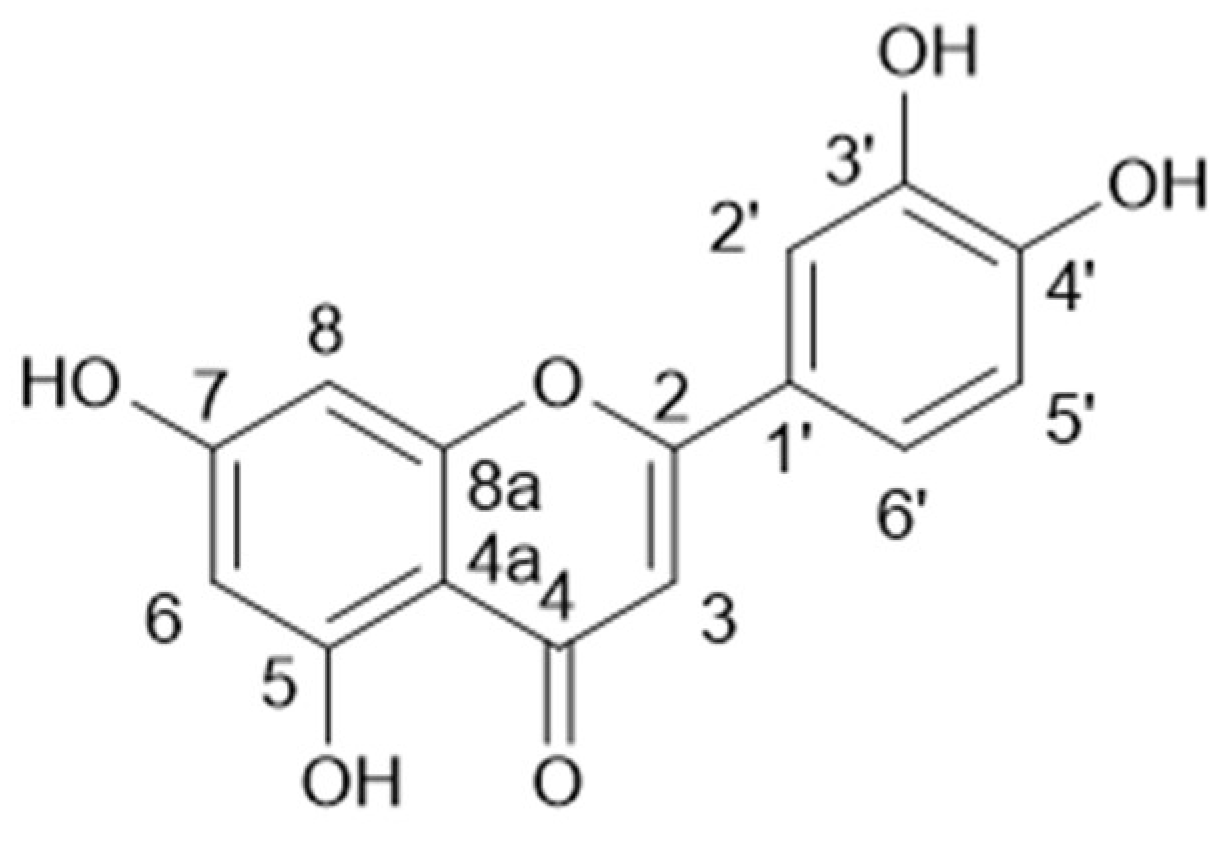
Compound 7 (4.6 mg, 77%) was isolated pure directly from FIIG fraction (6.0 mg) and was identified as luteolin by NMR spectrometry, by comparison of their spectroscopic data with the literature [30].
Luteolin (7): 1H-NMR (300 MHz, MeOD-d4): δ (ppm) 7.34 (d, 1H, H-2′), 6.87 (dd, 1H, J = 6.78 Hz, 2.08 Hz, H-5′), 7.36 (dd, 1H, J = 7.23 Hz, 2.29 Hz, H-6′), 6.41 (d, 1H, J = 2.16 Hz, H-8), 6.18 (d, 1H, J = 2.10 Hz, H-6), 6.51 (d, 1H, H-3). 13C-NMR (75 MHz, MeOD-d4): δ (ppm) 183.9 (C-4), 166.4 (C-2), 166.1 (C-7), 163.2 (C-5), 159.1 (C-8a), 151.0 (C-5′), 147.1 (C-4′), 123.7 (C-1′), 120.3 (C-2′), 116.8 (C-3′), 114.1 (C-6′), 105.3 (C-4a), 103.9 (C-3), 100.1 (C-6), 95.0 (C-8).
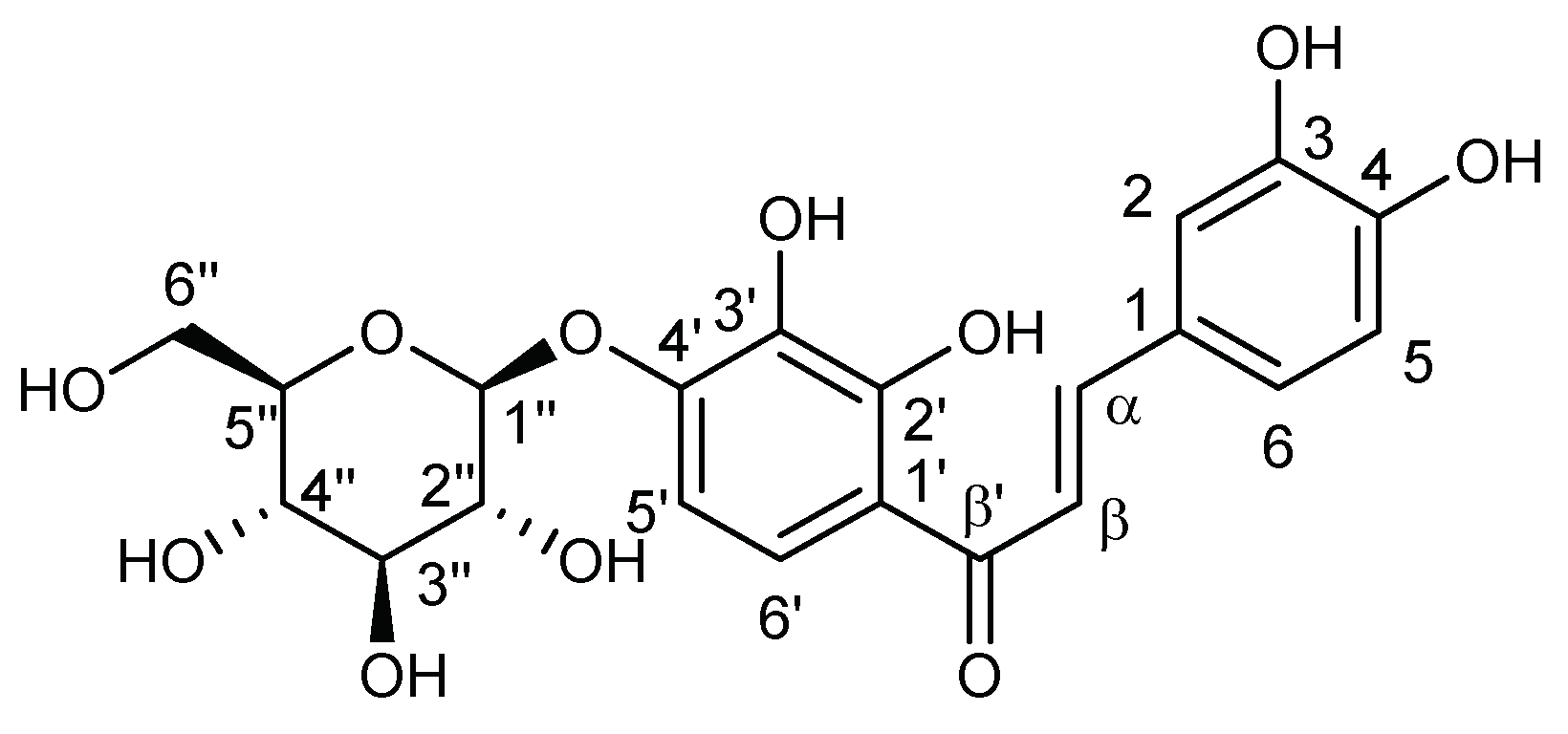
Chromatography on Sephadex LH-20 of a portion of the n-butanol sub-extract (FIII) (50 mg), eluted with methanol, yielded seven fractions (FIIIA–FIIIG). Chromatographic separation was monitored by TLC using the flavonoid buffer as mobile phase, and the fractions were pooled according to their chromatographic profile. A major compound (8) was observed in FIIIG. This fraction (14.0 mg) was subjected to polyamide chromatography, eluted with water/methanol mixtures, yielding three sub-fractions (SFIIIG1–SFIIIG3), of which the sub-fraction SFIIIG2 led to obtaining 8 in pure form (9.3 mg, 66.4%). Compound 8 was identified as marein by NMR spectrometry, in comparison to their spectroscopic data within the literature [31].
Marein (8): 1H-NMR (300 MHz, MeOD-d4): δ (ppm) 7.77 (d, 1H, J = 15.0 Hz, H-β), 7.63 (d, 1H, J = 9.0 Hz, H-6′), 7.56 (d, 1H, J = 15.0 Hz, H-α), 7.19 (d, 1H, J =1.8 Hz, H-2), 7.12 (dd, 1H, J = 8.3 Hz, 1.8 Hz, H-6), 6.84 (d, 1H, J = 9.0 Hz, H-5′), 6.82 (d, 1H, J = 8.3 Hz, H-5), 4.98 (d, 1H, J = 7.4 Hz, H-1″), 3.90 (dd, 1H, J = 12.0 Hz, 1.3 Hz, H-6″), 3.72 (dd, 1H, J = 12.0 Hz, 5.3 Hz, H-5″), 3.50 (m, 3H, H-2″, H-3″ and H-4″), 3.47 (dd, 1H, J = 12.0 Hz, 5.3 Hz, H-6″). 13C-NMR (75 MHz, MeOD-d4): δ (ppm) 194.6 (C-β′), 153.8 (C-2′), 151.7 (C-4′), 150.2 (C-4), 147.0 (C-α), 146.9 (C-3), 135.8 (C-3′), 128.3 (C-1), 124.0 (C-6), 122.6 (C-6′), 118.1 (C-β), 117.4 (C-1′), 116.6 (C-5), 115.9 (C-2), 108.1 (C-5′), 102.6 (C-1″), 78.5 (C-5″), 77.5 (C-4″), 74.7(C-2″), 71.3 (C-3″), 62.4 (C-6″).
2.4. Inhibition Assay on AChE and BChE In Vitro
Electric eel (Torpedo californica) AChE and horse serum BChE were used as source of both the cholinesterases. AChE and BChE inhibiting activities were measured in vitro by the spectrophotometric method developed by Ellman with slight modification [32]. The lyophilized enzyme, 500U AChE/300U BChE, was prepared in buffer A (8 mM K2HPO4, 2.3 mM NaH2PO4) to obtain 5/3 U/mL stock solution. Further enzyme dilution was carried out with buffer B (8mM K2HPO4, 2.3 mM NaH2PO4, 0.15 M NaCl, 0.05% Tween 20, pH 7.6) to produce 0.126/0.06 U/mL enzyme solution. Samples were dissolved in buffer B with 2.5% of MeOH as cosolvent. Enzyme solution (300 μL) and sample solution (300 μL) were mixed in a test tube and incubated for 60/120 min at room temperature. The reaction was started by adding 600 μL of the substrate solution (0.5 mM DTNB, 0.6 mM ATCI/BTCI, 0.1 M Na2HPO4, pH 7.5). The absorbance was read at 405 nm for 180 s at 27ºC. Enzyme activity was calculated by comparing reaction rates for the sample to the blank. All the reactions were performed in triplicate. IC50 values were determined with GraphPad Prism 5. Tacrine (99%) was used as reference AChE/BChE inhibitor.
2.5. Antioxidant Activity
The percentage of antioxidant activity (AA%) of each substance was assessed by DPPH free radical assay. The measurement of the DPPH radical scavenging activity was performed according to the methodology described by Brand-Williams et al. [33]. The samples were reacted with the stable DPPH radical in an ethanol solution. The reaction mixture consisted of adding 158 µL of the sample, 945 µL of ethanol and 95 µL of DPPH radical solution 0.5 mM in ethanol. When DPPH reacts with an antioxidant compound, which can donate hydrogen, it is reduced. The changes in colour (from deep violet to light yellow) were read [Absorbance (Abs)] at 517 nm after 100 min of reaction using a UV-VIS spectrophotometer. The mixture of ethanol (1040 µL) and sample (158 µL) serve as blank. The control solution was prepared by mixing ethanol (1100 µL) and DPPH radical solution (95 µL). The scavenging activity percentage (AA%) was determined according to:
AA% = 100 − {[(Abs sample-Abs blank) × 100]/Abs control}.
The Abs sample is the absorbance of the sample, Abs blank is the absorbance of the blank, and Abs control is the absorbance of the control. Trolox was used as a positive control. The IC50 values were obtained through extrapolation from linear regression analysis and denoted the concentration of sample required to scavenge 50% of DPPH radicals. All experiments were repeated at least three times.
3. Results and Discussion
3.1. Phytochemical Investigation of T. megapotamicum
Taking into account the scarce phytochemical information on T. megapotamicum available in the literature, and following the objectives proposed for this work, elucidate the structure of isolated major metabolites and evaluate them as potential inhibitors of cholinesterase enzymes and antioxidant agents, the phytochemical investigation from the fractionation of the ethanol extract of the aerial parts of T. megapotamicum was carried out (Scheme 1).
3.1.1. Chloroform Sub-Extract Composition
As described in the Materials and Methods sections, FIA was the first fraction obtained from the chloroform sub-extract. The analysis by GC-MS allowed identifying each volatile compound present in this fraction by comparing its mass spectrum with those of the database. Thus, were identified eugenol, 6,10,14-trimethyl-2-pentadecanone, methyl hexadecanoate, octyl acetate, and methyl linoleate. These secondary metabolites are well known and widely studied. They are reported for the first time as constituents of this species. Eugenol is a phenylpropene that belongs to the group of phenylpropanoids. This metabolite is one of the major constituents of essential oils obtained from plant species that belong to different families of plants [34].
FIB fraction displayed a major stain by TLC, therefore was analysed by GC-MS in the same analytical conditions used for the FIA analysis. The chromatogram revealed the presence of a major component (1), but its mass spectrum did not match with those stored in the MS database. Then, the compound was isolated to elucidate its structure. FIB was subjected to flash chromatography yielding three sub-fractions (SFIB1–SFIB3), of which the sub-fraction SFIB2 led to obtaining of 1 in pure form, and was identified as 1′-S-isobutyroxyeugenol isobutyrate (1) (Figure 1).
The structure was confirmed by NMR and GC-MS spectrometry. 1H-NMR and 13C-NMR data were in agreement with previously reported findings [27]. The mass spectrum of 1 showed an ion at m/z 320 which was assigned as the molecular ion for being the ion of greater m/z. The most important fragmentation corresponded to the successive loss of two fragments of 70 m/z to give m/z 250 (37%), and m/z 180 (100%) ions, respectively. The 13C NMR spectrum showed 18 signals, 10 of them corresponding to sp2 carbons at δC 151.28, 139.84, 137.86, 136.35, 122.86, 119.54, 117.00 and 111.40 ppm, and two of them to carbonylic carbons at δC 175.33 and 175.05 ppm; one signal of carbon bonded to an electronegative atom at δC 75.46 ppm; one signal of a methoxy group carbon at δC 56.05 ppm; two methynic carbons signals at δC 34.31 and 34.12 ppm and four methylic carbons signals at δC 19.17, 19.17, 19.06 and 19.06.
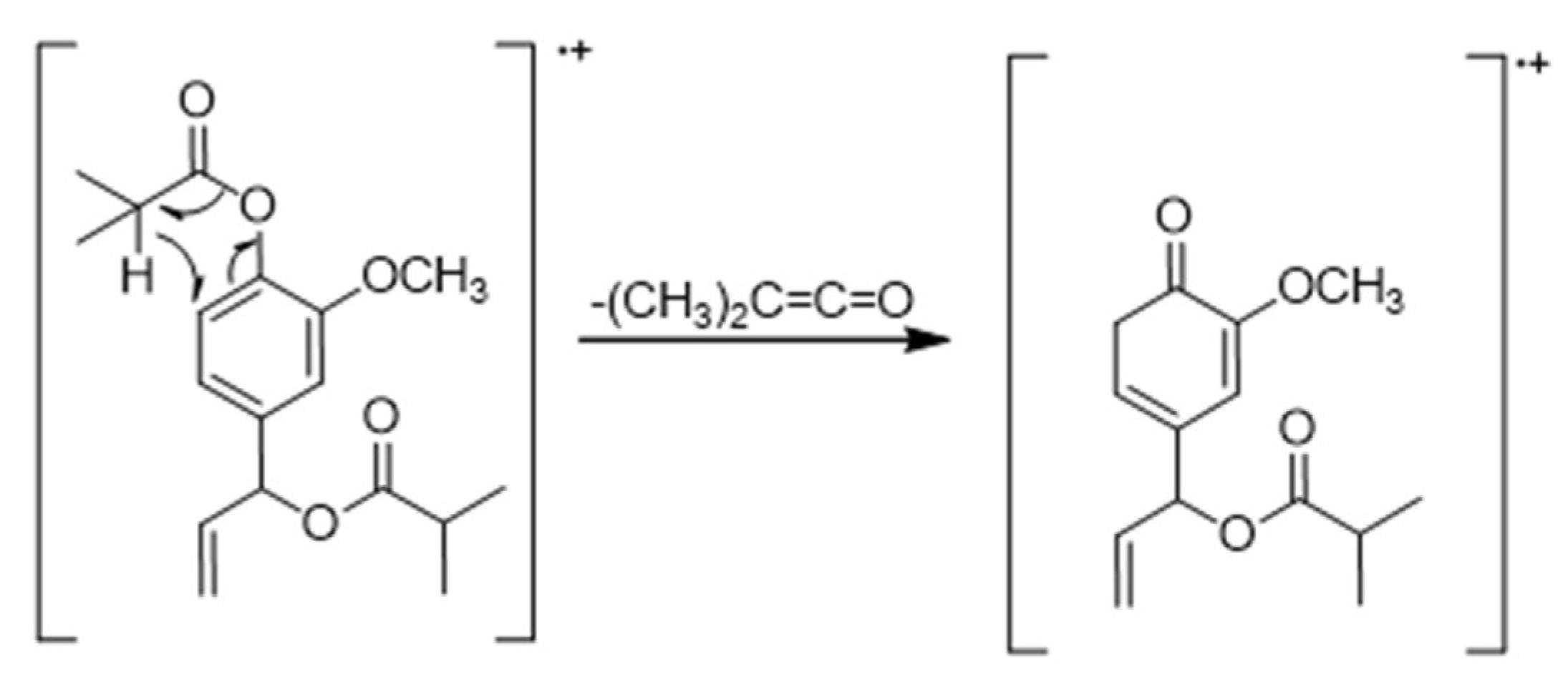
1H NMR displayed characteristic signals. A singlet of area 3 at δH 3.81 ppm, correlated with carbon signal at δC 56.05 ppm in the HSQC spectrum. The presence of a methoxy group was confirmed. In the aromatic region, a proton signal at δH 6.99 ppm was observed, correlated with δC 122.86 ppm and another signal corresponded with two protons at δH 6.93 ppm, correlated with δC 119.54, and 111.40 ppm, respectively, in the HSQC spectrum. A trisubstituted aromatic ring was confirmed. A spin system compatible with olefinic protons was observed: two doublet of doublet of doublets at δH 5.30 ppm (J = 17.0 Hz, 2.5 Hz, 1.2 Hz) and δH 5.27 ppm (J = 10.5 Hz, 1.2 Hz 2.5 Hz) and another doublet of doublet of doublets at δH 5.98 ppm (J= 17.0 Hz, 10.5Hz, 5.8 Hz) correlated with δC 117.00 and δC 136.35 ppm respectively, in HSQC spectrum compatible with a terminal alkene. Two multiples of area 1 at δH 2.83 and 2.62 ppm, and two doublets of area 6 each at δH 1.32 and 1.20 ppm, compatible with two spin-spin coupling systems between two equivalent methyl hydrogens and one methynic proton, correlated with δC 175.33 y, 175.05 ppm, respectively, in the HMBC spectrum, typical of two isobutyrate groups, were observed. This observation has been confirmed by the mass spectrum because the two successive fragments loss of m/z 70 from molecular ion, correspond to the loss of two ketenes formed from the two isobutyrate groups (Figure 2).
As can be seen in Figure 1, the C1’ in compound 1 is asymmetric. Previously, Mitsui et al. were able to assign the absolute configuration (S) to the single asymmetric carbon of a natural eugenol derivative similar to compound 1, with two acetate groups instead of two isobutyrate groups [35]. Although the spectroscopic data obtained from compound 1 were not sufficient to determine the absolute configuration of the asymmetric carbon, it is likely that as it is a biosynthesized natural product it has the same configuration.
The fraction FIC was subjected to flash chromatography. Two pure compounds, 2 and 3, were obtained from SFIC1, and SFIC3 sub-fractions, respectively. The GC-MS analysis of 2 allowed to identify the structure as lupeol (Figure 3). This result was confirmed by comparison of spectroscopic data obtained for this triterpene lupine with reported in the literature [28]. Lupeol had not been previously reported in this species, however, it was found in other plants of this family [36].
SFIC3 sub-fraction was injected in GC-MS, the chromatogram revealed the presence of a compound (3, tR = 10.81 min, 96.6%), and its mass spectrum did not match with those stored in the MS database. From its mass spectrum was found a m/z 292 (4%) molecular ion and the loss of two successive fragments were observed, of m/z 70 and m/z 42, giving a base peak of m/z 180. Both the base peak and the m/z ions originated by fragmentation of this major ion coincided with those observed for compound 1, thus, compound 3 could also correspond to a eugenol derivative. The loss of the two observed fragments, from the molecular ion (m/z 292), corresponds to the loss of one ketene formed from an isobutyrate group (m/z 70), and to the loss of one ketene formed from an acetate group (m/z 42). 1H NMR spectrum was close similar to compound 1. The only difference was the lack of the signals of one isobutyrate group and the present of a signal of an acetate group at δH 2.11ppm correlated by HMBC with a carbonyl carbon at δC 170.05 ppm. Compound 3 was identified as 1′-S-acetoxyeugenol isobutyrate and its structure was confirmed through the data reported in the literature [27] (Figure 4).
FID fraction was analysed by GC-MS in the same analytical conditions used for the FIA analysis. The chromatogram revealed the presence of two major compounds: 4 (tR = 20.64 min, 61.1%) and 5 (tR = 21.66 min, 38.8%). They were identified as stigmasterol (4) and β-sitosterol (5) by comparing its mass spectrums with those of the database. As mentioned in the Materials and Methods, the separation of both compounds was not achieved. By 1H and 13C NMR it was also possible to confirm the presence of the two phytosterols. Previous works have reported that these phytosterols are not efficient inhibitors of AChE and BChE enzymes [37].
3.1.2. Ethyl Acetate Sub-Extract Composition
Previous our phytochemical investigation, only one study of material from Northern Arizona, United States, has been reported, where three flavonoids (luteolin, luteolin 7-O-glucoside, and marein) present in the ethyl acetate sub-extract were identified [26].
Chromatography on silica gel of the ethyl acetate sub-extract (FII), yielded eight fractions: FIIA-FIIH. Two major compounds 6 and 7, were observed in FIIB and FIIG fractions, respectively, easily seen for their stain colour in the plate. FIIB fraction was subjected to flash chromatography, yielding three sub-fractions (SFIIB1–SFIIB3), of which the sub-fraction SFIIB3 led to obtaining of 6 in pure form. Compound 6 was identified by NMR spectrometry. 1H NMR spectrum displayed signals of an AX2 spin system according to a doublet of doublets at δH 5.25 ppm (J = 3.0 Hz, 12.6 Hz) assigned to an oxomethynic proton (H-2) that couples with two doublet of doublets a δH 2.66 ppm (J = 3.0, 17.4 Hz) and 2.88 pm (J = 12.6, 17.4 Hz) corresponding to methylenic protons, H-3a y H-3b, respectively. Five signals were observed in the aromatic region, two of them in an AB spin system (δH 5.87 ppm and 5.85 pm, J = 2.1 Hz) corresponding to two aromatic protons, H-6 and H-8, in meta-position; a singlet (δH 7.01 ppm, H-2’) and two doublets (δH 6.88 ppm and 6.75 ppm, J = 7.5) corresponding to H-6′ and H-5′, what did they indicate a 1′,3′,4′- trisubstituted ring. This evidence suggests a flavanone structure for compound 6, with two aromatic rings, one of them tetrasubstituted and another trisubstituted and four hydroxy groups bonded to the aromatic rings. In 13C NMR and DEPT experiments were observed 15 carbons signals, including one methylene carbon, six methynics carbons, and eight quaternary carbons. Given this data, and in comparison with previous reports in the literature, compound 6 was identified as 5,7,3’,4’-tetrahidroxyflavanone, known as eriodyctiol [29] (Figure 5). This flavonoid was not reported before in this species.
3.1.3. N-Butanol Sub-Extract Composition
Chromatography on Sephadex LH-20 of the n-butanol sub-extract (FIII) yielded seven fractions (FIIIA–FIIIG). A major compound (8) was observed in FIIIG. This fraction was subjected to polyamide chromatography yielding three sub-fractions (SFIIIG1–SFIIIG3), of which the sub-fraction SFIIIG2 led to obtaining 8 in pure form. Compound 8 was identified by NMR spectrometry, showing characteristic signals of sugars and a flavonoid skeleton, confirming the presence of a glycoside chalcone. The 1H NMR displayed two AB spin system. One of them, formed by two doublets at δH 7.77 ppm (J = 15.0 Hz) and 7.56 ppm (J = 15.0 Hz) is attributable to the olefinic protons H-α and H-β of the system C3 of the chalcone. The other system, formed by the signals at δH 7.63 ppm (J = 9.0 Hz) and 6.84 ppm (J = 9.0 Hz), of area one each, indicating the presence of two aromatic protons in ortho position, corresponding to the H-6’ and H-5’ of the A ring of the chalcone skeleton. Signals for three aromatic protons as two doublets at δH 7.19 ppm (J = 1.8 Hz, H-2) and 6.82 ppm (J = 8.3 Hz, H-5), and a double doublet at δH 7.12 ppm (J = 8.3 Hz, 1.8 Hz, H-6), indicated a tri-substituted B ring. Furthermore, a doublet belonging to a proton of an anomeric carbon at δH 4.98 ppm (J = 7.4 Hz, H-1’’) together with a set of signals between δH 3.4 and 3.8 ppm were observed. The above data were confirmed by 13C NMR and HSQC experiments, in which signals for twenty-one carbon atoms, including one methylenic carbon, twelve methylene carbons, and eight quaternary carbons were observed. The set of these signals confirmed the presence in the molecule of two aromatic rings and an open C3 system, characteristic of a chalcone, highlighting in its 13C NMR the signal corresponding to a carbonyl group at δc 194.6 ppm, and two olefinic carbons at δc 148.0 pm (C-α) and 118.1 pm (C-β). These data are in agreement with the structure of a glycoside chalcone, which has two aromatic rings, one tetra-substituted and the other trisubstituted, four hydroxyl groups attached to the two aromatic rings, a carbonyl group, and glucose bound to C-4’. Compound 8 was identified as marein (Figure 7) [31].
3.2. Inhibition Assay on AChE and BChE In Vitro and Antioxidant Capacity of the Sub-Extracts and of the Major Metabolites
Thelesperma megapotamicum (Spreng.) Kuntze (Asteraceae) is a species native to Argentina. It is commonly known as Indian tea or pampas tea and has been used in folk medicine as a digestive and antispasmodic agent [24]. Recent studies show its high antioxidant capacity, which is attributed to the total content of polyphenols present in the plant [25]. The phytochemical investigation from the fractionation of the ethanol extract of the aerial parts of T. megapotamicum, allowed to identify two phenylpropanoids (1 and 3), a triterpenoid (2), two phytosterols (4 and 5), and three flavonoids (6–8), as major secondary metabolites. Taking into account that polyphenols are attracting increasing attention due to the discovery of useful agents for the treatment of neurodegenerative diseases, studies were conducted into the neuroprotective activity of this species. These studies evaluated the inhibition in vitro of AChE and BChE, and the antioxidant capacity of the sub-extracts and of the major metabolites isolated from them. The AChE and BChE inhibition were determined by Ellman’s method and the antioxidant activity was evaluated through its ability as a free radical scavenger against DPPH.
The inhibitory activity of AChE and BChE, enzymes implicated in AD, was evaluated for the first time in this species. Sub-extracts showed no inhibition against AChE. The chloroform and n-butanol sub-extracts were active against BChE (Table 1). This result is interesting because BChE can delay the onset and decrease the rate of Aβ fibril formation in vitro, a central event in the pathogenesis of AD [38,39]. Phenylpropanoids 1 and 3, and the flavonoid 8, isolated from these sub-extracts, exhibited the highest activity against this enzyme, with IC50 values of 46.4 ± 1.2 μM, 59.0 ± 2.1 μM, and 51.6 ± 2.3 μM, respectively.
The ethyl acetate and n-butanol sub-extracts showed an interesting antioxidant activity (Table 1). Flavonoids 6, 7 and 8, obtained from these sub-extracts, were very active, with IC50 values of 17.9 ± 1.2 μM, 19.1 ± 2.8 μM, and 78.6 ± 1.7 μM, respectively.
It is concluded that the polyphenols (compounds 1, 3, 6–8) present in T. megapotamicum may be responsible for the in vitro inhibition against BChE, and for the antioxidant activity.
4. Conclusions
In summary, the phytochemical investigation from the fractionation of the ethanol extract of the aerial parts of T. megapotamicum, identified thirteen secondary metabolites, of which only two (7 and 8) had been reported for this species. Six of these compounds (1–3 and 6–8) were obtained in pure form. All the structures were identified from spectroscopic methods and consequently compared with literature data.
The neuroprotective activity of this species was studied by evaluating the inhibition in vitro of AChE and BChE, and antioxidant capacity of the sub-extracts and of the major metabolites isolated from them. The chloroform and n-butanol sub-extracts were the most active against BChE.
Phenylpropanoids 1 and 3, and the flavonoid 8, isolated from these sub-extracts, exhibited the highest activity against this enzyme. The ethyl acetate and n-butanol sub-extracts showed an interesting antioxidant activity. Flavonoids 6, 7 and 8, obtained from these sub-extracts, were very active.
The inhibitory activity against BChE and the antioxidant capacity of the polyphenols present in T. megapotamicum highlight this species as a promising source of active metabolites for the development of agents for the treatment of neurodegenerative diseases.
Funding
This research was funded by Universidad Nacional del Sur (UNS), grant number: PGI 24/Q105 and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), grant number: PICT-2017-1443 to MBF.
Acknowledgments
This work was supported by Universidad Nacional del Sur (UNS), grant number: PGI 24/Q105 and Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), grant number: PICT-2017-1443 to MBF. MBF is research member of CIC. BAS thanks the CONICET for a doctoral fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rao, A.V.; Balachandran, B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr. Neurosci. 2020, 5, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izaokovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Wen, L.L.; Huang, Y.N.; Chen, Y.T.; Ku, M.C. Dual effects of antioxidants in neurodegeneration: Direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr. Pharm. Des. 2006, 12, 3521–3533. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar] [CrossRef]
- Linseman, D.A. Targeting oxidative stress for neuroprotection. Antioxid Redox Signal. 2009, 11, 421–423. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Reynolds, A.; Laurie, C.; Mosley, R.L.; Gendelman, H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 297–325. [Google Scholar] [CrossRef]
- Maestri, D.M.; Nepote, V.; Lamarque, A.L.; Zygadlo, J.A. Natural products as antioxidants. In Phytochemistry: Advances in Research, 1st ed.; Imperato, F., Ed.; Research Signpost: Kerala, India, 2006; pp. 105–135. [Google Scholar]
- Mandel, S.A.; Amit, T.; Weinreb, O.; Reznichenko, L.; Youdim, M.B. Simultaneous manipulation of multiple brain targets by green tea catechins: A potential neuroprotective strategy for Alzheimer and Parkinson diseases. CNS Neurosci. Ther. 2008, 14, 352–365. [Google Scholar] [CrossRef]
- Mandel, S.A.; Amit, T.; Kalfon, L.; Reznichenko, L.; Weinreb, O.; Youdim, M.B. Cell signaling pathways and iron chelation in the neurorestorative activity of green tea polyphenols: Special reference to epigallocatechin gallate (EGCG). J. Alzheimers Dis. 2008, 15, 211–222. [Google Scholar] [CrossRef]
- Avramovich-Tirosh, Y.; Reznichenko, L.; Amit, T.; Zheng, H.; Fridkin, M.; Weinreb, O.; Mandel, S.; Youdim, M.B. Neurorescue activity, APP regulation and amyloid-b peptide reduction by novel multi-functional brain permeable iron- chelating- antioxidants, M-30 and green tea polyphenol, EGCG. Curr. Alzheimer Res. 2007, 4, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective–neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, L.; Amit, T.; Zheng, H.; Avramovich-Tirosh, Y.; Youdim, M.B.H.; Weinreb, O.; Mandel, S. Reduction of iron-regulated amyloid precursor protein and b-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: Implications for iron chelation in Alzheimer’s disease. J. Neurochem. 2006, 97, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Mandel, S.; Amit, T.; Youdim, M.B. Neurological mechanisms of green tea polyphenols in Alzheimer‘s and Parkinson’s diseases. J. Nutr. Biochem. 2004, 15, 506–516. [Google Scholar] [CrossRef]
- Levites, Y.; Amit, T.; Mandel, S.; Youdim, M.B. Neuroprotection and neurorescue against Ab toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. FASEB J. 2003, 17, 952–954. [Google Scholar] [CrossRef]
- Mancuso, C.; Bates, T.E.; Butterfield, D.A.; Calafato, S.; Cornelius, C.; Lorenzo, A.D.; Dinkova Kostova, A.T.; Calabrese, V. Natural antioxidants in Alzheimer’s disease. Expert Opin. Investig. Drugs 2007, 16, 1921–1931. [Google Scholar] [CrossRef]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Botanical phenolics and brain health. Neuromol. Med. 2008, 10, 259–274. [Google Scholar] [CrossRef]
- Schroeter, H.; Spencer, J.P.E.; Rice-Evans, C.; Williams, R.J. Flavonoids protect neurons from oxidized low-density-lipoprotein-induced apoptosis involving c-Jun N-terminal kinase (JNK), c-Jun and caspase-3. Biochem. J. 2001, 358, 547. [Google Scholar] [CrossRef]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef]
- Uriarte-Pueyo, I.; Calvo, M. Flavonoids as acetylcholinesterase inhibitors. Curr. Med. Chem. 2001, 18, 5289. [Google Scholar] [CrossRef]
- Qingbo, L.; Jie, W.; Bin, L.; Zhuo-Yang, C.; Ming, B.; Shaochun, S.; Xiao-Xiao, H.; Shao-Jiang, S. Phenylpropanoids and lignans from Prunus tomentosa seeds as efficient β-amyloid (Aβ) aggregation inhibitors. Bioorg. Chem. 2019, 84, 269–275. [Google Scholar] [CrossRef]
- Cantero, J.J.; Nuñez, C.O. Las Plantas Medicinales del sur de la Provincia de Córdoba, 1st ed.; Fundación Universidad Nacional de Río Cuarto: Río Cuarto, Argentina, 2000. [Google Scholar]
- Borneo, R.; Leon, A.E.; Aguirre, A.; Ribotta, P.; Cantero, J.J. Antioxidant capacity of medicinal plants from the Province of Cordoba (Argentina) and their in vitro testing in a model food system. Food Chem. 2009, 112, 664–670. [Google Scholar] [CrossRef]
- Ateya, A.M.; Okarter, T.U.; Knapp, J.E.; Schiff, P.L.; Slatkin, D.J. Flavonoids of Thelesperma megapotanicum. Planta Med. 1982, 45, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Thron, U.; Martin, R.; Reichling, J. Rareeugenol and Z-coniferyl alcohol derivatives in roots of three Coreopsis species. Z. Nat. Schung C 1989, 44, 7–11. [Google Scholar]
- Vela Gurovic, M.S.; Castro, M.J.; Richmond, V.; Faraoni, M.B.; Maier, M.S.; Murray, A.P. Triterpenoids with Acetylcholinesterase Inhibition from Chuquiraga erinacea D. Don. subsp. erinacea (Asteraceae). Planta Med. 2010, 76, 607–610. [Google Scholar] [CrossRef]
- Da Silva, L.A.L.; Faqueti, L.G.; Reginatto, F.H.; dos Santos, A.D.C.; Barison, A.; Biavatti, M.W. Phytochemical analysisof Vernonanthura tweedieana and a validated UPLC-PDA method for the quantification of eriodictyol. Rev. Bras. Farmacogn. 2015, 25, 375–381. [Google Scholar] [CrossRef]
- Nissler, L.; Gebhardt, R.; Berger, S. Flavonoid binding to a multi-drug-resistance transporter protein: An STD-NMR study. Anal. Bioanal. Chem. 2004, 379, 1045. [Google Scholar] [CrossRef]
- Redl, K.; Davis, B.; Bauer, R. Chalcone glycosides from Bidens campylotheca. Phytochemistry 1992, 32, 218–220. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. Lebenson Wiss Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Erdogan Orhan, I.; Kartal, M.; Kan, Y.; Sener, B. Activity of essential oils and individual components against acetyl and butyrylcholinesterase. Z. Nat. 2008, 63, 547–553. [Google Scholar] [CrossRef]
- Mitsui, S.; Kobayashi, S.; Nagahori, N.; Ogiso, A. Constituents from seeds of Alpinia galanga WILD and their anti-ulcer activities. Chem. Pharm. Bull. 1976, 24, 2377–2382. [Google Scholar] [CrossRef] [PubMed]
- Lajter, I. Biologically Active Secondary Metabolites from Asteraceae and Polygonaceae Species. Ph.D. Thesis, University of Szeged, Szeged, Hungary, 2015. [CrossRef]
- Gade, S.; Rajamanikyam, M.; Vadlapudi, V.; Madhav Nukala, K.; Aluvala, R.; Giddigari, C.; Jyothi Karanam, N.C.; Barua, N.; Pandey, R.; Saradhi, V.; et al. Acetylcholinesterase inhibitory activity of stigmasterol & hexacosanol is responsible for larvicidal and repellent properties of Chromolaena odorata. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1861, 541–550. [Google Scholar] [CrossRef]
- Podoly, E.Y.; Bruck, T.; Diamant, S.; Melamed-Book, N.; Weiss, A.; Huang, Y.; Livnah, O.; Langermann, S.; Wilgus, H.; Soreq, H. The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. Neurodegener. Dis. 2008, 5, 232. [Google Scholar] [CrossRef]
- Diamant, S.Z.; Podoly, E.; Friedler, A.; Ligumsky, H.; Livnah, O.; Soreq, H. Alzheimer’s disease and type 2 diabetes mellitus: The cholinesterase connection? Proc. Natl. Acad. Sci. USA 2006, 103, 8628. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Fractionation of the EE of T. megapotamicum. VC: volatile compounds.

Figure 1.
Structure of 1′-S-isobutyroxyeugenol isobutyrate (1).

Figure 2.
Loss of a neutral ketene from an isobutyrate group.

Figure 3.
Structure of lupeol (2).

Figure 4.
Structure of 1′-S-acetoxyeugenol isobutyrate (3).

Figure 5.
Structure of eriodyctiol (6).

Figure 6.
Structure of luteolin (7).

Figure 7.
Structure of marein (8).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
AChE, BChE inhibition and antioxidant activities of the sub-extracts.
| Sub-Extracts | IC50 AChE (μg/mL) | IC50 BChE (μg/mL) | IC50 DPPH (μg/mL) |
|---|---|---|---|
| Chloroform (FI) | 562.7 ± 8.0 | 30.7 ± 1.5 | >200 |
| Ethyl acetate (FII) | 168.0 ± 6.0 | 505.8 ± 9.4 | 19.6 ± 0.7 |
| n-butanol (FIII) | 337.1 ± 7.2 | 60.3 ± 2.3 | 49.5 ± 2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Siben, B.A.; Castro, M.J.; Faraoni, M.B. Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities. Chem. Proc. 2021, 3, 122. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08393
AMA Style
Siben BA, Castro MJ, Faraoni MB. Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities. Chemistry Proceedings. 2021; 3(1):122. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08393
Chicago/Turabian StyleSiben, Braian Alberto, María Julia Castro, and María Belén Faraoni. 2021. "Polyphenols from Thelesperma megapotamicum and Their Antioxidant and Neuroprotective Activities" Chemistry Proceedings 3, no. 1: 122. https://0-doi-org.brum.beds.ac.uk/10.3390/ecsoc-24-08393