Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer


Department of General, Visceral and Pediatric Surgery, University Medical Center Göttingen, 37075 Göttingen, Germany
*
Authors to whom correspondence should be addressed.
Epigenomes 2018, 2(2), 8; https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2020008
Submission received: 7 May 2018
/
Revised: 25 May 2018
/
Accepted: 28 May 2018
/
Published: 31 May 2018
(This article belongs to the Special Issue Epigenetics of Pancreatic Cancer)
{kind=link}
{kind=link}
Abstract
:While the mortality rates of cancer are generally declining, pancreatic cancer persists to be an exception with a 5-year-survival rate of less than 7%. Late diagnosis and resistance to conventional therapies contribute to high mortality rates in spite of the remarkable recent advances in cancer management and research. Consequently, there is an urgent need to find new and unconventional therapeutic targets to improve prognosis and survival of pancreatic cancer patients. In this review, we discuss the transcriptional effects of the most widely used epigenetic inhibitors in pancreatic cancer focusing on Bromodomain and Extraterminal domain (BET) and Histone Deacetylase (HDAC) inhibitors, which are currently highly promising therapeutic options. We suggest that these inhibitors can be better utilized at lower doses which exploit their transcriptional modulatory effects on pancreatic cancer transcriptional programs directed by specific factors such as MYC and Forkhead Box A1 (FOXA1), rather than simply based on their anti-proliferative effects. This approach can potentially help avoid the intolerable adverse events frequently elicited by the use of these treatments at higher doses. In particular, we underscore the crucial role of distal regulatory elements in mediating the specific effects of these epigenetic inhibitors and propose using them in a more selective and prudent manner.
1. Introduction
While the mortality rates of cancer are generally declining, pancreatic cancer persists to be an exception with a 5-year-survival rate of less than 7% [1,2]. Late diagnosis and resistance to conventional therapies contribute to high mortality rates in spite of the remarkable recent advances in cancer management and research [3]. Consequently, there is an urgent need to find new and unconventional therapeutic targets to improve prognosis and survival of pancreatic cancer patients.
In addition to various genomic mutations, such as KRAS and TP53, that play a role in the pathophysiology of pancreatic cancer, other mutations and signaling pathways play an equally important role by affecting transcription of entire subsets of genes, irrespective of genomic sequence [2,3,4,5,6,7,8]. Epigenetic pathways affect transcription either via modulation of histone modifications which can be activating or silencing, DNA methylation-mediated silencing, non-coding RNAs, and alteration of chromatin accessibility [9]. This meshwork provides the cells with various tools that can dramatically affect its transcriptome without the need to induce any irreversible changes at the level of the genome. Moreover, in contrast to the permanent and largely “all or nothing” effects of genomic mutations, modulation of the epigenome allows for more subtle, reversible changes in genome regulation.
Epigenetics represents a promising target in pancreatic cancer for various reasons. Firstly, epigenetic modifications are mainly mediated by enzymes and proteins whose activity can (at least in principle) be targeted by small molecule inhibitors. Secondly, many epigenetic pathways were found to be deregulated in pancreatic cancer, suggesting a crucial role of epigenetic regulation in this malignancy [4,5,6,7]. Additionally, pancreatic cancer, among others, was found to be addicted to the activation of aberrant transcriptional programs that not only drive the development and progression of cancer, but are also crucial for the maintenance of detrimental malignant characteristics such as metastasis and chemoresistance [10,11]. Epigenetic pathways were found to be major drivers of the perturbations of such programs in a highly intricate and context-specific manner [12]. Due to its tremendous pliability, epigenetic modulation provides an optimal tool to be hijacked in cancer development and progression creating a specific dependence of cancer cells on these pathways. Accordingly, many epigenetic inhibitors are currently under investigation for the treatment of numerous malignancies, including pancreatic cancer [3,13,14,15]. However, results from these studies so far have been unexpectedly modest and, in some cases, these inhibitors were associated with intolerable toxicities. Many of these studies use epigenetic inhibitors, whether alone or in combination, in a method akin to other more conventional drugs with defined targets such as chemotherapy and monoclonal antibodies. On the other hand, epigenetic inhibitors target transcriptional regulation in a complex and unconventional manner and this should be taken into consideration when investigating these drugs.
Numerous reviews have deeply discussed and extensively summarized the recent advances of targeting epigenetics in cancer, in general, as well as in pancreatic cancer in particular [3,9,16,17,18,19,20,21]. In this review, we focus on the transcriptional mechanisms of the most widely used epigenetic inhibitors in pancreatic cancer focusing on Bromodomain and Extraterminal (BET) and Histone Deacetylase (HDAC) inhibitors, which represent promising therapeutic options. We suggest that these inhibitors can be better utilized for their transcriptional modulation, rather than solely on their anti-proliferative effects, which can lead to intolerable adverse events. Moreover, we underscore the crucial role of distal regulatory elements in mediating the specific effects of these epigenetic inhibitors and propose using them in a more selective and prudent manner.
2. Bromodomain and Extraterminal Inhibitors in Pancreatic Cancer
The BET family of proteins consists of BRD2, BRD3, BRD4, and the testis-specific BRDT [22]. All BET family members contain two conserved bromodomains which enable them to recognize acetylation marks on the chromatin, in addition to an extraterminal domain which enables interactions with other proteins [23,24]. The BET family has attracted much attention due to its significant role in gene transcription regulation in addition to its implication in the development of the particularly aggressive Nuclear protein in Testis (NUT) midline carcinoma, which is characterized by the presence of a BRD4-NUT fusion gene and arises in the mediastinum, head, and neck of typically young patients [25,26,27,28]. This aberrant fusion gene includes the two bromo- and extraterminal domains of BRD4, in addition to a histone acetyltransferase (HAT) binding domain from the NUT gene. Thus, the fusion gene has the ability to aberrantly activate large chromatin domains [12]. The synthesis of the prototype BET inhibitor JQ1, which competitively binds to the acetylation-recognizing hydrophobic pockets in all BET members, has marked an explosion in the number of studies investigating the role of BET family members in gene transcription regulation due to their promising anti-proliferative effects in different cancer types [26,29]. However, many aspects are still unknown about the role of these factors in driving transcriptional activation and the best way to leverage their context-specific effects.
2.1. Role and Effects of Bromodomain and Extraterminal Inhibitors in Pancreatic Cancer
Interestingly, a general screen for limiting epigenetic regulators in pancreatic cancer identified two members of the BET family, BRD2 and BRD3, as major drivers in pancreatic cancer growth and progression [30]. In pancreatic cancer cell lines, BET inhibition exerts anti-proliferative effects by selectively targeting inflammatory and oncogenic pathways [31]. This effect was also observed even in pancreatic cells with chemotherapy-resistant and highly migratory phenotypes [32]. Concordantly, BET inhibitors were found to decrease tumor growth in patient-derived xenografts by attenuating inflammatory pathways in cancer cells and their associated fibroblasts [30,33,34]. Recently, Andricovich et al. [35] showed that pancreatic cells deficient for the lysine demethylase, KDM6A, are more sensitive to BET inhibition due to the activation of BET-dependent super enhancers. Moreover, the MYC proto-oncogene, perhaps the best known target for BET inhibition, was successfully used to subgroup 55 pancreatic cancer patient-derived xenografts based on its expression and accurately predicted sensitivity to BET inhibition via apoptosis with high expression of MYC correlating with more BET dependence [36].
Thus, it has become very clear that BET inhibitors have a perceptible anti-proliferative effect in pancreatic cancer and display a very promising potential as highly effective and selective therapeutic agents. These inhibitory effects may largely be due to BET members being crucial for driving aberrant transcriptional programs and can be heightened via specific deficiencies which lead to increased transcriptional dependence on BET family members. This supports the current efforts to identify certain subgroups of patients who may be more responsive to BET inhibition. However, it is important to note that the effects of BET inhibition are varied and highly specific for different tumor types and subgroups.
2.2. Bromodomain and Extraterminal Inhibition and Metastasis in Pancreatic Cancer
Metastasis is a major contributing factor to the very poor prognosis of the majority of pancreatic cancer patients [37]. Consequently, therapeutic agents that attenuate and/or prevent metastasis can be of extreme benefit to patients. Recently, it was uncovered that pancreatic mouse organoids from metastatic pancreatic ductal adenocarcinomas show a marked reprogramming in their enhancer landscape compared to organoids originated from normal pancreata, early neoplastic (PanIN) lesions or primary tumors [38]. The same pattern of reprogramming was observed in pancreatic cancer patients [39] and also in other cancer types including osteosarcoma, ependymoma, and rhabdomyosarcoma [40,41,42]. Therefore, it is clear that distal regulatory elements play a significant role in activating metastatic programs in different cancer types. This strongly suggests that targeting enhancers can be a highly efficient approach in managing metastatic pancreatic cancer and potentially preventing metastasis from primary sites.
Interestingly, recent studies implied that BET family members play an important role in modulating gene transcription through regulation of the 3D chromatin structure [43,44,45]. This structure creates specific compartmentalization which enable enhancers to interact with and affect specific target genes [46,47]. Moreover, we have previously shown that BET-dependent genes are not necessarily highly enriched for BRD4, the most studied member of the BET family [48]. Instead, while the genes that were highly affected by JQ1 treatment did not have a defined pattern of occupancy for BRD4 at their respective transcription starting site (TSS), they did display a better correlation with tissue-specific BRD4-occupied enhancers. Concordantly, Cao et al. [49] reported that expression of genes can be predicted in part by the activity of their enhancers alone. While this phenomena was observed in other systems, it is highly probable that gene dependence follows the same specificity paradigms in various contexts [50]. Indeed, BET inhibitors were observed to exert marked anti-proliferative effects in metastatic melanoma via deactivating the super enhancer of the oncogenic Adhesion Molecule With Ig Like Domain 2 (AMIGO2) [51]. The BET-dependent super enhancer activating Aldehyde Dehydrogenase 2 Family (ALDH), which promotes resistance to chemotherapy and disease recurrence, was identified as a promising target in ovarian cancer [52]. In the highly metastatic Merkel cell carcinoma, BET inhibition is highly effective as it downregulates MYC by targeting its putative super enhancer [53]. Altogether, BET inhibitors are strong candidates for treating metastatic pancreatic cancer and can potentially be used as adjuvant therapies to prevent metastasis and disease recurrence if found safe in further clinical studies. These inhibitors in particular can play this unique role because, as previously discussed, reprogramming of distal regulatory elements is a hallmark of metastatic phenotype and these enhancers are frequently particularly sensitive to BET inhibition.
2.3. Effect of Bromodomain and Extraterminal Inhibition on Distal Regulatory Elements
Recent efforts focusing on uncovering the mechanisms by which BET inhibitors affect the proliferation of cancer cells in different systems largely confirmed the implication of distal regulatory elements in mediating the observed effects. Interestingly, a particular subtype of these elements called “super enhancers” are remarkably associated with BET members and can be partly defined by the intensity of BRD4 occupancy at these regions [22,54]. We have previously tested the validity of the super enhancer concept which suggests that enhancers follow the rule of the vital few (the Pareto principle) where a low percentage of regulatory elements are responsible for most effects on the regulation of gene transcription [55]. While we have failed to discern such a pattern, many studies have indeed defined certain dependencies of crucial enhancers, whether “super” or “typical”, on BET members.
In diffuse large B-cell lymphoma, BRD4 was observed to be more preferentially enriched at enhancers. Interestingly, BET-dependent super enhancers of master regulatory transcription factors were correlated with the anti-proliferative effects of JQ1 observed in this system [56]. Metabolic changes promoting proliferation in a specific subgroup of melanoma were reported to occur via upregulation of the BET-dependent super enhancer for Peroxisome Proliferative Activated Receptor-γ (PPARG) Coactivator 1 α (PGC-1α) [57]. In castration-resistant prostate cancer, BRD4 was localized at the BET inhibitor-sensitive enhancer regions of the driver oncogene, Transmembrane Protease, Serine 2 (TMPRSS2) [58,59]. In general, these studies imply that specific BET-dependent enhancers are activated in cancerous cells, rendering them more sensitive to BET inhibition and providing a specific therapeutic target. Intriguingly, recent methods that can detect nascent RNA such as thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq) have shown that the effects of BET inhibition are dose-dependent and that high doses of BET inhibitors can lead to universal pausing of transcription while low concentrations affected specific subsets of genes in leukemia cells [60]. These hypersensitive genes were not necessarily controlled by super enhancers and correlated only in sub-clusters with other transcription factors and co-activators. This underscores the complexity of BET-orchestrated specific gene regulation, which may be associated with specific clusters of enhancers, but not exclusively explained by a “super enhancer” model.
In summary, investigating the efficacy of BET inhibitors in pancreatic cancer is highly justified given the promising anti-proliferative effects seen in different models. Additionally, recent data from pancreatic cancer and other systems identify a general pattern of BET-mediated activation of enhancers, or clusters thereof, that play a major role in driving detrimental aberrant transcriptional programs such as metastasis. However, the mechanism of action of BET inhibitors is still not fully understood and is likely highly complex and context-dependent. Many challenges exist in the investigation of the role of these inhibitors including the limitations of current techniques, measuring the anti-proliferative effects for BET inhibitors as a general read-out for efficacy, disregarding the bromodomain-independent roles of BETs, and the prejudiced focus of research on only one member of the BET family, BRD4. While BRD2 and BRD3 share a high homology with BRD4, their functions and specific roles are not well-defined [61,62]. BRD2 has garnered some attention due to recent reports that it frequently co-localizes with CCCTC-binding Factor (CTCF), an important insulator protein that demarcates transcriptional boundaries [45]. Both BRD2 and BRD3 affect gene transcription through different mechanisms than BRD4, namely by interaction with specific transcription factors such as E2F and GATA binding protein 1 (GATA1), respectively [63,64]. In leukemia, the knockdown of either BRD2 or BRD3 failed to recapitulate the effects of BET inhibition or BRD4 manipulation, implying that they have a lesser role in this system, in contrast to the reported role in pancreatic cancer [30,60]. Adding to the complexity is the proposed various roles of the different bromodomains within each member, namely BD1 and BD2 [65]. New targeting techniques utilizing protein-targeting chimeric molecules (PROTACs) can mark BET proteins for proteasomal degradation and will further shed light on the acetylation-independent effects of BETs [66,67]. A deeper understanding of each of the BET proteins, their bromodomains, as well as extraterminal domains will enable us to use these agents in a safe and effective manner. Generally, BET inhibitors exert promising synergistic effects in pancreatic cancer with other agents, such as chemotherapeutic drugs such as gemcitabine [33] and epigenetic inhibitors such as HDAC inhibitors [68].
3. Histone Deacetylase Inhibitors in Pancreatic Cancer
Histone modifications can be active marks such as acetylation of histone 3 at lysine 27 (H3K27ac) and methylation of histone 3 at lysine 4 (H3K4me1), or repressing marks such as trimethylation of histone 3 at lysine 27 (H3K27me3) or ubiquitination of histone 2A at lysine 119 (H2Aub) [69]. Histone acetylation is a marker associated with transcriptional activation and its manipulation can be beneficial in attenuating detrimental pathways in pancreatic cancer. HDAC inhibitors which inhibit the removal of protein lysine acetylation marks have a broader effect on gene expression [70] in comparison to BET inhibitors. HDAC inhibitors are classified based on their homology to yeast into different classes (Class I-IV) [71]. These classes differ in their domains, expression and effects, and their different roles are still to be clearly defined. HDAC inhibitors differ in their potency and selectivity but mainly show promising anti-proliferative effects in cancer with the main rationale of reactivating silenced tumor suppressor genes and reversing deregulated deacetylation of histones in cancer [72,73].
3.1. Role and Effects of Histone Deacetylase Inhibitors in Pancreatic Cancer
Histone deacetylase inhibitors were observed to induce p53-mediated pro-apoptotic effects in pancreatic cancer cells [74]. Additionally, they selectively inhibited proliferation of pancreatic cancer cells by affecting their aerobic metabolism and rendering them more sensitive to glycolytic inhibition [75,76]. In general, specific HDAC inhibitors that targeted certain HDAC classes showed variable effects with pan-HDAC inhibitors causing the most marked anti-proliferative effects [77]. While HDAC inhibition was found to potentiate the effects of gemcitabine in vitro and was reported to overcome its resistance, a clinical study combining HDAC inhibitors with gemcitabine in pancreatic cancer patients was prematurely terminated because the observed benefits did not outweigh the marked adverse events [78,79,80,81]. HDAC inhibitors were also reported to suppress metastasis as HDACs were described to mediate the repressor action of the Zinc Finger E-Box Binding Homeobox 1 (ZEB1) on the promoter of the known epithelial marker, calcium-dependent adhesion protein-1 (CDH1) [82,83,84]. Interestingly, we have also shown that HDAC inhibition attenuates epithelial-to-mesenchymal transition (EMT) and decreases stem-like properties in pancreatic cancer cells [85]. Given that both BET and HDAC inhibition show anti-proliferative and metastasis-suppressive effects in pancreatic cancer, Mazur et al. [68] combined these two agents in vitro and in vivo and observed a synergistic effect mediated by upregulation of the pro-apoptotic p57 protein. Synergy between those two agents appears at first glance to be paradoxical due to the fact that HDAC inhibitors stabilize a histone mark whose recognition is blocked by BET inhibition. However, both agents may work by attacking related transcriptional mechanisms. As previously mentioned, distal regulatory elements play a significant role in the mechanism of action of BET inhibitors. In contrast, the effects of HDAC inhibitors at these regions are less often reported. Recent studies have investigated the role of HDAC inhibitors on enhancer activity, which might further clarify the mechanisms by which these inhibitors act and identify new approaches to use them safely and effectively.
3.2. Histone Deacetylase Inhibition Role at Distal Regulatory Elements
As HDAC inhibitors stabilize a histone mark associated with active transcription, it is expected that it will lead mainly and directly to an upregulation of dependent genes. Surprisingly, we have detected a significant set of genes that are downregulated upon treatment with selective inhibitors of class I HDACs in the highly metastatic pancreatic cell line L3.6pl [85]. We have observed that while promoters of these downregulated genes gain acetylation as expected, individual associated distal regulatory elements of these genes show a dramatic loss of acetylation and better correlation with gene regulation. This has also been observed in the colorectal cancer cell line, HCT116, where treatment with an HDAC inhibitor also leads to the loss of H3K27ac at certain enhancer regions in a concentration-dependent manner [86]. However, this decrease is not universally observed at all enhancers as an increase of H3K27ac by HDAC inhibition at the enhancer of the pro-apoptotic B cell lymphoma-2-like 11 (BIM) gene was reported in triple negative breast cancer [87]. Consistent with a particular effect on enhancer activity, HDAC inhibition has been shown to repress enhancer RNA (eRNA) expression to a higher extent than BET inhibition in breast cancer cells [88].
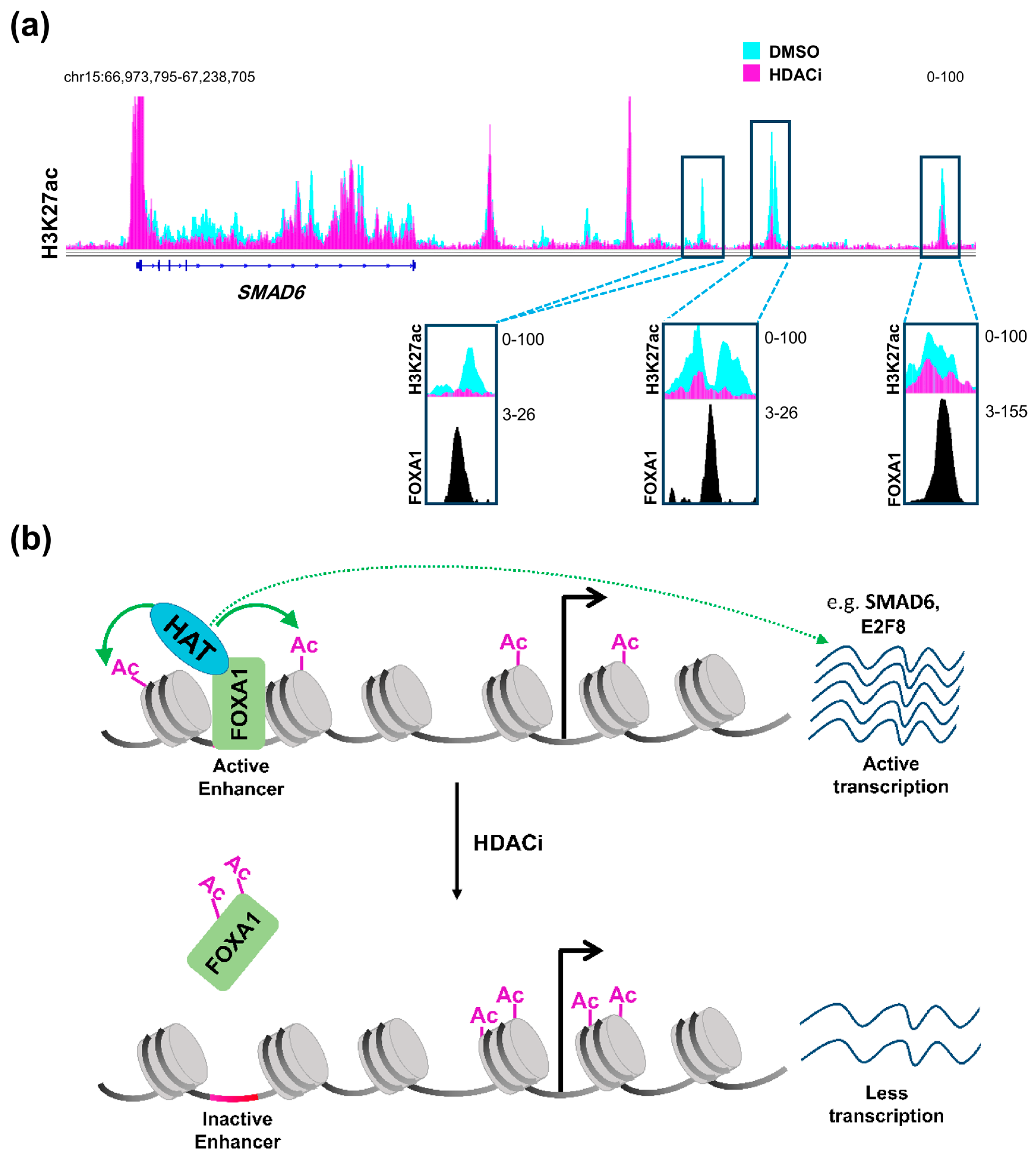
Therefore, it can be concluded that HDAC inhibitors affect distal regulatory elements in a manner that is distinct from promoters, probably decreasing acetylation at a subset of specific enhancers, thereby affecting gene transcription in a more complex and diverse manner. Such regulation can be partially explained by the status of activation of the enhancer itself prior to treatment [86]. Enhancers can either be in an active state and marked by H3K27ac and H3K4me1, inactive with only H3K27me3, or poised with a lower threshold to be activated via being marked by the H3K4me1 active mark and H3K27me3 repressive mark [89]. Sanchez et al. [86] demonstrated that enhancers which are already active are usually inactivated by HDAC inhibition while poised enhancers show a tendency to be activated. Another very important aspect that may play a central role in defining the response of an enhancer to HDAC inhibition is the repertoire of transcription factors expressed in the cellular system and their importance for cellular phenotype and tumorigenic gene expression. It was reported that HDAC inhibitors not only exert their effects on acetylated histones, but also on acetylated transcription factors [90,91]. Notably, acetylation of the pioneer transcription factor, Forkhead Box A1 (FOXA1) was shown to directly and negatively affect its ability to bind chromatin [92]. Remarkably, FOXA1 was recently shown to be specifically enriched in enhancer regions that are gained in metastatic pancreatic organoids [38]. Accordingly, we postulate that HDAC inhibitors can be used to attenuate the binding of FOXA1 to these enhancers leading to their deactivation. Indeed, enhancer regions that we identified as being lost following HDAC inhibitor treatment were found to be enriched for FOXA1 occupancy in another pancreatic cancer cell line [93] (Figure 1a). Interestingly, genes targeted by this mechanism were unaffected by BET inhibitor treatment [93]. Thus, combined treatment with HDAC and BET inhibitors can potentially simultaneously target different sets of activated enhancers to decrease the activation of reprogrammed enhancers synergistically and more effectively, activating aberrant transcriptional programs such as metastasis. This presents a model in which these two apparently counteracting agents can work together forming a successful therapeutic regimen in metastatic pancreatic cancer (Figure 1b).
4. Enhancers as an Optimal Paradigm for Therapeutic Targeting of Pancreatic Cancer
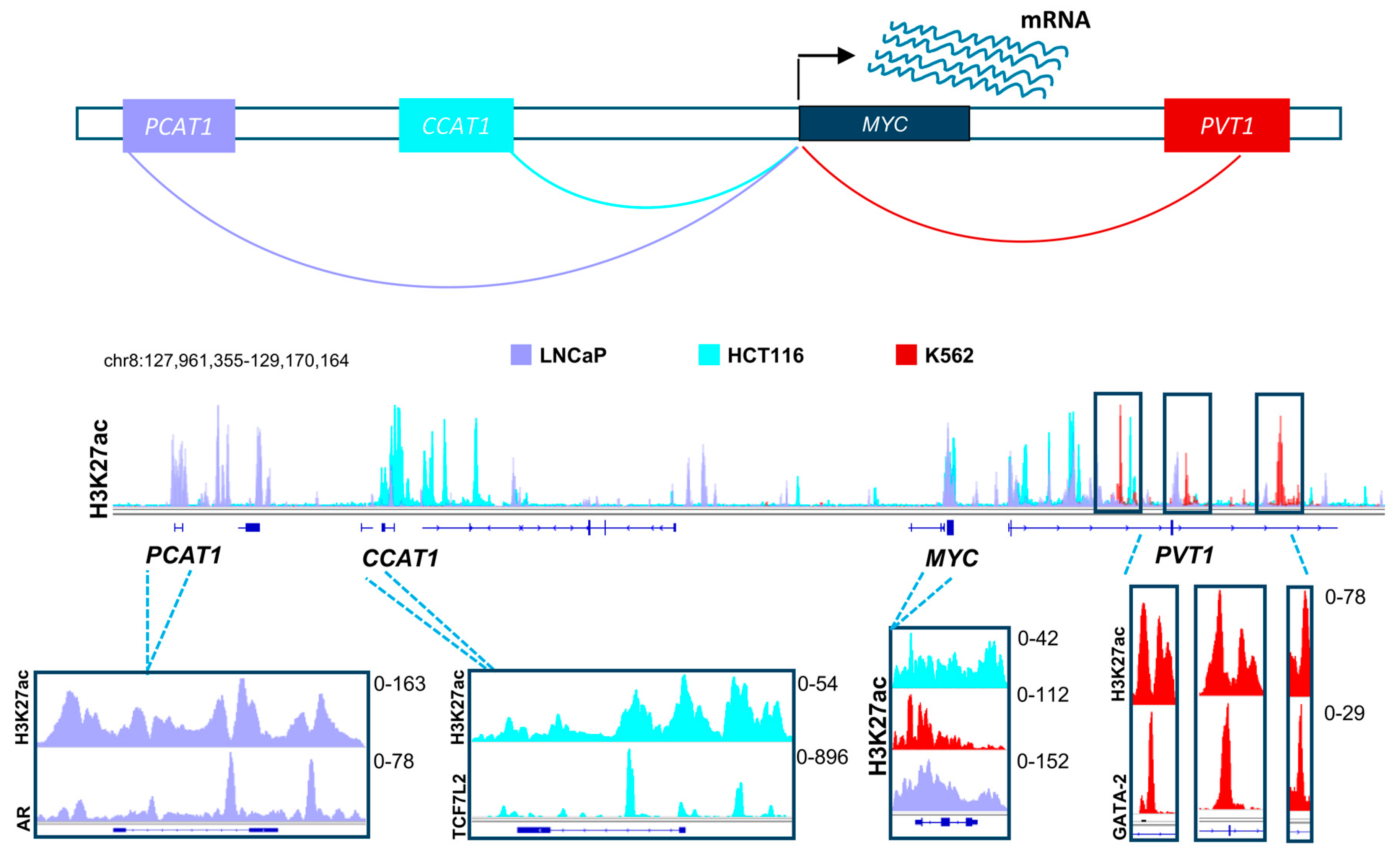
Based on the evidence discussed above, it is clear that distal regulatory elements play a special role in the scope of the effects of BET and HDAC inhibitors. Thus, it is highly probable that these elements will emerge as a major target of therapy in multiple diseases in the upcoming years. Many positive attributes contribute to the adequacy of enhancers as a target for therapy and manipulation. To activate a certain gene, the transcriptional machinery has to be recruited by transcription factors to the promoters of these genes [94]. Diversity in transcription factor recruitment and abundance are thus very important in regulating gene activation in different contexts and systems. Distal regulatory elements provide a platform with vast variability and substantial magnitude for recruitment of various transcription factors, thereby enabling regulation of gene transcription in a temporal and spatial manner. This means that in certain systems, driver oncogenic pathways can be activated by different transcription factors and enhancers, thus creating a dependence which can be specifically targeted. For example, different enhancers drive the activation of MYC in various systems. In colorectal cancer, the long non-coding RNA, Colon Cancer Associated Transcript 1 (CCAT1) is highly active and plays a significant role in MYC activation [95,96]. Consistent with the importance of the Wnt signaling pathway in colorectal cancer, we observed that this enhancer is highly occupied by Wnt-responsive β-catenin-dependent transcription factor TCF7L2 (Figure 2) [96]. This implies that Wnt-signaling mediated activation of MYC in colorectal cancer utilizes a specific mechanisms of activation which can be potentially be targeted by HDAC inhibitors, as they were reported to deplete TCF7L2 [97]. Interestingly, CCAT1 was also reported to be play a tumorigenic role in pancreatic cancer [98]. In other systems such as prostate cancer, MYC is activated by a different enhancer called Prostate Cancer Associated Transcript 1 (PCAT1). Consistent, with the androgen receptor (AR) being a primary driver of prostate cancer, we observed this enhancer to be particularly occupied by AR in LNCaP prostate cancer cells [95,99,100,101]. Alternatively, the PVT1 oncogene is active in many other systems such as leukemia and plays a similar role as an enhancer of the MYC gene [102]. Analogous to the tumor- and context-specific effects shown for TCF7L2 and AR in colorectal and prostate cancers, respectively, we observed an enrichment of the hematopoietic transcription factor GATA-2 on the MYC enhancer within the PVT1 gene [96]. Together, these examples show the complexity by which diverse distal regulatory elements utilize specific transcription factor repertoires to induce common oncogenic pathways. We postulate that distinct, but similar transcription factor networks will also be discovered in pancreatic cancer which can be specifically targeted by inhibitors of BET, HDAC or other epigenetic regulators. Importantly, activation of oncogenes such as MYC by alternative BET-independent transcriptional pathways can lead to resistance to BET inhibitors in leukemia cells [103]. Thus, identifying which enhancers are specifically active in pancreatic cancer cells and identifying and targeting their dependencies will play an important role in the optimal application of BET inhibitors in the treatment of pancreatic cancer. Furthermore, targeting tumor-specific enhancer regions will be more likely to spare normal cells and may possibly lead to less long term adverse effects [104]. Development of highly specific BET inhibitors supports this theory by showing selective anti-proliferative effects in certain cancer cell lines while not affecting other systems [105]. Future studies will test the validity of this rationale upon successful prolongation of patient survival.
5. Targeting Transcription Factors in Pancreatic Cancer: A Code for Specificity
Enhancers are known to be highly-bound by specific transcription factors that mediate transcriptional activation of target gene expression through the recruitment of other activators and transcription initiators [106]. When transcription factors co-localize with a factor of interest, BETs for example, these factors will naturally play a significant role in its mechanism and effects. Interestingly, the BET context-specific effects discerned in the vast body of literature available can probably be explained by the different interacting factors at specific sites where BET members co-localize with other transcription factors. BET-dependent enhancers require BET members and certain transcription factors for their activation, while other BET-independent enhancers might have other factors and activation pathways which make them tolerant to the loss of one of many activators upon BET inhibition. Furthermore, the different expression levels of these transcription factors in different systems, whether absent or highly or lowly expressed, in addition to their pioneering potential can also play a role in enhancer dependency and activity.
Paradoxically, super enhancers are by definition normally highly enriched for transcription factor binding, which can theoretically render them less dependent on one particular factor [107]. However, this is not usually the case given that, as previously discussed, BET inhibition has the ability to turn off certain BET-dependent super enhancers but deletion of individual components of “super enhancers” does not equally impair the activation of the target gene [56,57,95,108,109]. Thus, it appears that BET proteins serve to integrate the activity of transcription factors at BET-dependent enhancers in a manner such that the sum of the activity of the transcription factor binding is greater than that of the components. Subsequently, once a better understanding of transcription factor function at enhancers and promoters and the dependence of each on BET proteins has been achieved, targeting specific transcription factors or their upstream signaling pathways, possibly alone or in combination with BET inhibitors, can provide us with a new layer of specific gene transcriptional manipulation.
However, therapeutically targeting transcription factors can be quite challenging. One approach can be targeting signaling pathways that control the activity of the transcription factor of interest. For example, the transcription factor Endothelial PAS Domain Protein 1 (EPAS1 or HIF2A) was demonstrated to play a role in promoting pancreatic cancer in cells and mice [110]. A crosstalk with Wnt-signaling was identified in this system which uncovers a new target that can potentially be inhibited. Furthermore, Transforming Growth Factor-β (TGFβ) signaling was reported to cooperate with mutant p53 to mediate distal metastasis in pancreatic cancer mouse models [111]. The activation of Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB), which also promotes EMT in pancreatic cancer, can also be inhibited by blocking its activation via NF-κB inhibitor-α (IκBα) phosphorylation [112]. Another approach to target transcription factors is to attenuate their recruitment by affecting their ability to bind chromatin as previously mentioned for FOXA1 and its acetylation. This can also be achieved by designing specific inhibitors that prevent the binding of DNA to a certain transcription factor [113]; however this approach has proven to be very difficult for therapeutic application. Another approach can be targeting cooperating transcription factors. For example, NF-κB cooperates with E26 transformation–specific (ETS) transcription factors to recruit BET members to activate genes, rendering these sensitive to BET inhibition [114,115]. In general, transcription factors can play specific roles in a context-dependent manner based on the combinatorial repertoire of transcription factors expressed, thereby enabling a given transcription factor to activate a different set of genes and programs, dependent upon the expression of other factors. Therefore, identifying important transcription factors playing a role in aberrant transcriptional activation may uncover specific targets that can be manipulated by available inhibitors.
6. Conclusions: Unconventional Epigenetic Agents Should Be Used in Unconventional Ways
Epigenetic agents are a special subclass of drugs whose targets and effects are dependent on the epigenetic and transcriptional landscape of each system. In general, a major trend is seen where low concentrations of these agents affect hypersensitive dependent genes and higher concentrations frequently display a more universal effect [60]. So far, gene transcription modulatory agents have been used to initiate cell cycle arrest and/or apoptosis and mainly administered at high levels just under their maximum tolerable dose (MTD). We speculate that administering these agents at these doses likely influences their specificity and probably promotes many of the intolerable adverse effects that might lead to premature termination of clinical studies. In fact, higher doses of such inhibitors, which lead to cell cycle arrest, may in fact impede the activity of many chemotherapeutic agents, while lower concentrations which elicit specific transcriptional reprogramming may have minimal effects on their own, but significantly synergize with other therapies. To ensure the maximum utilization of these agents, concentrations lower than MTD should be taken into consideration when performing pharmacodynamic and pharmacokinetic studies in vivo. However, these studies will face major challenges as markers that can be detected ex vivo to measure efficacy are still not clearly defined. Ideally, a marker should be easily detectable and highly sensitive, for example enhancer RNA of BET-dependent genes or stable messenger RNA of a highly dependent and hypersensitive gene. Using profoundly regulated gene levels in blood as a marker for BET inhibition efficacy uncovered that BET inhibitors exert their effects at lower doses than MTD in progressive lymphoma patients [116].
In this review, we summarized data pertaining to the effects of BET and HDAC inhibitors, two of the most promising epigenetic agents, in pancreatic cancer. We draw the conclusion that these agents likely mediate their specific effects through distal regulatory elements. By investigating the potential utility of these agents in lower concentrations, we may be able to uncover their potential as safe adjuvant therapies in combination with other standard of care treatments to manage and prevent recurrence of pancreatic cancer and various malignancies in general.
Author Contributions
F.H.H. and S.A.J. have conceived the ideas for this work and wrote the manuscript.
Acknowledgments
F.H.H. was funded by the German Academic Exchange Service (DAAD). This work was funded by grants from the Deutsche Krebshilfe (PiPAC Consortium) [70112505], Deutsche Forschungsgemeinschaft (DFG) [JO 815/3-2], and the Roggenbuck Foundation to S.A.J.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Parameters for Data Analysis Used in Figure Generation
Chromatin immune-precipitation profiles which are shown in Figure 1 and Figure 2 as examples were downloaded from the Encyclopedia of DNA Elements (ENCODE) consortium when available (H3K27ac in HCT116: GSM2534277; TCF7L2 in HCT116: GSM782123; H3K27ac in K562: GSM733656; GATA-2 in K562: GSM935373) [96]. Other profiles were downloaded from the European Nucleotide Archive (H3K27ac in LNCaP: SRR2566837 [99]; AR in LNCaP: SRR4025870 [100]; H3K27ac in L3.6pl: SRR5042516,18-21 [85]; FOXA1 in CFPAC1: SRR1736462 [93]). Reads were mapped to the hg19 genome using BOWTIE/2.2.5 [117] and converted to bam using SAMTOOLS/1.4 [118]. DEEPTOOLS/2.4.0 [119] was used to produce bigwig files with ignoring the duplicates and extending the reads for 200 base pairs. Bigwig files were viewed using IGV 2.4 [120,121].
References
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann. Oncol. 2017, 28, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Jakel, C.; Bergmann, F.; Toth, R.; Assenov, Y.; van der Duin, D.; Strobel, O.; Hank, T.; Kloppel, G.; Dorrell, C.; Grompe, M.; et al. Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability. Nat. Commun. 2017, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Parikh, H.; Xiao, W.; Hoskins, J.W.; Pflicke, H.; Liu, X.; Collins, I.; Zhou, W.; Wang, Z.; Powell, J.; et al. An integrated transcriptome and epigenome analysis identifies a novel candidate gene for pancreatic cancer. BMC Med. Genom. 2013, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- McCleary-Wheeler, A.L.; Lomberk, G.A.; Weiss, F.U.; Schneider, G.; Fabbri, M.; Poshusta, T.L.; Dusetti, N.J.; Baumgart, S.; Iovanna, J.L.; Ellenrieder, V.; et al. Insights into the epigenetic mechanisms controlling pancreatic carcinogenesis. Cancer Lett. 2013, 328, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Iguchi, E.; Safgren, S.L.; Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Pancreatic cancer, a mis-interpreter of the epigenetic language. Yale J. Biol. Med. 2016, 89, 575–590. [Google Scholar] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Gene Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Gene Dev. 2015, 29, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of valproic acid in combination with S-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014, 34, 5187–5191. [Google Scholar] [PubMed]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, E.; Kerner, Z.; Nanda, N.; Ahuja, N. Epigenetic therapy in gastrointestinal cancer: The right combination. Ther. Adv. Gastroenterol. 2016, 9, 560–579. [Google Scholar] [CrossRef] [PubMed]
- Lomberk, G.A.; Iovanna, J.; Urrutia, R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics 2016, 8, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef] [PubMed]
- Silverman, B.R.; Shi, J. Alterations of epigenetic regulators in pancreatic cancer and their clinical implications. Int. J. Mol. Sci. 2016, 17, 2138. [Google Scholar] [CrossRef] [PubMed]
- Quilichini, E.; Haumaitre, C. Implication of epigenetics in pancreas development and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Klieser, E.; Swierczynski, S.; Mayr, C.; Schmidt, J.; Neureiter, D.; Kiesslich, T.; Illig, R. Role of histone deacetylases in pancreas: Implications for pathogenesis and therapy. World J. Gastrointest. Oncol. 2015, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Garmpis, N.; Karatzas, T.; Nikolidakis, L.; Kostakis, I.D.; Garmpi, A.; Karamaroudis, S.; Boutsikos, G.; Damaskou, Z.; Kostakis, A.; et al. Histone deacetylase (HDAC) inhibitors: Current evidence for therapeutic activities in pancreatic cancer. Anticancer Res. 2015, 35, 3129–3135. [Google Scholar] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [PubMed]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed]
- French, C.A. Pathogenesis of NUT midline carcinoma. Ann. Rev. Pathol. 2012, 7, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Al Diffalha, S.; Al Aukla, N.; Hasan, S.; Dickinson, S.; Khalil, F. NUT midline carcinoma: A rare malignancy. Cancer Control 2017, 24, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.G.; French, C.A.; Cameron, M.J.; Fletcher, C.D.; Jackman, D.M.; Lathan, C.S.; Sholl, L.M. Pathologic characteristics of NUT midline carcinoma arising in the mediastinum. Am. J. Surg. Pathol. 2012, 36, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.L.; Tian, M.; Li, X.; Li, J.J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nahar, S.; Nakagawa, A.; Fernandez-Barrena, M.G.; Mertz, J.A.; Bryant, B.M.; Adams, C.E.; Mino-Kenudson, M.; Von Alt, K.N.; Chang, K.; et al. Regulation of GLI underlies a role for BET bromodomains in pancreatic cancer growth and the tumor microenvironment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4259–4270. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.S.; Williams, C.R.; Royce, D.B.; Pioli, P.A.; Sporn, M.B.; Liby, K.T. Bromodomain inhibitors, JQ1 and I-BET 762, as potential therapies for pancreatic cancer. Cancer Lett. 2017, 394, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Kumar, K.; Knab, L.M.; Chow, C.R.; Raza, S.S.; Bentrem, D.J.; Ebine, K.; Munshi, H.G. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014, 13, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tateishi, K.; Kudo, Y.; Hoshikawa, M.; Tanaka, M.; Nakatsuka, T.; Fujiwara, H.; Miyabayashi, K.; Takahashi, R.; Tanaka, Y.; et al. Stromal remodeling by the BET bromodomain inhibitor JQ1 suppresses the progression of human pancreatic cancer. Oncotarget 2016, 7, 61469–61484. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Tseng, T.W.; Sousa, C.M.; Liu, S.; Truitt, M.L.; He, N.; Ding, N.; Liddle, C.; Atkins, A.R.; et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc. Natl. Acad. Sci. USA 2017, 114, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.; Bigonnet, M.; Gayet, O.; Loncle, C.; Maignan, A.; Gilabert, M.; Moutardier, V.; Garcia, S.; Turrini, O.; Delpero, J.R.; et al. Gene expression profiling of patient-derived pancreatic cancer xenografts predicts sensitivity to the BET bromodomain inhibitor JQ1: Implications for individualized medicine efforts. EMBO Mol. Med. 2017, 9, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Uccello, M.; Moschetta, M.; Mak, G.; Alam, T.; Henriquez, C.M.; Arkenau, H.T. Towards an optimal treatment algorithm for metastatic pancreatic ductal adenocarcinoma (PDA). Curr. Oncol. 2018, 25, e90–e94. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 2017, 170, 875–888. [Google Scholar] [CrossRef] [PubMed]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.J.; Bayles, I.; Funnell, A.P.W.; Miller, T.E.; Saiakhova, A.; Lizardo, M.M.; Bartels, C.F.; Kapteijn, M.Y.; Hung, S.; Mendoza, A.; et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat. Med. 2018, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; De Leo, A.; Wang, Z.; Kerekovic, A.; Hills, R.; Lieberman, P.M. BET-inhibitors disrupt Rad21-dependent conformational control of KSHV latency. PLoS Pathog. 2017, 13, e1006100. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Zhang, F.; Jaganathan, A.; Sharma, R.; Zhang, Q.; Konuma, T.; Shen, T.; Lee, J.Y.; Ren, C.; Chen, C.H.; et al. Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol. Cell 2017, 65, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.C.; Gilgenast, T.G.; Bartman, C.R.; Edwards, C.R.; Stonestrom, A.J.; Huang, P.; Emerson, D.J.; Evans, P.; Werner, M.T.; Keller, C.A.; et al. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 2017, 66, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Oh, S.; Yoo, K.H. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol. Cells 2017, 40, 169–177. [Google Scholar] [PubMed]
- Kaiser, V.B.; Semple, C.A. When TADs go bad: Chromatin structure and nuclear organisation in human disease. F1000Research 2017, 6, 314. [Google Scholar] [CrossRef] [PubMed]
- Najafova, Z.; Tirado-Magallanes, R.; Subramaniam, M.; Hossan, T.; Schmidt, G.; Nagarajan, S.; Baumgart, S.J.; Mishra, V.K.; Bedi, U.; Hesse, E.; et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017, 45, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Anyansi, C.; Hu, X.; Xu, L.; Xiong, L.; Tang, W.; Mok, M.T.S.; Cheng, C.; Fan, X.; Gerstein, M.; et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017, 49, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Nagarajan, S.; Baumgart, S.J.; Kosinsky, R.L.; Najafova, Z.; Kari, V.; Hennion, M.; Indenbirken, D.; Bonn, S.; Grundhoff, A.; et al. RNF40 regulates gene expression in an epigenetic context-dependent manner. Genome Biol. 2017, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Fontanals-Cirera, B.; Hasson, D.; Vardabasso, C.; Di Micco, R.; Agrawal, P.; Chowdhury, A.; Gantz, M.; de Pablos-Aragoneses, A.; Morgenstern, A.; Wu, P.; et al. Harnessing BET inhibitor sensitivity reveals AMIGO2 as a melanoma survival gene. Mol. Cell 2017, 68, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Kannan, A.; Kern, M.; Moreno, M.A.; Vural, E.; Stack, B., Jr.; Suen, J.Y.; Tackett, A.J.; Gao, L. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 2015, 10, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.H.; Johnsen, S.A. Super enhancers—New analyses and perspectives on the low hanging fruit. Transcription 2018, 9, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Gelato, K.A.; Schockel, L.; Klingbeil, O.; Ruckert, T.; Lesche, R.; Toedling, J.; Kalfon, E.; Heroult, M.; Lejeune, P.; Monning, U.; et al. Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene 2018, 37, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Muhar, M.; Ebert, A.; Neumann, T.; Umkehrer, C.; Jude, J.; Wieshofer, C.; Rescheneder, P.; Lipp, J.J.; Herzog, V.A.; Reichholf, B.; et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 2018, 360, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harbor Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Vancurova, I.; Uddin, M.M.; Zou, Y.; Vancura, A. Combination therapies targeting HDAC and IKK in solid tumors. Trends Pharmacol. Sci. 2018, 39, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Wang, W.H.; Wu, W.Y.; Hsu, C.C.; Wei, L.R.; Wang, S.F.; Hsu, Y.W.; Liaw, C.C.; Tsai, W.C. Novel histone deacetylase inhibitor AR-42 exhibits antitumor activity in pancreatic cancer cells by affecting multiple biochemical pathways. PLoS ONE 2017, 12, e0183368. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Qiao, Z.; Wang, Y.; Kuai, Q.; Li, C.; Wang, Y.; Jiang, X.; Wang, X.; Li, W.; He, M.; et al. Chidamide inhibits aerobic metabolism to induce pancreatic cancer cell growth arrest by promoting Mcl-1 degradation. PLoS ONE 2016, 11, e0166896. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Manfredi, M.; Brandi, J.; Buzzi, A.; Conte, E.; Pacchiana, R.; Cecconi, D.; Marengo, E.; Donadelli, M. Trichostatin A alters cytoskeleton and energy metabolism of pancreatic adenocarcinoma cells: An in depth proteomic study. J. Cell. Biochem. 2018, 119, 2696–2707. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, J.; Zhao, J.; Yun, W.; Xie, C.; Taub, J.W.; Azmi, A.; Mohammad, R.M.; Dong, Y.; Kong, W.; et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS ONE 2012, 7, e52095. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.H.; Xu, X.G.; Yan, S.L.; Sun, Z.; Ying, Y.; Wang, B.K.; Tu, Y.X. Depletion of HDAC1, 7 and 8 by histone deacetylase inhibition confers elimination of pancreatic cancer stem cells in combination with gemcitabine. Sci. Rep. 2018, 8, 1621. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Chiorean, E.G.; O’Dwyer, P.J.; Gabrail, N.Y.; Alcindor, T.; Potvin, D.; Chao, R.; Hurwitz, H. Phase I/II study of mocetinostat in combination with gemcitabine for patients with advanced pancreatic cancer and other advanced solid tumors. Cancer Chemother. Pharmacol. 2018, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7, 41615. [Google Scholar] [CrossRef] [PubMed]
- Samulitis, B.K.; Pond, K.W.; Pond, E.; Cress, A.E.; Patel, H.; Wisner, L.; Patel, C.; Dorr, R.T.; Landowski, T.H. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol. Ther. 2015, 16, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, L.; Kang, S.G.; Lu, Y.; Yang, Z.; Huynh, T.; Chen, C.; Zhou, R.; Guo, M.; Zhao, Y. Gd-metallofullerenol nanomaterial suppresses pancreatic cancer metastasis by inhibiting the interaction of histone deacetylase 1 and metastasis-associated protein 1. ACS Nano 2015, 9, 6826–6836. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.J.; Richmond, P.A.; Bunker, E.N.; Karman, S.S.; Azofeifa, J.; Garnett, A.T.; Xu, Q.; Wheeler, G.E.; Toomey, C.M.; Zhang, Q.; et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018, 46, 1756–1776. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.P.; Ling, K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys27 acetylation in the BIM gene promoter and enhancers. Oncol. Lett. 2017, 14, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.B.; Tanaka, Y.; Kim, Y.J.; Xie, P.; Zhang, M.Q.; Park, I.H.; Kim, T.H. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 2015, 13, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Zhang, B.; Li, Y.; King, J.L.; Tian, R.; Xia, S.; Schiavon, C.R.; Dong, J.T. HDAC-mediated deacetylation of KLF5 associates with its proteasomal degradation. Biochem. Biophys. Res. Commun. 2018, 500, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Beier, U.H.; Akimova, T.; Dahiya, S.; Han, R.; Samanta, A.; Levine, M.H.; Hancock, W.W. Histone/protein deacetylase inhibitor therapy for enhancement of Foxp3+ T-regulatory cell function posttransplantation. Am. J. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Cirillo, L.A. Stable chromatin binding prevents FoxA acetylation, preserving FoxA chromatin remodeling. J. Biol. Chem. 2010, 285, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis-regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Gotze, S.; Coersmeyer, M.; Muller, O.; Sievers, S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int. J. Oncol. 2014, 45, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhou, X.; Xia, Q.; Shen, J.; Yan, J.; Zhu, J.; Li, X.; Shu, M. Long non-coding RNA CCAT1 that can be activated by c-Myc promotes pancreatic cancer cell proliferation and migration. Am. J. Transl. Res. 2016, 8, 5444–5454. [Google Scholar] [PubMed]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Cyrta, J.; Murphy, D.A.; Walczak, E.G.; Ran, L.; Agrawal, P.; Xie, Y.; Chen, Y.; Wang, S.; Zhan, Y.; et al. Aberrant Activation of a gastrointestinal transcriptional circuit in prostate cancer mediates castration resistance. Cancer Cell 2017, 32, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.L.; Tuzova, A.V.; Bolton, E.M.; Lynch, T.H.; Perry, A.S. Long noncoding RNAs and prostate carcinogenesis: The missing ‘linc’? Trends Mol. Med. 2014, 20, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Yu, X.; Lai, J.; Yang, L.; Chen, S.; Li, Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 2015, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. Epigenetics: The first 25 centuries. Philos. Trans. R. Soc. B 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Huang, X.; Bellin, R.; Faivre, E.J.; Hessler, P.; Lam, L.; Bui, M.-H.; Wilcox, D.M.; Uziel, T.; Ferguson, D.C.; et al. ABBV-744, a first-in-class and highly selective inhibitor of the second beomodomain of BET family proteins, displays robust activities in preclinical models of acute myelogenous leukemia. In Proceedings of the AACR Annual Meeting, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Peng, X.L.; So, K.K.; He, L.; Zhao, Y.; Zhou, J.; Li, Y.; Yao, M.; Xu, B.; Zhang, S.; Yao, H.; et al. MyoD- and FoxO3-mediated hotspot interaction orchestrates super-enhancer activity during myogenic differentiation. Nucleic Acids Res. 2017, 45, 8785–8805. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.; Hughes, J.R.; Babbs, C.; Davies, J.O.J.; Graham, B.J.; Hanssen, L.; Kassouf, M.T.; Marieke Oudelaar, A.M.; Sharpe, J.A.; Suciu, M.C.; et al. Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 2016, 48, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y.; Willi, M.; HyunYoo, K.; Zeng, X.; Wang, C.; Metser, G.; Hennighausen, L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016, 48, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Zhang, J.; Fu, Q.; Wei, T.; Sun, X.; Chen, Q.; Yang, J.; Bai, X.; Liang, T. Hypoxia-inducible factor-2αpromotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol. Cancer 2017, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Macgregor-Das, A.; Saunders, T.; Whittle, M.C.; Makohon-Moore, A.; Kohutek, Z.A.; Poling, J.; Herbst, B.T.; Javier, B.M.; Cope, L.; et al. Mutant p53 together with TGFβ signaling influence organ-specific hematogenous colonization patterns of pancreatic cancer. Clin. Cancer Res. 2017, 23, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-κ B pathway leads to deregulation of epithelial-mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. J. Tech. Methods Pathol. 2016, 96, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Nhili, R.; Peixoto, P.; Depauw, S.; Flajollet, S.; Dezitter, X.; Munde, M.M.; Ismail, M.A.; Kumar, A.; Farahat, A.A.; Stephens, C.E.; et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013, 41, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zeng, S.; Zou, Y.; Shi, M.; Qiu, Q.; Xiao, Y.; Chen, G.; Yang, X.; Liang, L.; Xu, H. The suppression of bromodomain and extra-terminal domain inhibits vascular inflammation by blocking NF-κB and MAPK activation. Br. J. Pharmacol. 2017, 174, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Hensel, T.; Giorgi, C.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Buch, T.; Niggli, F.K.; Schafer, B.W.; Burdach, S.; Richter, G.H. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget 2016, 7, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Blum, K.A.; Younes, A.; Abramson, J.S.; Maris, M.B.; Flinn, I.W.; Goy, A.; Border, D.R.; Cooper, M.R.; Sims, R.J. Pharmacodynamic assessment in whole blood for the BET bromodomain inhibitor CPI-0610 of target engagement in patients with progressive lymphoma. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, 5. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
HDAC inhibition and FOXA1 at distal regulatory elements. (a) H3K27ac profiles in L3.6pl cells at SMAD6, a gene that is downregulated upon treatment with class I HDAC inhibitor (HDACi) showing distal regulatory regions that dramatically lose H3K27ac and co-localize with FOXA1 in the CFPAC1 pancreatic cancer cell line; (b) A schematic model showing an enhancer region activated by FOXA1 leading to acetylation of histones by a histone acetyltransferase (HAT), which leads to the activation of the gene. Upon treatment with HDACi, increased FOXA1 acetylation attenuates its binding to chromatin leading to downregulation of the gene. DMSO: Dimethylsulfoxide; HDAC: histone deacetylase; FOXA1: Forkhead Box A1. (See Appendix A for details of data analysis).
Figure 1.
HDAC inhibition and FOXA1 at distal regulatory elements. (a) H3K27ac profiles in L3.6pl cells at SMAD6, a gene that is downregulated upon treatment with class I HDAC inhibitor (HDACi) showing distal regulatory regions that dramatically lose H3K27ac and co-localize with FOXA1 in the CFPAC1 pancreatic cancer cell line; (b) A schematic model showing an enhancer region activated by FOXA1 leading to acetylation of histones by a histone acetyltransferase (HAT), which leads to the activation of the gene. Upon treatment with HDACi, increased FOXA1 acetylation attenuates its binding to chromatin leading to downregulation of the gene. DMSO: Dimethylsulfoxide; HDAC: histone deacetylase; FOXA1: Forkhead Box A1. (See Appendix A for details of data analysis).

Figure 2.
A schematic model showing distal regulatory mechanisms controlling the expression of the MYC gene in different systems, namely in prostate cancer (shown in light purple), colorectal cancer (shown in turquoise), and leukemic cell lines (shown in red). The H3K27ac layered profile in these different cell lines uncover differential activation of unique enhancers in each system. These enhancers are specifically enriched with driver transcription factors (androgen receptor, AR; TCF7L2 and GATA2), which are hallmarks of these tumor types. TCF7L2: Transcription Factor 7-like 2; GATA2: GATA binding protein-2. (See Appendix A for details of data analysis).
Figure 2.
A schematic model showing distal regulatory mechanisms controlling the expression of the MYC gene in different systems, namely in prostate cancer (shown in light purple), colorectal cancer (shown in turquoise), and leukemic cell lines (shown in red). The H3K27ac layered profile in these different cell lines uncover differential activation of unique enhancers in each system. These enhancers are specifically enriched with driver transcription factors (androgen receptor, AR; TCF7L2 and GATA2), which are hallmarks of these tumor types. TCF7L2: Transcription Factor 7-like 2; GATA2: GATA binding protein-2. (See Appendix A for details of data analysis).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hamdan, F.H.; Johnsen, S.A. Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer. Epigenomes 2018, 2, 8. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2020008
AMA Style
Hamdan FH, Johnsen SA. Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer. Epigenomes. 2018; 2(2):8. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2020008
Chicago/Turabian StyleHamdan, Feda H., and Steven A. Johnsen. 2018. "Epigenetic Targeting of Aberrant Transcriptional Modulation in Pancreatic Cancer" Epigenomes 2, no. 2: 8. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes2020008