LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells
by
 and
and
Alessandro Fiorenzano
1, 
Emilia Pascale
2,3,
Eduardo Jorge Patriarca
2,3,
Gabriella Minchiotti
2,3 and
Annalisa Fico
2,3,* 1
Department of Experimental Medical Science and Lund Stem Cell Center BMC, Lund University, 22632 Lund, Sweden
2
Stem Cell Fate Laboratory, Institute of Genetics and Biophysics “A. Buzzati-Traverso”, CNR, 80131 Naples, Italy
3
Institute of Genetics and Biophysics “A. Buzzati-Traverso”, CNR, 80131 Naples, Italy
*
Author to whom correspondence should be addressed.
Epigenomes 2019, 3(3), 14; https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes3030014
Submission received: 27 June 2019
/
Revised: 2 August 2019
/
Accepted: 3 August 2019
/
Published: 6 August 2019
(This article belongs to the Special Issue Epigenome, Epitranscriptome and Single Cell Analysis in Cell Fate Choice)
Abstract
:The power of embryonic stem cells (ESCs) lies in their ability to self-renew and differentiate. Behind these two unique capabilities is a fine-tuned molecular network that shapes the genetic, epigenetic, and epitranscriptomic ESC plasticity. Although RNA has been shown to be functionally important in only a small minority of long non-coding RNA genes, a growing body of evidence has highlighted the pivotal and intricate role of lncRNAs in chromatin remodeling. Due to their multifaceted nature, lncRNAs interact with DNA, RNA, and proteins, and are emerging as new modulators of extensive gene expression programs through their participation in ESC-specific regulatory circuitries. Here, we review the tight cooperation between lncRNAs and Polycomb repressive complex 2 (PRC2), which is intimately involved in determining and maintaining the ESC epigenetic landscape. The lncRNA-PRC2 partnership is fundamental in securing the fully pluripotent state of ESCs, which must be primed to differentiate properly. We also reflect on the advantages brought to this field of research by the advent of single-cell analysis.
1. Introduction
A hallmark of embryonic stem cells (ESCs) is their ability to maintain a pluripotent and self-renewal network while retaining the capacity to promptly respond to a variety of microenvironmental signals activating the expression of gene differentiation programs [1,2]. ESC cellular plasticity is preserved by an intricate transcriptional regulatory circuitry involving the interplay between transcription factors (TFs), epigenetic machinery, and non-coding RNAs. Over the past two decades, our understanding of how the non-coding genome impacts on cell fate decisions and tissue patterning during development has vastly expanded [3,4,5]. Recent insights into long non-coding RNAs (lncRNAs) have challenged the conventional paradigm that RNA is merely a messenger passing information between DNA and protein, shifting the focus away from the protein-coding genome. A large portion of the mammalian genome encodes genes that transcribe lncRNAs without any apparent protein-coding potential [6,7]. Although little is yet known about the dynamic regulation of their transcription, biogenesis, and degradation, lncRNAs cannot be dismissed as transcriptional ‘noise’. Rather, they are emerging as fundamental regulators in a wide range of molecular biological processes, thereby defining a new and crucial layer of regulation in stem cell biology [8,9]. LncRNAs are broadly classified as transcripts longer than 200 nucleotides, typically transcribed by RNA polymerase II, spliced, and polyadenylated [10]. It is estimated that the human genome contains approximately 16,000 lncRNA genes, which can be further profiled into five distinct subgroups: (1) long intergenic non-coding RNAs (lincRNAs), which do not overlap with the annotated coding genes; (2) sense lncRNAs, which are transcribed from the sense strand of protein-coding genes and can even overlap with entire introns; (3) natural antisense transcripts (NATs), transcribed from the opposite strand of the protein-coding sense transcripts [11]; (4) enhancer RNAs, which stabilize the interaction between enhancers and promoters of nearby target genes [12]; and (5) transcribed ultra-conserved elements (T-UCEs), derived from DNA sequences and with the remarkable feature of retaining extended perfect sequence identity between human, mouse, and rat genomes [13]. LncRNAs are developmentally and temporally regulated by gene expression [14,15,16,17,18]. Since lncRNA expression is more cell- and tissue-specific than mRNA expression [19,20,21,22], stem cells offer a useful system for studying their critical functions during stem cell differentiation and somatic cell reprogramming [23]. The highly multifaceted nature of lncRNAs in exhibiting distinct cytotopic localizations (i.e., nucleus/cytosol) and in operating via different modes of action and with different target specificity is advancing the idea that they participate in many different regulatory pathways, reflecting their multi-functional role in cells [19,20,21,24]. By interplaying with different types of molecular factors, including DNA, RNA, and proteins, they generate an intricate network of interactions with transcripts, enhancers, and chromatin-modifier complexes [25]. This network endows lncRNAs with the capacity to transduce higher-order regulatory networks during stem cell differentiation by orchestrating the patterning of cells into tissues during development [13,26]. Although growing evidence indicates that lncRNAs exert critical physiological functions involved in genome integrity, cellular homeostasis, and metabolic responses in adult tissue stem cells, as well as in cell lineage commitment and the pluripotency network, their mode of action remains elusive and few have been mechanistically characterized to any great extent. To date, several annotated lncRNAs are preferentially localized in the nucleus, though very few have been functionally characterized as regulators of gene expression in different cellular contexts and events, including transcriptional and post-transcriptional regulation and reorganization of chromatin architecture. However, the biological role of several lncRNAs in vertebrate development such as Hotair [27], MIAT/Gumafu [28], Evx1-as [29], Braveheart, and Haunt [30] is still debated. Mutant studies in mouse models reported the dispensable role of some lncRNAs in viability, embryogenesis, and fertility, raising the possibility of their phenotypical redundancy and challenging the paradigm that RNA itself does not play a role in the observed functions [31,32]. Moreover, some lncRNAs were shown to exert a local regulation by acting as promoters for cis-regulation of neighboring genes [19,33]. Here, we summarize recent advances that have revealed the functions and molecular mechanisms involved in transcriptional regulation of annotated nuclear-retained lncRNA in stem cell biology, focusing on pluripotency and stem cell differentiation. This review addresses the principles of how lncRNAs cross-talk with chromatin by recruiting, tethering, and modulating the activity of epigenetic factors. In particular, we discuss the functional and intimate partnership between the chromatin modifier Polycomb repressive complex (PRC) 2 and lncRNAs as a classic example of how these molecules shape the epigenetic landscape and how this impacts on cell-fate programming and reprogramming. We also examine the latest insights into lncRNA functional roles, due in part to the advent of single-cell analysis, as well as future perspectives and current limitations in the field.
2. Nuclear-Retained LncRNAs and Their Functions
Among the well-studied lncRNAs, many reside in the nucleus where they play a variety of roles in basal cellular processes, including direct interaction with the transcription machinery, RNA splicing and maturation, chromatin remodeling, and the organization of nuclear domains [19,34]. LncRNAs perform these functions through different mechanisms: (1) They can act as transcriptional regulators with other nucleic acids forming DNA/RNA duplexes able to activate or repress transcription via cis-acting regulation of the expression of genes near their site of synthesis on the same chromosome, or trans-acting regulation of the expression of distally located genes across multiple chromosomes. LncRNAs can also interact directly with TFs, facilitating their localization and the association of TFs or other co-factors with chromatin in a cell- or tissue-specific manner [35,36]; (2) They participate in post-transcriptional events by modulating alternative splicing of sets of genes and controlling the stability of coding transcripts [37,38]; (3) They are involved in the organization of nuclear structure, interacting with the nuclear matrix either by acting as a ‘scaffold’ to modulate interchromosomal interactions or by rearranging nuclear speckles, structures involved in processing, maturation, and the export of precursor mRNA (pre-mRNA) [39,40]; (4) They regulate methylation and epigenetic states, guiding chromatin-remodeling factors to specific genomic loci by cooperating with histone readers, writers, and erasers—chromatin modifier enzymes that add, recognize, or remove post-translational modifications on histone proteins [41,42]. Among all the functions assigned to nuclear lncRNAs to date, epigenetic modifiers are the most frequent coupled protein partners [42]. Recent technologies probing lncRNA and chromatin interactions include RNA immunoprecipitation (RIP), cross-linking and immunoprecipitation (CLIP), chromatin isolation by RNA purification (ChIRP), capture hybridization analysis of RNA targets (CHART), and RNA antisense purification (RAP) [43]. These approaches, together with high-throughput tiling microarrays and innovations in next-generation sequencing (NGS)-based techniques applied to transcribed regions, have generated a genome-wide map of lncRNA-chromatin interactions providing direct evidence that a plethora of lncRNAs are associated with specific genomic regions, that they fold into domains, and that by recruiting epigenetic modulators, they mediate chromatin loop formation and control gene expression. Specifically, several lncRNAs were shown to interact with repressing and activating complexes, such as PRCs [43], involved in chromatin compaction and transcriptional silencing, and the SWitch/Sucrose NonFermentable (SWI/SNF) chromatin-remodeling complex [44]. In the following sections, we discuss the latest insights into lncRNA-mediated epigenetic regulations in the balance between ESC self-renewal and differentiation, and in particular, the modification of chromatin states via lncRNA-PRC2 interplay acting as a repressor of gene expression [44].
3. Polycomb Repressive Complex 2 in Embryonic Stem Cells
ESCs show the remarkable property of retaining pluripotency gene expression programs while enhancing cell fate transitions. Polycomb proteins, initially identified in Drosophila melanogaster as repressors of Hox homeotic genes, are evolutionary conserved from invertebrates to mammals. These proteins play a critical role in developmental pathways ensuring the proper activation of differentiation programs [45,46,47]. In fruit flies, Polycomb complexes are recruited to chromatin by conserved and extended 1 kb DNA domains called Polycomb response elements (PREs) through the activity of PRE-binding proteins [48]. PRE-like elements have not yet been defined in mammals. However, the function of, and the epigenetic mechanism by which PRCs act as repressor complexes—facilitating chromatin oligomerization that leads to transcriptional silencing via inhibition of chromatin remodeling activity—are evolutionarily conserved in eukaryotic cells [49]. Two specific complexes, PRC1 and PRC2, have been extensively studied [50,51]. Emerging evidence indicates that these distinct complexes can work both individually and synergistically by sharing common target genes in order to increase the level of gene repression. Specifically, PRC2 catalyzes histone H3 lysine 27 (H3K27) methylation and is thought to recruit PRC1 at the promoter level by working as a docking site for the PRC1, which catalyzes ubiquitylation of histone H2A on lysine 119 (H2AK119ub), thus maintaining transcriptional repression [51,52]. The mammalian PRC2 core complex contains four key proteins: embryonic ectoderm development (EED), suppressor of zeste 12 (SUZ12), RBAP48, and the catalytic enhancer of the zeste homolog 1/2 (EZH1/2) (Figure 1).
EZH1/2 contain a putative RNA-binding domain. These histone methyltransferases are responsible for the enzymatic activity of PRC2 by catalyzing mono-, di-, and trimethylation of H3K27 (H3K27me1, H3K27me2, and H3K27me3, respectively) [53,54,55]. PRC2 is tightly involved in the early steps of embryogenesis. Although EZH1/2 and EED knockout (KO) mice exhibit embryonic lethality after implantation, PRC2 deficiency seems to be dispensable in ESC self-renewal since it alters global H3K27me3 patterns without modifying the expression level of pluripotency-associated genes. Nevertheless, EZH2 KO ESCs are primed to spontaneous differentiation, leading to an increase in the expression of PRC2-targeted genes biased to differentiation and lineage commitment [56,57]. PRC2 is responsible for establishing cell type-specific gene expression programs by associating several regulatory subunits to the core complex. These accessory proteins, also required for a proper enzymatic activity or able to increase the affinity with a putative RNA binding, could directly influence and modify the interaction between PRC2 and chromatin and modulate the transcription of selected genes driving stem cell differentiation [58]. Metal response element binding transcription factor 2 (MTF2) and Jumonji AT-rich interactive domain 2 (JARID2), are newly identified regulatory subunits that interact with PRC2 in ESCs [59]. MTF2 depletion is accompanied by an increase in expression of the core pluripotent genes maintaining ESCs in an undifferentiated state, thereby preventing differentiation [60,61]. JARID2, a member of the Jumonji protein family of histone demethylases but without catalytic activity, plays an essential role during the early stages of development, interacting with and regulating PRC2 activity and facilitating its recruitment to promoters of target loci [62]. JARID2 expression is regulated by OCT4, which together with NANOG and SOX2 constitutes a triad of TFs crucial for ESC self-renewal and pluripotency, and is rapidly downregulated upon exposure to proper differentiation signals [63]. ESCs lacking JARID2 are prone to differentiate, enabling the cell response to signals of differentiation [64]. The unique property of ESCs that enables them to maintain a stable pluripotent phenotype while simultaneously remaining poised to differentiate upon the occurrence of external stimuli is finely tuned by chromatin-based mechanisms in which key developmental regulatory genes are dynamically marked in promoter regions by bivalent epigenetic signatures. Bivalent chromatin domains are genomic loci characterized by simultaneous occupancy of both H3K4me3 and H3K27me3 epigenetic modifications in the same region and were shown to endow mouse ESCs with a unique epigenetic landscape [65,66] (Figure 1). The balance between positive and negative histone modifications determines whether RNA polymerase II, pre-loaded and phosphorylated at serine 5 of its C-terminal domain on the bivalent region, is paused or released from pause, thus facilitating recruitment of the positive transcription elongation factor (P-TEFb) and preparing genes for rapid activation [67]. Emerging findings show that the repressive H3K27me3 mark is deposited exclusively by PRC2 and that it is stabilized by JARID2 interaction, which also protects it from the activity of H3K27 demethylases and is crucial in establishing the poised RNA polymerase II. Upon terminal differentiation, the bivalent chromatin domain could be converted into either a stable active (H3K4me3) or stable repressed (H3K27me3) state by the action of H3K27 or H3K4 demethylases, respectively [13,68] (Figure 1).
4. LncRNA-Directed Chromatin Regulation via PRC2 in Cell Fate
Recent breakthrough discoveries postulate a mechanistic model in which PRC2 identifies its site-specific target genes via regulatory RNAs, thus pointing to a functional interplay between non-coding RNA and chromatin modifiers [69,70]. RIP sequencing technology provided for the first time genome-wide profiles of non-coding RNAs including antisense, intergenic, and promoter-associated transcripts that directly interact with PRC2 in ESCs, putting forward the concept that nuclear lncRNAs are key players in chromatin epigenetic status regulation [43]. They act as cofactors to recruit Polycomb proteins to target loci prevalently via the EZH2 subunit, introducing epigenetic changes to chromatin, in order to direct stem cell patterning in tissues and organs during development, thus raising the question of how PRC2 discriminates between distinct RNAs in physiological contexts. In the following subsections, we describe some well-studied examples of lncRNAs that modulate chromatin structure and function in stem cells and differentiation by interplaying with PRC2.
4.1. X Inactive-Specific Transcript (Xist)
Xist was one of the first regulatory lncRNAs to be identified that challenged the simple recruitment model of epigenetic machinery to chromatin. This 17 kb lncRNA functions as a master regulator of chromatin epigenetic status and exemplifies the importance and versatility of lncRNAs in multiple physiological cellular processes [71,72]. In the last two decades, the study of Xist has provided a greater understanding of how lncRNAs guide histone modifiers to modulate epigenetic control of gene transcription and institute stable repressive chromatin states [73]. These insights led to the identification of novel interactors and new molecular mechanisms underlying this intricate partnership. In mammals, Xist is responsible for the entire inactivation of one X-chromosome in female somatic cells by forming a transcriptionally repressed heterochromatic structure (termed inactive X, Xi, or Barr body) in order to equalize X-chromosome dosages between the sexes. Transcribed from the X-inactivation center, Xist initiates, propagates, and maintains repression by physically coating the X-chromosome in cis, thus triggering extensive histone methylation and chromatin compaction via recruitment of chromatin modifiers [74,75,76,77] (Figure 2).
Xist levels are finely tuned by two nearby genes transcribed from the active X-chromosome, one of which is Jpx. The Jpx gene transcribes a lncRNA inducing Xist transcription and the antisense lncRNA Tsix, which enforces Xist silencing either by recruiting DNA methyltransferase 3a (Dnmt3a) on the Xist promoter or via RNA antisense targeting [78,79]. PRC2 is recruited for initiation of the X-chromosome inactivation (XCI) by direct and specific binding with RepA, a 1.6 kb lncRNA located within the 5′ end of the Xist gene spanning the evolutionary conserved A-repeat domain of Xist [80,81]. By RIP-seq, it was demonstrated that EZH2 and JARID 2 mediate the binding of PRC2 to Xist [43] (Table 1) and facilitate PRC2 recruitment on chromatin, which lays down the repressive H3K27me3 mark. Tsix hinders RepA-PRC2 function by titrating and preventing RepA-PRC2 transfer to chromatin and modulating PRC2 activity. RepA depletion abolishes full-length Xist induction and H3K27me3 deposition, while PRC2 silencing affects Xist upregulation [82,83]. Using an unbiased proteomic approach, a recent study identified an RNA-binding protein, ATRX, with high affinity for PRC2, which mediates direct binding of the epigenetic complex to Xist. ATRX silencing leads to spatial redistribution of PRC2 occupancy by altering Polycomb target gene regulation at the global level on the genome [84]. PRC1 is also involved in chromosome silencing induced by Xist. The noncanonical Polycomb group RING finger 3/5 (PCGF3/5)-PRC1 complex was shown to initiate recruitment of both PRC1 and PRC2 during XCI by catalyzing post-translational modifications of core histone proteins via mono-ubiquitylation of histone H2A on lysine 119 (H2AK119ub1) and H3K27me3, respectively. An RNA-binding protein is required to recruit PCGF3/5-PRC1 and guide binding with Xist via a specific Xist-Polycomb interaction domain, a 600 nucleotide sequence containing the Xist B-repeat element. PCGF3/5 depletion leads to female-specific embryo lethality and abolishes Xist-mediated gene repression [85]. Biallelic X-chromosome activation is considered an important hallmark of ESC pluripotency, as it is able to stabilize the naïve state of pluripotent stem cells by arresting differentiation. Indeed, the grade of pluripotency distinguishing the naïve and primed pluripotency of female ESCs and epiblast-derived stem cells, as well as the identification of fully reprogrammed colonies in induced pluripotent stem cell (iPSC) cultures, can be defined by the X-chromosome methylation status [86,87]. The presence of double X-chromosome activation inhibits MAPK and GSK3 signaling, known to be key pathways promoting the exit from the pluripotent state, and stimulates LIF/STAT3 and PI3K/AKT pathways required to maintain self-renewal and pluripotency. On adopting a specific cell fate, ESCs exit from pluripotency and the transition toward a differentiated state is characterized by dramatic chromatin changes leading to XCI. In this scenario, the germline factor PRDM14 exerts a critical function in self-renewal and ESC pluripotency by playing a dual role in repressing Xist during X-chromosome reactivation (XCR). PRDM14 silences the Xist activator Rnf12 via recruitment of PRC2 and directly targets Xist by binding intron 1. Gain-of-function experiments showed that PRMD14 overexpression accelerates XCR during the conversion of epiblast-derived stem cells to ESC transition and affects iPSC derivation and maintenance [88]. iPSC reprogramming is also associated with XCR, underscoring the need for chromatin remodeling to achieve a completely undifferentiated state. Female iPSCs are less stable in long-term culture compared to male-derived iPSCs and undergo erosion of dosage compensation, which compromises H3K27me3 foci in XCI and Xist, leading to ectopic expression of genes from the inactive X-chromosome [89]. As in Xist-mediated XCI, the 91 kb-long lncRNA Kcnq1ot1, exclusively localized in the nucleus, is important for bidirectional repression of genes in the Kcnq1 domain [90,91]. Kcnq1ot1 induces and maintains transcriptional silencing by recruiting the histone methyltransferase G9a and PRC2 via a lineage-specific mechanism. Specifically, its interaction with chromatin and subsequent repressive patterns were found predominantly in extraembryonic compared to other embryonic tissues. Within the Kcnq1 domain, repressive histone marks span both nearby genes located within 200 kb of the Kcnq1 imprinting control region and distantly located imprinted genes only in the placenta, underlying lineage-specific silencing modes of action [92].
4.2. Maternally Expressed 3 (Meg3)
Meg3, also known as gene trap locus 2, is a lncRNA encoded in the imprinted Dlk1-Dio3 gene region on chromosome 14q32.2 [98,99]. The cis-elements regulating Meg3 expression comprise two differentially methylated regions (DMRs): intergenic (IG)-DMR and Meg3-DMR [100]. Meg3, together with multiple non-coding RNAs and small non-coding RNAs, including the largest mammalian miRNA cluster, is expressed from the maternally inherited allele only. IG-DMR and Meg3-DMR are unmethylated on the maternal allele, whereas they are methylated on the paternally inherited allele, from which three protein-coding genes (Dlk1, Rtl1, Dio3) are expressed [100]. Meg3 gene deletion obtained by targeting either Meg3 or IG-DMR is embryonically lethal and different phenotypes are observed depending on the KO model [101,102,103]. Furthermore, the Dlk1-Dio3 gene locus expression is linked to the optimal cellular reprogramming process and efficiency of the iPSC generation. RNA expression patterns of ESCs and iPSCs were in fact shown to be essentially indistinguishable with the exception of few maternally expressed non-coding transcripts originating from the Dlk1-Dio3 cluster, which is silenced in the majority of iPSC lines, thus compromising the ability of cells to generate entirely iPSC-derived adult mice (known as ‘all-iPSC mice’) [104]. However, this remains a controversial issue, and further studies will be needed to fully define the tight correlation between the loss of imprinting at the Dlk1-Dio3 locus and reduced pluripotency [105].
Mechanistic studies reported a role for Meg3 in epigenetic regulation through its interaction with chromatin-modifying complexes such as PRC2, thereby guiding these complexes to genomic sites via DNA/RNA triplex formation [106]. The physical interaction between Meg3 and EZH2 was described [93] (Table 1) and Meg3 was shown to be indispensable for the proper recruitment and assembly of PRC2 in a subset of target genes in human iPSCs [94]. In addition, native and cross-linked RIP of JARID2 revealed that Meg3, as well as other lncRNAs deriving from the imprinted Dlk1-Dio3 locus, interacts with PRC2 via JARID2 [94]. In the absence of JARID2, the interaction between Meg3 and PRC2 is much weaker, suggesting that the contact point of JARID2 gives the major contribution to the affinity for Meg3 [94]. However, in vitro binding analysis of Meg3 suggests that JARID2 does not bind to RNA in a sequence-specific manner, in line with the finding that the primary sequences of lncRNAs are not generally well conserved [24,107].
Of note, although the mechanistic details of epigenetic alteration of the imprinted Dlk1-Dio3 locus during reprogramming remain elusive, it is plausible that the interaction between Meg3 and PRC2 plays an important role [108].
4.3. Trans-Spliced Rhabdomyosarcoma 2 Associated Transcript (tsRMST)
Alternative splicing, which arises from post-transcriptional events, can lead to the generation of multiple transcript isoforms from a single gene, thus providing an essential source of diversity for the transcriptome and proteome [109,110,111,112,113]. Splicing can occur either in cis or in trans [114,115]. A single pre-mRNA is originated by cis-splicing of exons, whereas two or more separate pre-mRNAs are derived either from the same gene or from two or more different genes by intragenic and intergenic trans-splicing, respectively. Trans-splicing is largely detected by comparing reference genomes with expressed sequence tags (ESTs)/mRNAs [116,117,118,119] or by the NGS of mRNAs (RNA-seq) [120,121,122,123]. However, these kinds of approaches generate a significant number of false positives. To tackle this issue, a computational pipeline named TSscan, based on transcriptome sequencing data from different NGS platforms and several undifferentiated hESC lines was developed, and confirmed that four trans-splicing events (tsCSNK1G3, tsARHGAP5, tsFAT1, tsRMST) occur in hESCs [95]. All four trans-spliced RNAs are also expressed in human iPSCs, suggesting that these events tend to occur in human pluripotent stem cells. Of particular interest is tsRMST, the first trans-spliced lincRNA to be identified, which is highly expressed in both hESCs and iPSCs, and whose expression is reduced after in vitro hESC differentiation, suggesting the possibility that tsRMST is specifically expressed in pluripotent stem cells, and may thus play a role in pluripotency maintenance. Microarray-based genome-wide expression profiling performed on tsRMST knockdown hESCs showed that the expression levels of pluripotency-associated genes such as NANOG, POU5F1, and SOX2 were significantly decreased, whereas key lineage-specific TFs such as GATA6 (endoderm) and PAX6 (neuroectoderm) were induced [95]. At the mechanistic level, RIP analysis showed that tsRMST interacts directly with SUZ12 (Figure 3 and Table 1) and NANOG. In line with these results, chromatin immunoprecipitation qPCR experiments on tsRMST promoters repressing lineage-specific genes (GATA6 and PAX6) further showed the loss of NANOG and SUZ12 occupancy, as well as H3K27me3 modification in tsRMST knockdown hESCs (Figure 3) [95]. A model was proposed in which tsRMST suppresses lineage differentiation in hESCs via recruitment of NANOG and PRC2 [95] and plays a regulatory role in noncanonical Wnt signaling [124].
4.4. Long Intergenic Noncoding HOXA1 (Linc-HOXA1)
Linc-HOXA1 (also known as Haunt, Halr1, and Gm15055) is located approximately 50 kb downstream of the Hoxa gene cluster. By single-cell transcript counting performed in mESCs, there emerged a considerable heterogeneity of linc-HOXA1 and Hoxa1 RNA in pluripotent cultures at single-cell resolution and an inverse correlation between linc-HOXA1 RNA and Hoxa1 mRNA abundance (Figure 4) [96]. Single-cell multiplex transcript counting analysis is crucial to see the effects that, as will be discussed in ‘State of the Art and Future Perspectives’, might be hard to observe using bulk measurements.
At the molecular level, linc-HOXA1 recruits the protein PURB as a transcriptional cofactor, thereby repressing Hoxa1 transcription in cis [125]. PRC2 is recruited to the Hoxa locus and is part of a complex mechanism for fine-tuning Hoxa gene regulation in mESCs. The linc-HOXA1 gene locus contains an OCT4-responsive positive cis-regulatory element, which potentially regulates the lincRNA itself and Hoxa gene activation. Further studies based on the generation of linc-HOXA1 KO ESCs, aimed at investigating the regulation of Hoxa gene expression and chromatin status in mESCs, highlighted by RIP the interaction between linc-HOXA1 and PRC2 [125]. Specifically, linc-HOXA1 recruits PRC2 to the Hoxa gene cluster to maintain the repressive histone mark H3K27me3 on Hoxa gene promoters in mESCs. Alkaline phosphatase (a stem cell membrane marker) staining, as well as immunofluorescence and RT-qPCR analysis of the pluripotency markers SSEA1 and OCT4, confirmed that pluripotency of mESCs was not disrupted by linc-HOXA1 knockdown, and excluded the possibility that the increased Hoxa gene expression was due to mESC differentiation [96]. In contrast, H3K27me3 modification and SUZ12 binding (Table 1) were decreased at the Hoxa gene promoters. However, no significant changes in H3K4me3 were observed at Hoxa gene promoters in KO ESCs [96].
4.5. The Transcribed Ultraconserved LncRNA T-UCstem1
The ultraconserved lncRNA T-UCstem1 was recently identified and its role in ESC self-renewal maintenance was elucidated. T-UCstem1 exerts a dual but distinct function in the nucleus and in the cytosol. In the nucleus, as demonstrated by RIP, T-UCstem1 directly binds PRC2, recruiting the complex to bivalent chromatin-associated genes. In T-UCstem1-depleted cells, PRC2 displacement leads to an increased H3K4me3/H3K27me3 ratio at bivalent chromatin domains, committing ESCs to exit from pluripotency and to differentiation [13]. Interestingly, absence of T-UCstem1 determines a cellular phenotype similar to that resulting from the withdrawal of PRC2 activity from ESCs, and a global gene derepression of bivalent chromatin-associated genes (Table 1) leading to spontaneous differentiation [67,126,127].
4.6. Heart-Associated LncRNA Braveheart (Bvht)
Bvht is a heart-associated lncRNA in the mouse. Using multiple ESC differentiation strategies, Bvht was shown to be essential for nascent mesoderm progression towards cardiac fate. Specifically, Bvht triggers a cardiovascular gene network core upstream the mesoderm posterior 1 (MesP1) (Table 1), a master regulator of multipotent cardiovascular progenitors. At the molecular level, by native RIP, Bvht was shown to interact with SUZ12 at numerous stages during ESC-cardiac differentiation directly. In cells lacking Bvht expression, SUZ12 and the repressive modification H3K27me3 are enriched at promoters of cardiac-associated genes, such as MesP1. Interestingly, these genes still exhibit bivalent chromatin domains in Bvht-depleted cells, similar to their initial configuration in ESCs, in line with the inability of these cells to trigger cardiac cell commitment [97]. Of note, Bvht is an example of lncRNA involved in differentiation while all the others we mentioned play a role in the stemness maintenance.
Of note, LncRNAs can also influence the interaction between PRC2 and other epigenetic modifying complexes, working as a scaffold to bind multiple chromatin-modifier structures. For instance, the lateral mesoderm-specific lncRNA Fendrr, critical for cardiac mesoderm specification, positively modulates chromatin state to activate a subset of developmental regulators by binding both PRC2 and Trithorax group/MLL protein complexes (TrxG/MLL) [128]. Recent studies also described lncRNA binding in the regulation of active chromatin configuration states. TrxG/MLL, which catalyzes methylation of the activation epigenetic mark histone H3 at lysine 4, could be recruited to the HoxA promoter by the lncRNAs HOTTIP and Mistral through the interaction with WDR5, a specific subunit of the MLL complex, or the lncRNA HoxBlinc, which is associated with active chromatin states by regulating cardiac/hematopoietic differentiation [129].
5. State of the Art and Future Perspectives
LncRNAs participate in a wide variety of stem cells and developmental physiological processes such as transcriptional regulation, chromatin modification, imprinting, DNA methylation, and splicing, introducing a novel layer of biological regulation. The functional role of lncRNAs in chromatin remodeling in a temporal and cell type-specific manner has become a hot topic. LncRNAs act as important cofactors in mediating PRC2 recruitment to specific target loci, thus determining specific chromatin states corresponding to different stages of embryonic development and stem cell differentiation. Although promiscuous RNA binding to PRC2 is described [83], several studies underscore the importance of lncRNA binding to this chromatin remodeling complex, which then mediates cofactor recruitment conferring its specificity of action. While ESCs have provided a powerful model for investigating the developmental role of these multifaceted molecules, research into the molecular mechanism by which lncRNAs guide PRC2 to chromatin is still in its infancy and current knowledge is greatly limited by methodological shortcomings. In addition, the extremely cell-specific mode of action of lncRNAs makes the understanding of their molecular mechanisms even more challenging.
5.1. Current and Future Methodologies
Several methods have recently been developed to map lncRNAs on chromatin, shedding considerable light on their biological importance in epigenetic regulation. Similar to the well-established ChIP technique, RIP has proved to be a powerful tool to study the direct binding between individual proteins and RNA molecules in close proximity [130,131]. Using a specific antibody against the protein of interest, the RNA-binding protein is pulled down and further analyzed by PCR-based approaches, hybridization, or sequencing. RIP can be carried out either in a native condition to identify lncRNAs directly associated with the protein and to quantify their abundance within immunoprecipitates, or by using CLIP strategies where whole tissues or cells are treated with UV-B irradiation. Cross-linking generates a covalent bond, which serves to precisely map the direct and indirect binding sites between RNA-protein complexes. Recent technologies directly probing lncRNAs have greatly advanced our understanding of lncRNA function in ESC gene regulation. CHART is able to identify where lncRNAs localize, as well as their protein partners, providing a genomic map of lncRNA targets [130]. The biotinylated complementary oligonucleotides are designed to have a high affinity for the lncRNA of interest. Similarly, RAP identifies regions of the target RNA through hybrid capture via antisense oligonucleotides, allowing sequencing of RNA and DNA from purification samples, but like CHART, requires prior knowledge of the lncRNA sequence of interest. ChIRP is another useful and rapid method to study the role of lncRNA in epigenetic regulation. ChIRP maps genomic binding sites of chromatin-associated lncRNAs, identifying lncRNA-chromatin interactions by including complementary oligonucleotides that hybridize to the target lncRNA [130]. All these techniques can be extended genome-wide using sequencing methodologies and integrated to identify the genomic localization of RNA and its coupled protein, generating a more comprehensive profile of lncRNA function, including recruitment of chromatin-modifiers.
5.2. The Advent of Single-Cell Technologies: Limits and Potential
Nascent NGS technologies at single-cell resolution could provide valuable insights into the complex role of lncRNA in stem cell differentiation. Single-cell RNA sequencing (scRNAseq) correlates the transcriptional profile of individual cells to the complexity of biological systems within a population [132,133,134]. ScRNAseq provides a better understanding of the function of a single cell within the context of its microenvironment by identifying novel cell types and characterizing heterogeneous subtypes (Figure 3), highlighting regulatory relationships between coding and non-coding genes, and tracing the trajectories of distinct cell lineages during differentiation. High-resolution at the single-cell level could overcome the current limitation in bulk RNA sequencing due to the low abundance of lncRNAs and the difficulty in distinguishing them from transcriptional noise. A recent study reported the direct comparison of lncRNA profiles by combining bulk tissue sequencing and single-cell RNAseq of the human neocortex at different stages of development. Single-cell transcriptomic analysis provided high-resolution data revealing that lncRNAs generally detected at low levels in bulk-seq are in fact abundantly expressed in individual cells, showing remarkable cell-type specificity. This may also explain why some lncRNAs display reduced expression in tissues [135]. Single-cell sequencing also addresses another major hurdle related to the tight connection between lncRNAs and the biological milieu in which they function [96]. However, precisely, how transcriptional heterogeneity and changes in the transcriptome reflect pluripotent states and differentiation can only be clarified by investigating the epigenome at the single-cell level. Since transcriptional changes precede or follow epigenetic marks, single-cell epigenome sequencing provides a high-resolution map of chromatin accessibility, including open or closed chromatin states and nucleosome positioning. Taken alone, scRNAseq results leave unanswered questions that might be solved by integrating multiple levels of information. For instance, to better unravel the molecular mechanisms underlying lncRNAs and their role in tethering the PRC2 complex and other chromatin modifiers, an integrative analysis combining ChIPseq and scRNAseq experiments could be useful. Despite being a challenging task, the possibility of obtaining an integrated analysis of multi-omic profiles at the single-cell level would undoubtedly be a powerful tool. Combined single-cell RNA and epigenome sequencing could break up intricate regulatory systems into individual layers, providing new insights into how genetic variation is related to transcriptional variation. The technological advancements for efficiently identifying open chromatin regions and regulatory elements across the genome are ongoing. A variety of assays have been designed to assess how chromatin compaction and DNA-binding proteins regulate gene expression at single-cell resolution since epigenetic regulation comes in different forms. Nevertheless, epigenome-based methods have several limitations when used at the single-cell level, yielding lower throughput and scalability compared to scRNAseq.
Currently, the use of bisulfite sequencing to study nucleosome occupancy and methylome at the single-cell level limits the capture rates and does not provide extensive and functional mapping of chromatin states at single-cell resolution. In contrast, the assay for transposase-accessible chromatin sequencing (ATAC-seq) will likely become a routinely used strategy in single-cell analysis and represents an important step forward in assessing epigenetic diversity. ATAC-seq identifies genome-wide chromatin accessible regions by using a transposase enzyme to insert and tag open chromatin. Via a microfluidic approach, this technique uses barcoding to successfully process tens of thousands of single cells in parallel, providing a regulatory landscape of the genome at the epigenetic level. A common drawback in both single-cell transcriptomics and epigenomics is technical variability. Although the level of technical noise in scRNAseq could be estimated and normalized, such strategies have not yet been developed in epigenome sequencing [136].
6. Conclusions
Their extremely tissue-specific expression and exclusive mode of action further complicate our understanding of the role of lncRNA in physiological processes. Ongoing studies are, therefore, combining genome-wide transcriptional analysis of single cells with the complexity and heterogeneity of tissue structures by using in situ transcriptomics technology to map lncRNAs based on their localization. Thanks to their cell type- and organ-specific expression patterns, lncRNAs might be employed as candidate targets for stem cell-based therapies and other clinical applications such as the screening of new drugs to introduce chromatin changes. While epigenetic drugs produced to date function mainly as enzyme inhibitors by attenuating their activity and inducing drastic chromatin changes in the genome, manipulating lncRNA expression that normally targets one gene or a small set of genes may reduce the aberrant expression of off-target genes and facilitate the development of small-molecule drugs with more specific modes of action. Loss-of-function studies using both small interfering RNA and antisense oligonucleotides, as well as gain-of-function experiments describe only local alterations without detecting changes in overall chromatin configuration. Moreover, deciphering the ‘epigenetic code’ of lncRNA could unravel the intricate network of these different molecule species, elucidating the molecular mechanisms controlling stem cell differentiation and their potential involvement in tissue regeneration.
Funding
This work was supported by the TRNSCAN-2 Project BeFIT; AIRC (IG 20736), Epigenomics Flagship Project (EPIGEN) MIUR-CNR, Italian Ministry of Education-University-Research (grant CTN01_00177 Cluster ALISEI_IRMI).
Acknowledgments
We apologize to all the authors, whose work could not be cited due to space limitation. We thank Maria Rosaria Matarazzo for useful discussions. We acknowledge C. Fisher for English editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. Microrna-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Scholer, H.R. Regulatory networks in embryo-derived pluripotent stem cells. Nat. Rev. Mol. Cell Biol. 2005, 6, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode v7 catalog of human long noncoding rnas: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human hox loci by noncoding rnas. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding rnas. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Zheng, H.; Li, J.; Liu, D.; Li, H.; Samudrala, R.; Yu, J.; Wong, G.K. Mouse transcriptome: Neutral evolution of ‘non-coding’ complementary dnas. Nature 2004, 431, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Ballarino, M. Long noncoding rna regulation of pluripotency. Stem Cells Int. 2016, 2016, 1797692. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.Y.; Johnson, R.; Stanton, L.W. Human long non-coding rnas promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. Embo J. 2012, 31, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding rnas. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef]
- Wight, M.; Werner, A. The functions of natural antisense transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Orom, U.A.; Shiekhattar, R. Long noncoding rnas usher in a new era in the biology of enhancers. Cell 2013, 154, 1190–1193. [Google Scholar] [CrossRef] [PubMed]
- Fiorenzano, A.; Pascale, E.; Gagliardi, M.; Terreri, S.; Papa, M.; Andolfi, G.; Galasso, M.; Tagliazucchi, G.M.; Taccioli, C.; Patriarca, E.J.; et al. An ultraconserved element containing lncrna preserves transcriptional dynamics and maintains esc self-renewal. Stem Cell Rep. 2018, 10, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding rnas. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Dostie, J. Reciprocal regulation of chromatin state and architecture by hotairm1 contributes to temporal collinear hoxa gene activation. Nucleic Acids Res. 2017, 45, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, Z.; Xu, X.; Liu, G.; Song, X.; Wang, R.; Sui, X.; Liu, T.; Chang, X.; Huang, D. As1dhrs4, a head-to-head natural antisense transcript, silences the dhrs4 gene cluster in cis and trans. Proc. Natl. Acad. Sci. USA 2012, 109, 14110–14115. [Google Scholar] [CrossRef]
- Sarropoulos, I.; Marin, R.; Cardoso-Moreira, M.; Kaessmann, H. Developmental dynamics of lncrnas across mammalian organs and species. Nature 2019, 571, 510–514. [Google Scholar] [CrossRef]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncrnas at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Ulitsky, I.; Bartel, D.P. Lincrnas: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding rnas in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding rna-ror modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Fico, A.; Fiorenzano, A.; Pascale, E.; Patriarca, E.J.; Minchiotti, G. Long non-coding rna in stem cell pluripotency and lineage commitment: Functions and evolutionary conservation. Cell. Mol. Life Sci. 2019, 76, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Scarola, M.; Comisso, E.; Pascolo, R.; Chiaradia, R.; Marion, R.M.; Schneider, C.; Blasco, M.A.; Schoeftner, S.; Benetti, R. Epigenetic silencing of oct4 by a complex containing suv39h1 and oct4 pseudogene lncrna. Nat. Commun. 2015, 6, 7631. [Google Scholar] [CrossRef]
- Pal, D.; Rao, M.R.S. Long noncoding rnas in pluripotency of stem cells and cell fate specification. Adv. Exp. Med. Biol. 2017, 1008, 223–252. [Google Scholar]
- Amandio, A.R.; Necsulea, A.; Joye, E.; Mascrez, B.; Duboule, D. Hotair is dispensible for mouse development. PLoS Genet. 2016, 12, e1006232. [Google Scholar] [CrossRef]
- Ip, J.Y.; Sone, M.; Nashiki, C.; Pan, Q.; Kitaichi, K.; Yanaka, K.; Abe, T.; Takao, K.; Miyakawa, T.; Blencowe, B.J.; et al. Gomafu lncrna knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci. Rep. 2016, 6, 27204. [Google Scholar] [CrossRef]
- Bell, C.C.; Amaral, P.P.; Kalsbeek, A.; Magor, G.W.; Gillinder, K.R.; Tangermann, P.; di Lisio, L.; Cheetham, S.W.; Gruhl, F.; Frith, J.; et al. The evx1/evx1as gene locus regulates anterior-posterior patterning during gastrulation. Sci. Rep. 2016, 6, 26657. [Google Scholar] [CrossRef]
- Han, X.; Luo, S.; Peng, G.; Lu, J.Y.; Cui, G.; Liu, L.; Yan, P.; Yin, Y.; Liu, W.; Wang, R.; et al. Mouse knockout models reveal largely dispensable but context-dependent functions of lncrnas during development. J. Mol. Cell Biol. 2018, 10, 175–178. [Google Scholar] [CrossRef]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Goff, L.A.; Lodato, S.; Bonev, B.; Groff, A.F.; Gerhardinger, C.; Sanchez-Gomez, D.B.; Hacisuleyman, E.; Li, E.; Spence, M.; et al. Multiple knockout mouse models reveal lincrnas are required for life and brain development. eLife 2013, 2, e01749. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear long noncoding rnas: Key regulators of gene expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding rnas: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ye, B.; Yang, L.; Zhu, X.; Huang, G.; Zhu, P.; Du, Y.; Wu, J.; Qin, X.; Chen, R.; et al. Long noncoding rna lnckdm2b is required for ilc3 maintenance by initiation of zfp292 expression. Nat. Immunol. 2017, 18, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. Meg3 long noncoding rna regulates the tgf-beta pathway genes through formation of rna-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, I.; Munita, R.; Agirre, E.; Dittmer, T.A.; Gysling, K.; Misteli, T.; Luco, R.F. A lncrna regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat. Struct. Mol. Biol. 2015, 22, 370–376. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, A.; Ho, T.T.; Zhang, Z.; Zhou, N.; Ding, X.; Zhang, X.; Xu, M.; Mo, Y.Y. Linc-ror promotes c-myc expression through hnrnp i and auf1. Nucleic Acids Res. 2016, 44, 3059–3069. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding rna hotair in protein ubiquitination. Nat. Commun. 2013, 4, 2939. [Google Scholar] [CrossRef]
- Puvvula, P.K.; Desetty, R.D.; Pineau, P.; Marchio, A.; Moon, A.; Dejean, A.; Bischof, O. Long noncoding rna panda and scaffold-attachment-factor safa control senescence entry and exit. Nat. Commun. 2014, 5, 5323. [Google Scholar] [CrossRef]
- Chalei, V.; Sansom, S.N.; Kong, L.; Lee, S.; Montiel, J.F.; Vance, K.W.; Ponting, C.P. The long non-coding rna dali is an epigenetic regulator of neural differentiation. eLife 2014, 3, e04530. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.S.; Ruthenburg, A.J. Nuclear fractionation reveals thousands of chromatin-tethered noncoding rnas adjacent to active genes. Cell Rep. 2015, 12, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated rnas by rip-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Tanigawa, A.; Naganuma, T.; Ohkawa, Y.; Souquere, S.; Pierron, G.; Hirose, T. Swi/snf chromatin-remodeling complexes function in noncoding rna-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4304–4309. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef]
- Lewis, E.B. A gene complex controlling segmentation in drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Cavalli, G. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 2009, 136, 3531–3542. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The polycomb complex prc2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Chan, C.S.; Rastelli, L.; Pirrotta, V. A polycomb response element in the ubx gene that determines an epigenetically inherited state of repression. EMBO J. 1994, 13, 2553–2564. [Google Scholar] [CrossRef]
- Muller, J.; Kassis, J.A. Polycomb response elements and targeting of polycomb group proteins in drosophila. Curr. Opin. Genet. Dev. 2006, 16, 476–484. [Google Scholar] [CrossRef]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of h2a restrains poised rna polymerase ii at bivalent genes in mouse es cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Endo, T.A.; Endoh, T.; Isono, K.; Sharif, J.; Ohara, O.; Toyoda, T.; Ito, T.; Eskeland, R.; Bickmore, W.A.; et al. Histone h2a mono-ubiquitination is a crucial step to mediate prc1-dependent repression of developmental genes to maintain es cell identity. PLoS Genet. 2012, 8, e1002774. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone h3 lysine 27 methylation in polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Czermin, B.; Melfi, R.; McCabe, D.; Seitz, V.; Imhof, A.; Pirrotta, V. Drosophila enhancer of zeste/esc complexes have a histone h3 methyltransferase activity that marks chromosomal polycomb sites. Cell 2002, 111, 185–196. [Google Scholar] [CrossRef]
- Muller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a drosophila polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef]
- O’Carroll, D.; Erhardt, S.; Pagani, M.; Barton, S.C.; Surani, M.A.; Jenuwein, T. The polycomb-group gene ezh2 is required for early mouse development. Mol. Cell. Biol. 2001, 21, 4330–4336. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Yee, D.; Magnuson, T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 2008, 26, 1496–1505. [Google Scholar] [CrossRef]
- Landeira, D.; Sauer, S.; Poot, R.; Dvorkina, M.; Mazzarella, L.; Jorgensen, H.F.; Pereira, C.F.; Leleu, M.; Piccolo, F.M.; Spivakov, M.; et al. Jarid2 is a prc2 component in embryonic stem cells required for multi-lineage differentiation and recruitment of prc1 and rna polymerase ii to developmental regulators. Nat. Cell Biol. 2010, 12, 618–624. [Google Scholar] [CrossRef]
- Shen, X.; Kim, W.; Fujiwara, Y.; Simon, M.D.; Liu, Y.; Mysliwiec, M.R.; Yuan, G.C.; Lee, Y.; Orkin, S.H. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 2009, 139, 1303–1314. [Google Scholar] [CrossRef]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. Jarid2 regulates binding of the polycomb repressive complex 2 to target genes in es cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef]
- Zhang, Z.; Jones, A.; Sun, C.W.; Li, C.; Chang, C.W.; Joo, H.Y.; Dai, Q.; Mysliwiec, M.R.; Wu, L.C.; Guo, Y.; et al. Prc2 complexes with jarid2, mtf2, and esprc2p48 in es cells to modulate es cell pluripotency and somatic cell reprogramming. Stem Cells 2011, 29, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kasinath, V.; Faini, M.; Poepsel, S.; Reif, D.; Feng, X.A.; Stjepanovic, G.; Aebersold, R.; Nogales, E. Structures of human prc2 with its cofactors aebp2 and jarid2. Science 2018, 359, 940–944. [Google Scholar] [CrossRef]
- Landeira, D.; Bagci, H.; Malinowski, A.R.; Brown, K.E.; Soza-Ried, J.; Feytout, A.; Webster, Z.; Ndjetehe, E.; Cantone, I.; Asenjo, H.G.; et al. Jarid2 coordinates nanog expression and pcp/wnt signaling required for efficient esc differentiation and early embryo development. Cell Rep. 2015, 12, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/jumonji coordinates control of prc2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Tian, S.; Nie, J.; Yang, C.; Ruotti, V.; Wei, H.; Jonsdottir, G.A.; Stewart, R.; Thomson, J.A. Whole-genome analysis of histone h3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 2007, 1, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Aloia, L.; Di Stefano, B.; Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 2013, 140, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.A.; Chang, H.Y. Long noncoding rnas in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding rna as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Epigenetic regulation by long noncoding rnas. Science 2012, 338, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The xist lncrna exploits three-dimensional genome architecture to spread across the x chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Pinter, S.F.; Fang, R.; Sarma, K.; Rutenberg-Schoenberg, M.; Bowman, S.K.; Kesner, B.A.; Maier, V.K.; Kingston, R.E.; Lee, J.T. High-resolution xist binding maps reveal two-step spreading during x-chromosome inactivation. Nature 2013, 504, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Mak, W.; Nesterova, T.B.; de Napoles, M.; Appanah, R.; Yamanaka, S.; Otte, A.P.; Brockdorff, N. Reactivation of the paternal x chromosome in early mouse embryos. Science 2004, 303, 666–669. [Google Scholar] [CrossRef] [PubMed]
- da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 is implicated in the initial xist-induced targeting of prc2 to the inactive x chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef]
- Pinter, S.F.; Sadreyev, R.I.; Yildirim, E.; Jeon, Y.; Ohsumi, T.K.; Borowsky, M.; Lee, J.T. Spreading of x chromosome inactivation via a hierarchy of defined polycomb stations. Genome Res. 2012, 22, 1864–1876. [Google Scholar] [CrossRef]
- Cerase, A.; Smeets, D.; Tang, Y.A.; Gdula, M.; Kraus, F.; Spivakov, M.; Moindrot, B.; Leleu, M.; Tattermusch, A.; Demmerle, J.; et al. Spatial separation of xist rna and polycomb proteins revealed by superresolution microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 2235–2240. [Google Scholar] [CrossRef]
- Tian, D.; Sun, S.; Lee, J.T. The long noncoding rna, jpx, is a molecular switch for x chromosome inactivation. Cell 2010, 143, 390–403. [Google Scholar] [CrossRef]
- Sun, S.; Del Rosario, B.C.; Szanto, A.; Ogawa, Y.; Jeon, Y.; Lee, J.T. Jpx rna activates xist by evicting ctcf. Cell 2013, 153, 1537–1551. [Google Scholar] [CrossRef]
- Saldana-Meyer, R.; Gonzalez-Buendia, E.; Guerrero, G.; Narendra, V.; Bonasio, R.; Recillas-Targa, F.; Reinberg, D. Ctcf regulates the human p53 gene through direct interaction with its natural antisense transcript, wrap53. Genes Dev. 2014, 28, 723–734. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat rna to the mouse x chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of xist rna. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Zheng, L.; Goodrich, K.J.; Cech, T.R. Promiscuous rna binding by polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013, 20, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Cifuentes-Rojas, C.; Ergun, A.; Del Rosario, A.; Jeon, Y.; White, F.; Sadreyev, R.; Lee, J.T. Atrx directs binding of prc2 to xist rna and polycomb targets. Cell 2014, 159, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. Pcgf3/5-prc1 initiates polycomb recruitment in x chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.G.; Meisig, J.; Nakamura, T.; Okamoto, I.; Sieber, A.; Picard, C.; Borensztein, M.; Saitou, M.; Bluthgen, N.; Heard, E. The two active x chromosomes in female escs block exit from the pluripotent state by modulating the esc signaling network. Cell Stem Cell 2014, 14, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Anguera, M.C.; Sadreyev, R.; Zhang, Z.; Szanto, A.; Payer, B.; Sheridan, S.D.; Kwok, S.; Haggarty, S.J.; Sur, M.; Alvarez, J.; et al. Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 2012, 11, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Payer, B.; Rosenberg, M.; Yamaji, M.; Yabuta, Y.; Koyanagi-Aoi, M.; Hayashi, K.; Yamanaka, S.; Saitou, M.; Lee, J.T. Tsix rna and the germline factor, prdm14, link x reactivation and stem cell reprogramming. Mol. Cell 2013, 52, 805–818. [Google Scholar] [CrossRef]
- Tchieu, J.; Kuoy, E.; Chin, M.H.; Trinh, H.; Patterson, M.; Sherman, S.P.; Aimiuwu, O.; Lindgren, A.; Hakimian, S.; Zack, J.A.; et al. Female human ipscs retain an inactive x chromosome. Cell Stem Cell 2010, 7, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Kanduri, C.; Thakur, N.; Pandey, R.R. The length of the transcript encoded from the kcnq1ot1 antisense promoter determines the degree of silencing. EMBO J. 2006, 25, 2096–2106. [Google Scholar] [CrossRef]
- Lewis, A.; Mitsuya, K.; Umlauf, D.; Smith, P.; Dean, W.; Walter, J.; Higgins, M.; Feil, R.; Reik, W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 2004, 36, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding rna mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Son, J.; Shen, S.S.; Reinberg, D.; Bonasio, R. Prc2 binds active promoters and contacts nascent rnas in embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Bonasio, R.; Saldana-Meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between jarid2 and noncoding rnas regulate prc2 recruitment to chromatin. Mol. Cell 2014, 53, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.S.; Yu, C.Y.; Chuang, C.Y.; Hsiao, M.; Kao, C.F.; Kuo, H.C.; Chuang, T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Zhao, G.N.; Chen, X.F.; Hao, D.L.; Zhao, X.; Lv, X.; Liu, D.P. The long noncoding rna gm15055 represses hoxa gene expression by recruiting prc2 to the gene cluster. Nucleic Acids Res. 2016, 44, 2613–2627. [Google Scholar] [CrossRef] [PubMed]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a long noncoding rna required for cardiovascular lineage commitment. Cell 2013, 152, 570–583. [Google Scholar] [CrossRef]
- Benetatos, L.; Vartholomatos, G.; Hatzimichael, E. Meg3 imprinted gene contribution in tumorigenesis. Int. J. Cancer 2011, 129, 773–779. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Klibanski, A. Meg3 noncoding rna: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef]
- da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian dlk1-dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef]
- Takahashi, N.; Okamoto, A.; Kobayashi, R.; Shirai, M.; Obata, Y.; Ogawa, H.; Sotomaru, Y.; Kono, T. Deletion of gtl2, imprinted non-coding rna, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 2009, 18, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.P.; Youngson, N.; Takada, S.; Seitz, H.; Reik, W.; Paulsen, M.; Cavaille, J.; Ferguson-Smith, A.C. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the dlk1-gtl2 imprinted cluster on mouse chromosome 12. Nat. Genet. 2003, 35, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qf1 in mouse induced pluripotent stem cells. Nature 2010, 465, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Markoulaki, S.; Hanna, J.H.; Faddah, D.A.; Buganim, Y.; Kim, J.; Ganz, K.; Steine, E.J.; Cassady, J.P.; Creyghton, M.P.; et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell 2011, 9, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Sherpa, C.; Rausch, J.W.; Le Grice, S.F. Structural characterization of maternally expressed gene 3 rna reveals conserved motifs and potential sites of interaction with polycomb repressive complex 2. Nucleic Acids Res. 2018, 46, 10432–10447. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincrnas in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef]
- Sanli, I.; Lalevee, S.; Cammisa, M.; Perrin, A.; Rage, F.; Lleres, D.; Riccio, A.; Bertrand, E.; Feil, R. Meg3 non-coding rna expression controls imprinting by preventing transcriptional upregulation in cis. Cell Rep. 2018, 23, 337–348. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Maniatis, T.; Tasic, B. Alternative pre-mrna splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef]
- Black, D.L.; Grabowski, P.J. Alternative pre-mrna splicing and neuronal function. Prog. Mol. Subcell. Biol. 2003, 31, 187–216. [Google Scholar] [PubMed]
- Blencowe, B.J. Alternative splicing: New insights from global analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Kryukov, K.; Clemente, J.C.; Komiyama, T.; Suzuki, Y.; Imanishi, T.; Ikeo, K.; Gojobori, T. The evolutionary relationship between gene duplication and alternative splicing. Gene 2008, 427, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Aigaki, T. Alternative trans-splicing: A novel mode of pre-mrna processing. Biol. Cell 2006, 98, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Gingeras, T.R. Implications of chimaeric non-co-linear transcripts. Nature 2009, 461, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, X.; Shepelev, V.; Fedorov, A. Bioinformatic analysis of exon repetition, exon scrambling and trans-splicing in humans. Bioinformatics 2006, 22, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, L.; Jiang, H.; Wang, W. Short homologous sequences are strongly associated with the generation of chimeric rnas in eukaryotes. J. Mol. Evol. 2009, 68, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Herai, R.H.; Yamagishi, M.E. Detection of human interchromosomal trans-splicing in sequence databanks. Brief. Bioinform. 2010, 11, 198–209. [Google Scholar] [CrossRef]
- Kim, P.; Yoon, S.; Kim, N.; Lee, S.; Ko, M.; Lee, H.; Kang, H.; Kim, J.; Lee, S. Chimerdb 2.0—A knowledgebase for fusion genes updated. Nucleic Acids Res. 2010, 38, D81–D85. [Google Scholar] [CrossRef]
- McManus, C.J.; Duff, M.O.; Eipper-Mains, J.; Graveley, B.R. Global analysis of trans-splicing in drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 12975–12979. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.; Hu, X.; Zhang, Y.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X.; et al. Deep rna sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Al-Balool, H.H.; Weber, D.; Liu, Y.; Wade, M.; Guleria, K.; Nam, P.L.; Clayton, J.; Rowe, W.; Coxhead, J.; Irving, J.; et al. Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 2011, 21, 1788–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.; Wei, Y.; Kang, Y.; Landweber, L.F. Detection of a common chimeric transcript between human chromosomes 7 and 16. Biol. Direct 2012, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Kuo, H.C. The trans-spliced long noncoding rna tsrmst impedes human embryonic stem cell differentiation through wnt5a-mediated inhibition of the epithelial-to-mesenchymal transition. Stem Cells 2016, 34, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Cabili, M.N.; Rinn, J.; Raj, A. Linc-hoxa1 is a noncoding rna that represses hoxa1 transcription in cis. Genes Dev. 2013, 27, 1260–1271. [Google Scholar] [CrossRef] [PubMed]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jorgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncrna fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef]
- Deng, C.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.; Bungert, J.; Qiu, Y.; Huang, S. Hoxblinc rna recruits set1/mll complexes to activate hox gene expression patterns and mesoderm lineage development. Cell Rep. 2016, 14, 103–114. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, J.; Hu, G. Genome-wide methods for investigating long noncoding rnas. Biomed. Pharmacother. 2019, 111, 395–401. [Google Scholar] [CrossRef]
- Petri, R.; Jakobsson, J. Identifying mirna targets using ago-ripseq. Methods Mol. Biol. 2018, 1720, 131–140. [Google Scholar] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Tsang, J.C.; Ilicic, T.; Henriksson, J.; Natarajan, K.N.; Tuck, A.C.; Gao, X.; Buhler, M.; Liu, P.; et al. Single cell rna-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015, 17, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Lukowski, S.W.; Chiu, H.S.; Senabouth, A.; Bruxner, T.J.C.; Christ, A.N.; Palpant, N.J.; Powell, J.E. Single-cell rna-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018, 28, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Tiklova, K.; Bjorklund, A.K.; Lahti, L.; Fiorenzano, A.; Nolbrant, S.; Gillberg, L.; Volakakis, N.; Yokota, C.; Hilscher, M.M.; Hauling, T.; et al. Single-cell rna sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun. 2019, 10, 581. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding rnas in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Pivotal role of PRC2 in stemness and differentiation. At bivalent domains, the simultaneous presence of repressive and active chromatin marks counterbalance each other. PRC2, guided and stabilized by the cooperation of a variety of lncRNAs, confers repression by H3K27me3 catalyzed by the subunit SUZ12. Many of these bivalently marked genes are bound by a non-processive form of RNA polymerase II (poised RNA Pol II). Upon differentiation, bivalent chromatin genes that remain inactive in differentiated cells lose the active H3K4me3 mark. By contrast, activated bivalent chromatin genes lose the repressive H3K27me3 mark due to the displacement of PRC2.
Figure 1.
Pivotal role of PRC2 in stemness and differentiation. At bivalent domains, the simultaneous presence of repressive and active chromatin marks counterbalance each other. PRC2, guided and stabilized by the cooperation of a variety of lncRNAs, confers repression by H3K27me3 catalyzed by the subunit SUZ12. Many of these bivalently marked genes are bound by a non-processive form of RNA polymerase II (poised RNA Pol II). Upon differentiation, bivalent chromatin genes that remain inactive in differentiated cells lose the active H3K4me3 mark. By contrast, activated bivalent chromatin genes lose the repressive H3K27me3 mark due to the displacement of PRC2.

Figure 2.
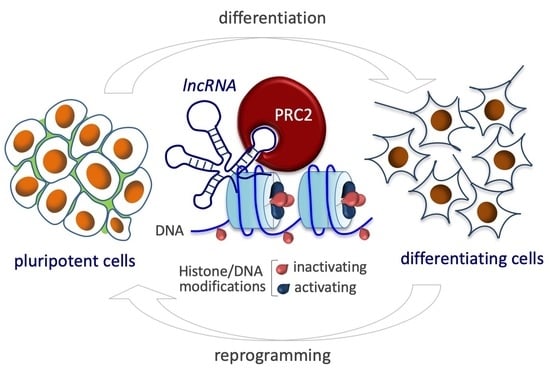
The X-chromosome state in females is linked to cell differentiation. Pluripotent cells have two active X-chromosomes (XaXa) and undergo X-chromosome inactivation when differentiated, resulting in one active and one inactive X-chromosome (XaXi). The lncRNA Xist is the main player in such process. The expression of the Xist gene on one of the X-chromosomes leads to the recruitment of the PRC2 complex to that chromosome, establishing its inactivation (top panel). During cell reprogramming, the X-chromosome state is reversed by X-chromosome Reactivation (bottom panel).
Figure 2.
The X-chromosome state in females is linked to cell differentiation. Pluripotent cells have two active X-chromosomes (XaXa) and undergo X-chromosome inactivation when differentiated, resulting in one active and one inactive X-chromosome (XaXi). The lncRNA Xist is the main player in such process. The expression of the Xist gene on one of the X-chromosomes leads to the recruitment of the PRC2 complex to that chromosome, establishing its inactivation (top panel). During cell reprogramming, the X-chromosome state is reversed by X-chromosome Reactivation (bottom panel).

Figure 3.
The lncRNA tsRMST suppresses lineage differentiation in hESCs. A scheme representing the exon order of tsRMST (upper panel). In pluripotent cells, the interaction of tsRMST with NANOG and the PRC2 complex promotes occupancy on inactive genes, such as GATA6 and PAX6 (middle panel). In differentiating cells, the absence of tsRMST leads to the displacement of PRC2 with the consequent induction of the lineage-specific genes (bottom panel) [95].
Figure 3.
The lncRNA tsRMST suppresses lineage differentiation in hESCs. A scheme representing the exon order of tsRMST (upper panel). In pluripotent cells, the interaction of tsRMST with NANOG and the PRC2 complex promotes occupancy on inactive genes, such as GATA6 and PAX6 (middle panel). In differentiating cells, the absence of tsRMST leads to the displacement of PRC2 with the consequent induction of the lineage-specific genes (bottom panel) [95].

Figure 4.
Comparison of single-cell versus bulk analysis results in lncRNA studies. ESCs show a cellular heterogeneity of linc-HOXA1 and Hoxa1 RNA within the population. A single-cell gene expression analysis highlighted an inverse correlation between linc-HOXA1 RNA and Hoxa1 mRNA abundance. Before the advent of the single-cell approach, the bulk analysis were showing an average result, losing the real feature of cells and masking the close interaction between linc-HOXA1 and Hoxa1 [96,125].
Figure 4.
Comparison of single-cell versus bulk analysis results in lncRNA studies. ESCs show a cellular heterogeneity of linc-HOXA1 and Hoxa1 RNA within the population. A single-cell gene expression analysis highlighted an inverse correlation between linc-HOXA1 RNA and Hoxa1 mRNA abundance. Before the advent of the single-cell approach, the bulk analysis were showing an average result, losing the real feature of cells and masking the close interaction between linc-HOXA1 and Hoxa1 [96,125].


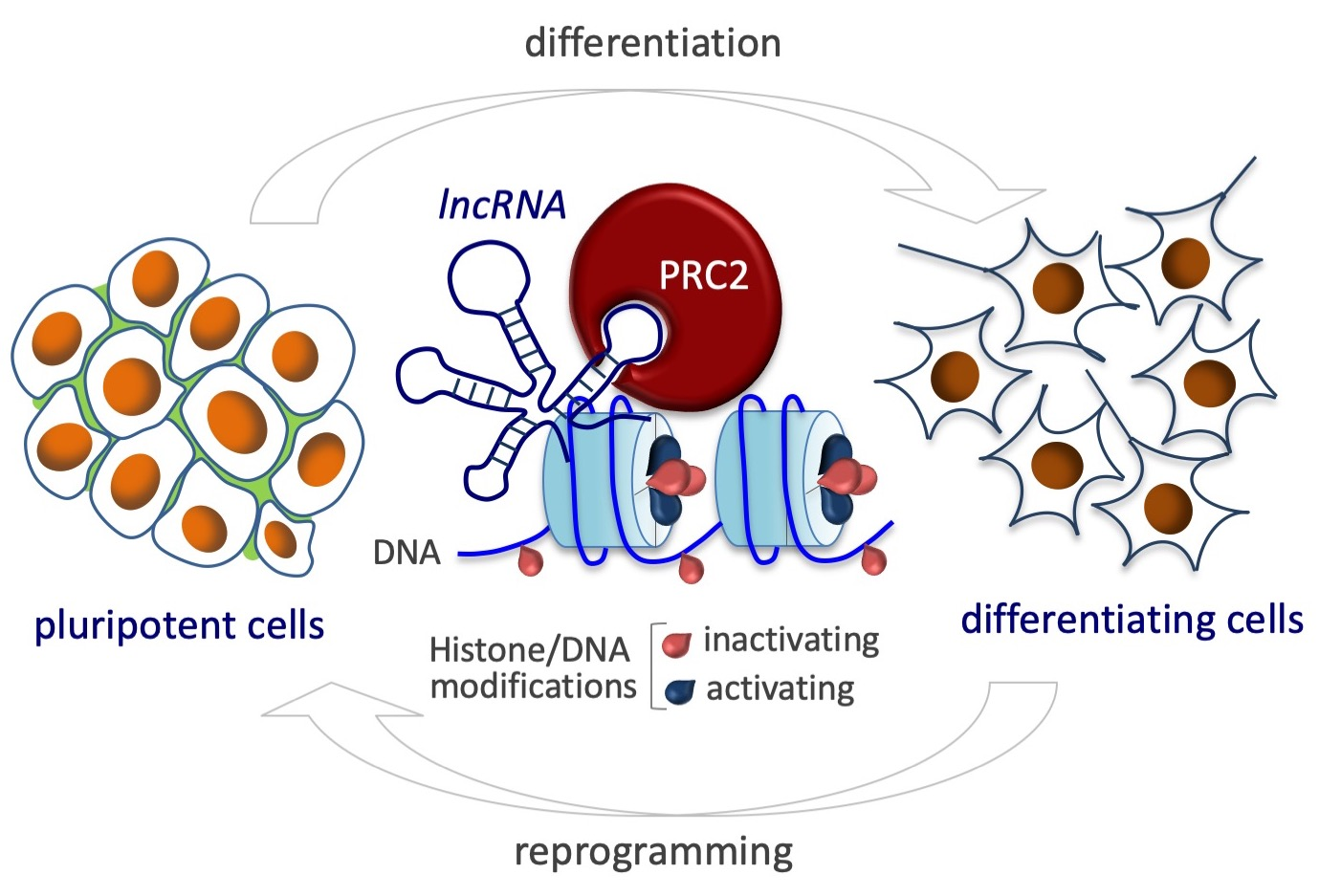{kind=link}
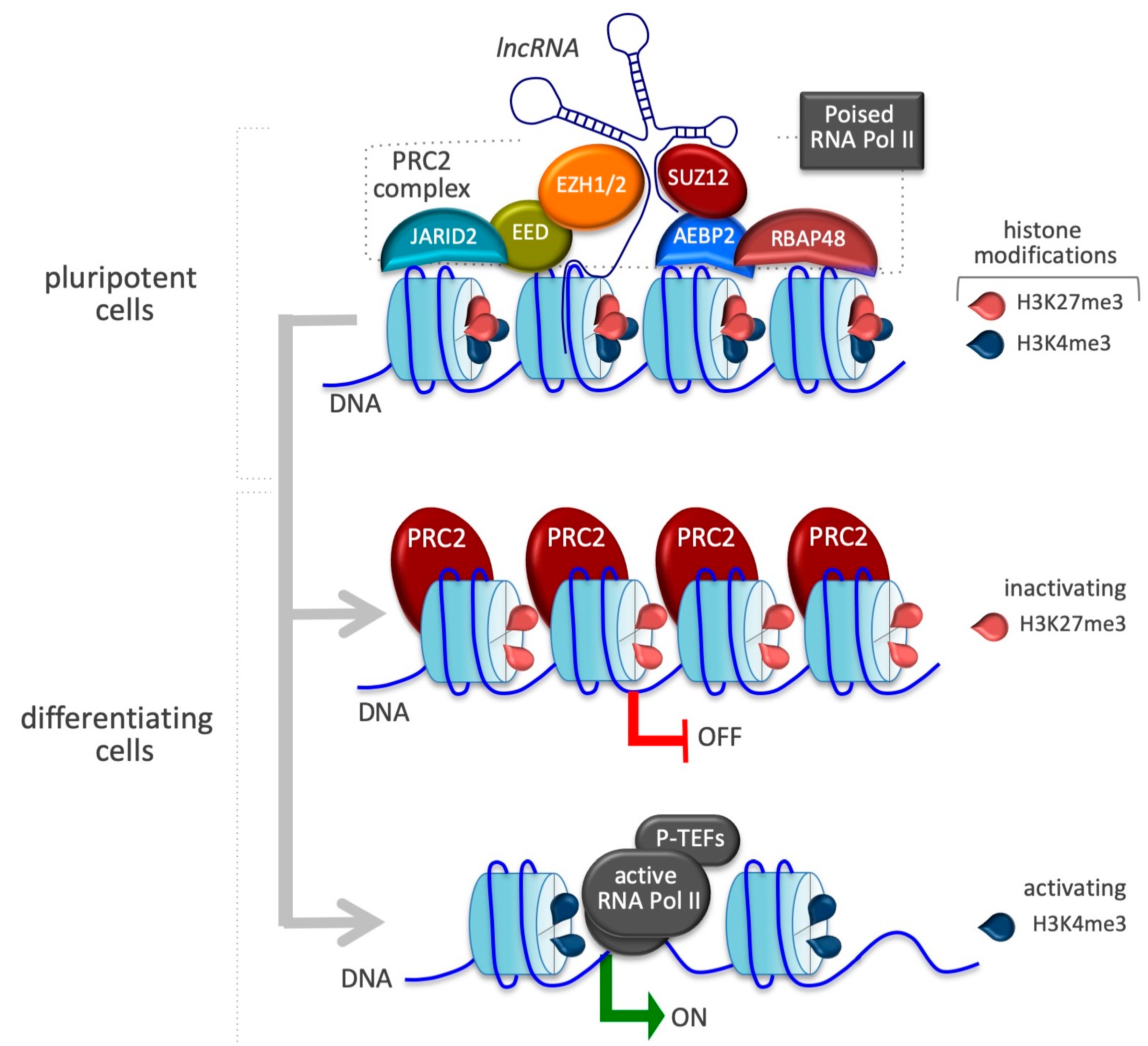{kind=link}
{kind=link}
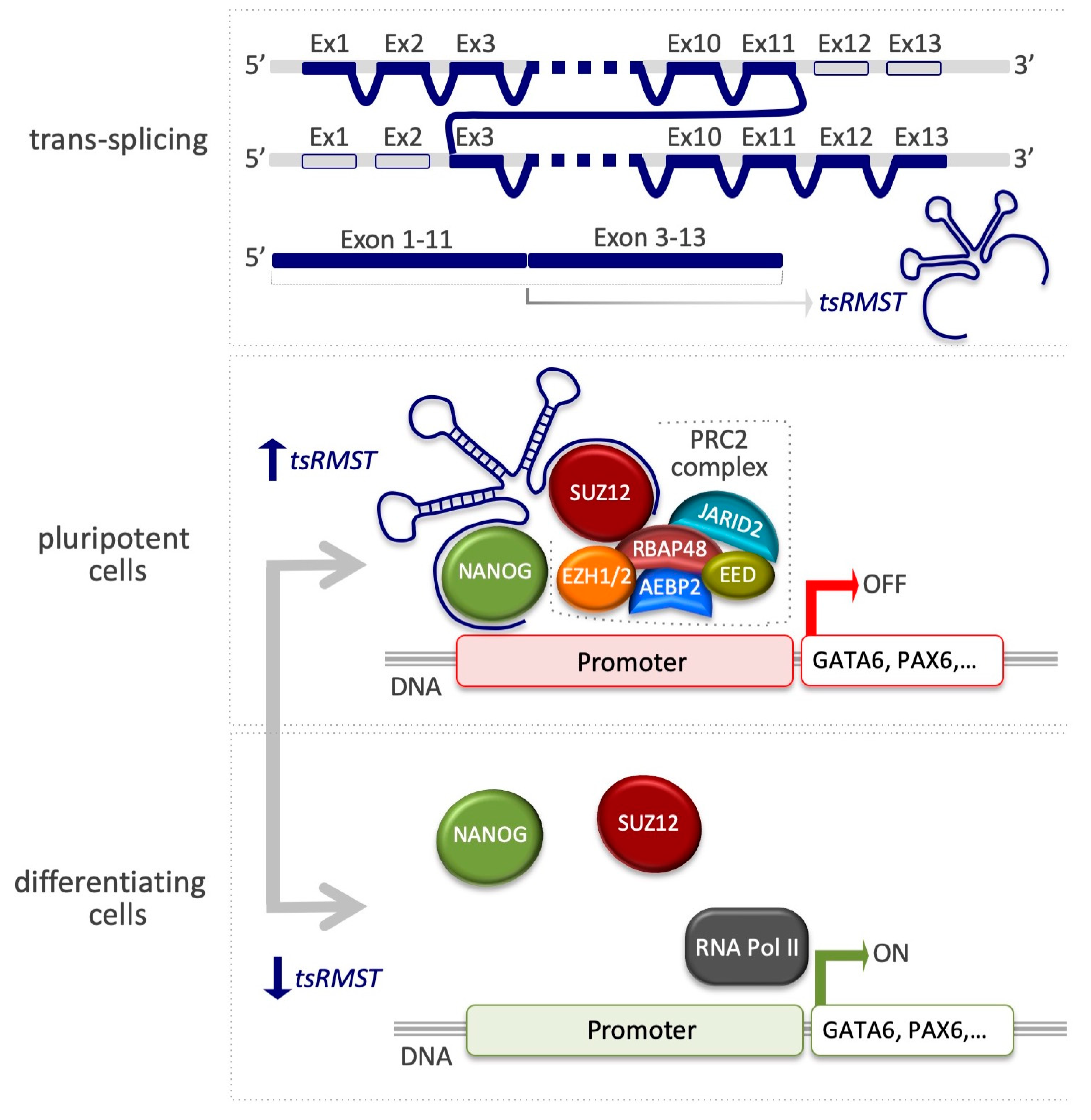{kind=link}
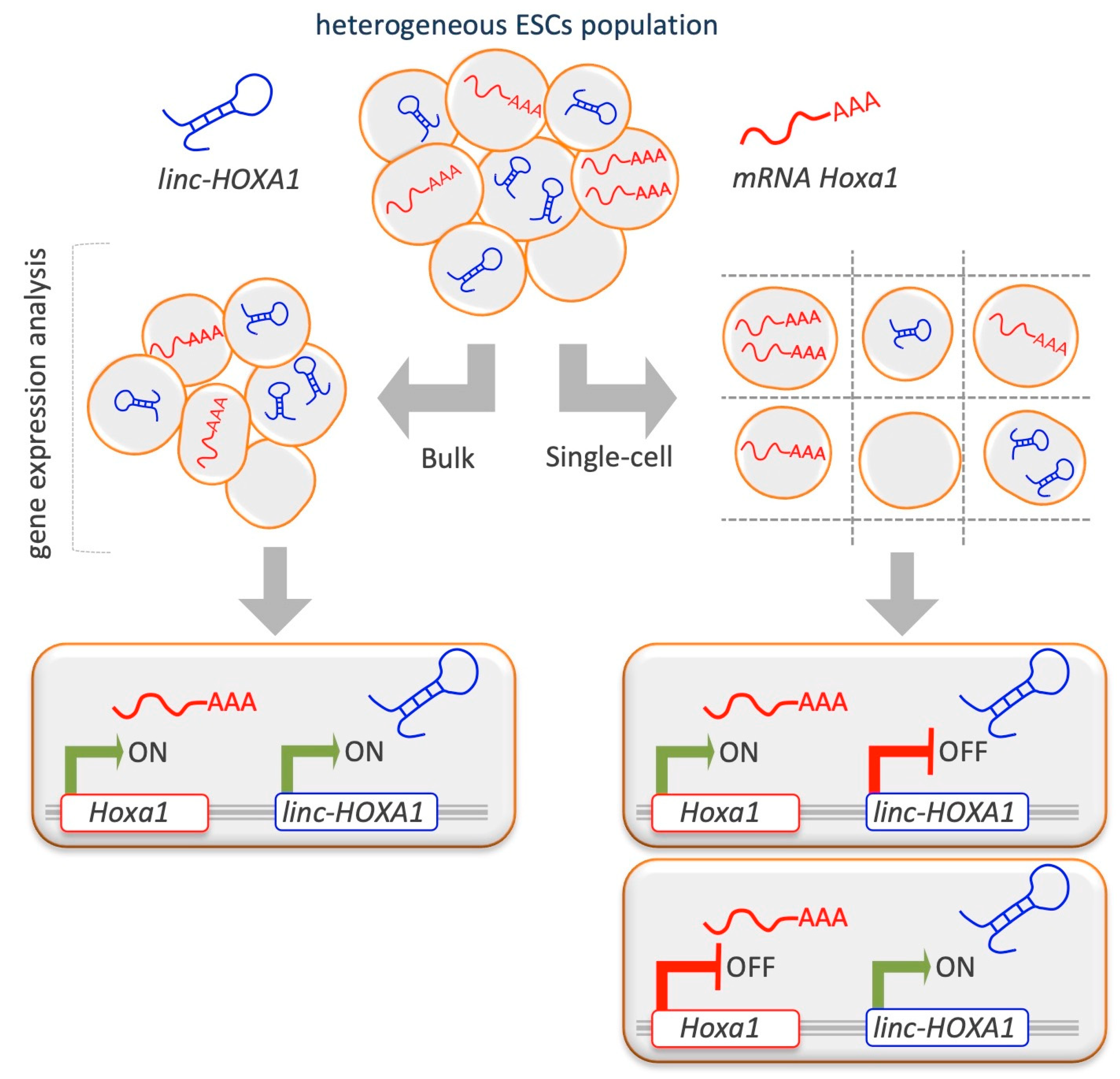{kind=link}
Table 1.
PRC2 protein partners and the regulatory function of lncRNAs.
| LncRNAs | PRC2 Protein Partners | Regulatory Function | Refs. |
|---|---|---|---|
| Xist | EZH2, JARID 2 | X-chromosome inactivation | [82] |
| Kcnq1ot1 | EZH2, SUZ12 | Kcnq1 inactivation | [92] |
| Meg3 | EZH2, JARID2 | ESC subset of genes inactivation | [93,94] |
| tsRMST | SUZ12 | Pluripotency-associated genes activation (NANOG, POU5F1, SOX2) and cell commitment genes inhibition (GATA6, PAX6) | [95] |
| Linc-HOXA1 | SUZ12 | Hoxa inactivation | [96] |
| T-UCstem1 | SUZ12 | Global de-repression of bivalent chromatin-associated genes | [13] |
| Bvht | SUZ12 | Mesp1 activation | [97] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fiorenzano, A.; Pascale, E.; Patriarca, E.J.; Minchiotti, G.; Fico, A. LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells. Epigenomes 2019, 3, 14. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes3030014
AMA Style
Fiorenzano A, Pascale E, Patriarca EJ, Minchiotti G, Fico A. LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells. Epigenomes. 2019; 3(3):14. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes3030014
Chicago/Turabian StyleFiorenzano, Alessandro, Emilia Pascale, Eduardo Jorge Patriarca, Gabriella Minchiotti, and Annalisa Fico. 2019. "LncRNAs and PRC2: Coupled Partners in Embryonic Stem Cells" Epigenomes 3, no. 3: 14. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes3030014