The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma


1
Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center, Smithville, TX 78957, USA
2
Graduate Program in Genetics & Epigenetics, UTHealth Graduate School of Biomedical Sciences, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA
*
Author to whom correspondence should be addressed.
Epigenomes 2021, 5(1), 2; https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010002
Submission received: 13 December 2020
/
Revised: 31 December 2020
/
Accepted: 3 January 2021
/
Published: 5 January 2021
(This article belongs to the Special Issue Recent Advances in Biological Methylation)
Abstract
:Arginine methylation is an essential post-translational modification (PTM) deposited by protein arginine methyltransferases (PRMTs) and recognized by Tudor domain-containing proteins. Of the nine mammalian PRMTs, PRMT5 is the primary enzyme responsible for the deposition of symmetric arginine methylation marks in cells. The staphylococcal nuclease and Tudor domain-containing 1 (SND1) effector protein is a key reader of the marks deposited by PRMT5. Both PRMT5 and SND1 are broadly expressed and their deregulation is reported to be associated with a range of disease phenotypes, including cancer. Hepatocellular carcinoma (HCC) is an example of a cancer type that often displays elevated PRMT5 and SND1 levels, and there is evidence that hyperactivation of this axis is oncogenic. Importantly, this pathway can be tempered with small-molecule inhibitors that target PRMT5, offering a therapeutic node for cancer, such as HCC, that display high PRMT5–SND1 axis activity. Here we summarize the known activities of this writer–reader pair, with a focus on their biological roles in HCC. This will help establish a foundation for treating HCC with PRMT5 inhibitors and also identify potential biomarkers that could predict sensitivity to this type of therapy.
1. Introduction
Signal transduction is the process by which information is relayed through a cell. Extracellular signals, such as growth factors or contact points with other cells, stimulate receptors on the cell surface to initiate this process by converting one stimulus (ligand binding) into another (phosphorylation). This signal initiation event is then propelled through the cytoplasm and into the nucleus using a series of sequential PTM events that rely on “reader” proteins, or effector molecules, to dock onto a specific PTM and then promote the deposition of a new PTM downstream, which in turn is read and relayed by another effector. There is a vast array of different PTMs including, but not limited to, phosphorylation, acetylation, ubiquitination and methylation, which can all be read by specialized protein domains in effector molecules [1]. These globular domain types include SH2s (phosphor-tyrosine readers), FHAs/14-3-3s/BRCTs (phosphor-serine/threonine readers), Bromo/YEATS domains (acetyl-lysine readers), UBA/UIM/GAT/CUE domains (ubiquitinated-lysine readers), Chromo/PHD/Tudor/BAH domains (methylated-lysine readers), and Tudors (methylated-arginine readers). In this review, we will focus on just one single thread in this hairball of signaling networks: the PRMT5–SND1 axis.
Arginine residues are frequently methylated post-translationally, and these modifications come in one of three flavors: monomethylarginine (MMA), asymmetric dimethylarginine (ADMA), or symmetric dimethylarginine (SDMA) (Figure 1). These methyl-mark additions occur at the peripheral omega nitrogen of arginine guanidine moieties and are commonly seen at glycine- and arginine-rich (GAR) motifs. Protein arginine methyltransferases (PRMTs) are the family of nine closely-related enzymes which deposit all three of these marks [2]. There is a tenth arginine methyltransferase called NDUFAF7, which is not a member of PRMT family, and seems to be a dedicated mitochondrial enzyme [3]. PRMT1 is the primary Type I PRMT, which deposits the majority of ADMA marks. Other Type I PRMTs include PRMT2, PRMT3, PRMT4/CARM1, PRMT6 and PRMT8. PRMT7 is capable of depositing only MMA marks, and it is referred to as a Type III enzyme. That leaves the Type II enzymes, which deposit SDMA marks. PRMT9 has just one known substrate (SAP145) [4], and NDUFAF7 is also dedicated to just one mitochondrial substrate (NDUFS2) [3]. All the remaining proteins marked with SDMA are thought to be PRMT5 substrates. Arginine methylation is a relatively abundant PTM and over the years, a number of studies have reported similar rations of the different types of arginine methylation in both tissue and cell lines [5,6,7], which generally breaks down to 1500:3:2:1 for Arginine:ADMA:SDMA:MMA. So, approximately 0.6% of all arginine residues found in proteins are arginine methylated.
Tudor domains are the only globular folds known to bind methylated arginine motifs [8]. This domain type was first identified in the Drosophila melanogaster Tudor protein which contains repeating domains that are present in a number of other proteins in many different species [9]. A detailed understanding of how Tudor domains interact with SDMA motifs was first gleaned from biochemistry and structure studies involving the human survival motor neuron (SMN) protein [10,11], which is mutated in spinal muscular atrophy syndrome. All in all, eight Tudor domain-containing proteins have been reported to be methyl-arginine readers. The vast majority of methyl-arginine readers recognize SDMA motifs, including SMN, SPF30, and SND1, which are ubiquitously expressed, and TDRD1, TDRD2, TDRD6, and TDRD9, which are all germ cell-specific proteins. TDRD3 is currently the only known ADMA motif effector protein that is also ubiquitously expressed (Figure 1).
PRMT5 has emerged as an important player in HCC [12], the fourth leading cause of cancer mortality in the world [13]. This link to HCC is strengthened by the fact that a downstream reader of the PRMT5-deposited SDMA marks—SND1—has been identified as a driver of HCC formation [14], though the precise molecular mechanism of action remains poorly understood. This review focuses on summarizing key biological functions of the PRMT5–SND1 reader–writer pair (Figure 2), then surveys what is known about these proteins as they relate to HCC, and concludes with speculation on unexplored avenues of therapeutic modulation of methylarginine levels in HCC as a potential form of treatment.
2. Biological Roles of PRMT5—The Primary Depositor of SDMA Marks
PRMT5 is the predominant Type II arginine methyltransferase, indicating that it confers both MMA and SDMA marks on target substrates in a distributive manner [15] (Figure 1). The primary targets of PRMT5 methylation are RNA-binding proteins, epigenetic modulators and core histones, which has implicated this enzyme in transcriptional regulation and the control of faithful alternative splicing [16]. PRMT5 is not enzymatically active on its own, and is found in a protein complex called the methylosome.
2.1. PRMT5 Forms a Stable Complex with MEP50
Regardless of the methylation target, PRMT5 requires the co-factor methylosome protein 50 (MEP50) for stability and enzymatic activity [17]. MEP50 is also referred to as WDR77. Loss of MEP50 results in the destabilization of the PRMT5 protein and vice versa [18]. Enzymatic activity is further dependent on hetero-octamerization of these two proteins to form a complex of four PRMT5 molecules and four MEP50 molecules [19,20,21]. MEP50 has also been identified as a potential coactivator of the androgen receptor [22], but it is unclear whether PRMT5 is recruited with MEP50 in this context, or whether it functions independently. Importantly, HeLa cell fractionation studies from a sucrose gradient indicate that PRMT5 and MEP50 only occur together and are not found in a complex without the other, nor do they exist in the free un-complexed form [17]. Similar fractionation experiments using Xenopus egg extracts and gel filtration also reveal the existence of a single PRMT5–MEP50 complex and no free monomeric form of either protein [23]. Thus, these two proteins are tightly complexed and likely do not function independently.
2.2. The Methylosome is Targeted to Distinct Substrates by Adaptor Proteins
The PRMT5–MEP50 protein complex requires additional adaptor proteins to aid in identifying substrates that will be targeted for symmetric methylation. There are five known adaptors that link the methylosome to its substrates, and these are pICln, RIOK1, COPR5, Sharpin and OXR1A. pICln is a spliceosome assembly chaperone, which recruits PRMT5 to facilitate the efficient methylation of SmB/B’ and SmD1/2 [24,25], as well as a number of ribosomal proteins [26]. A second adaptor is RIOK1, which is critical for the methylation of nucleolin by PRMT5–MEP50 [27], and is important for pre-rRNA transcription and processing. RIOK1 and pICln compete for binding, suggesting that there may be a common pocket for adaptor protein binding, either on PRMT5 or MEP50 [27]. COPR5 is the third adaptor to be identified, and it recruits PRMT5 activity to nucleosomes to support the deposition of H3R8me2s and H4R3me2s marks, in its role as an epigenetic regulator [28]. The fourth adaptor is Sharpin, and this interaction targets PRMT5 to methylate the transcription factor SKI [29]. Finally, OXR1A also regulates PRMT5′s ability to methylate histones, and it is the H3R2me2s methylation that is stimulated by this adaptor [30]. OXR1A and PRMT5 interact in the pituitary gland and regulate growth hormone expression, which in turn impacts liver metabolism. Both RIOK1 and pICln were identified in independent shRNA screens that also identified PRMT5 as a vulnerability in MTAP-null tumors [31,32,33], further supporting the key role that these adaptors play in helping the PRMT5–MEP50 methylosome find its targets for methylation.
2.3. The Identification of PRMT5 Substrates Implicate It in the Regulation of Transcription, Splicing, Signal Transduction and the Repair of DNA Damage
The initial characterization of PRMT5 as an arginine methyltransferase revealed that it methylates H2A and H4, using an in vitro methylation assay [34]. Importantly, the first five residues of H2A and H4 are identical (SGRGK…), and it is the arginine in position 3 that is methylated by PRMT5. Knockout studies showed that the H2AR3me2s modification is particularly sensitive to PRMT5 loss in vivo [35]. Sm proteins were also shown to be methylated by PRMT5 early on in the study of this PRMT [24]. Since then, over the last twenty years, a large number of PRMT5 substrates have been identified [16]. These studies have been spurred on by the development of efficient pan-substrate antibodies that recognize Rme2s marks on different substrates, and can be used to enrich for methylated peptides from tissue and cell extracts, which can then be identified by mass spectrometry. The first such substrate screens were performed by the Richard lab [36], and subsequent screening studies have dramatically expanded on the number of known symmetrically methylated proteins into the 100s [37]. Gene ontology (GO) analysis of the identified PRMT5 substrates reveals strong enrichment of RNA splicing and processing, as well as PTM regulated gene expression pathways and, to a lesser extent, translation.
2.4. PRMT5 Functional Misdirection Due to Cross-Reactivity with the FLAG Antibody
The PRMT5 field has been confounded by the occurrence of a major artifact of tandem affinity protein (TAP) complex purifications that use the FLAG-tag. Over the years, it is well established that when purifying a FLAG tagged protein using FLAG-M2 beads, a major contaminant is the PRMT5–MEP50 protein complex, because the M2 antibody binds directly to PRMT5 and purifies both it and its associated proteins. This was first reported by Danny Reinberg’s lab over 18 years ago [38]. Subsequent studies by the Siekhattar lab reported the same thing [39]. PRMT5 is also listed as a common contaminant of FLAG immunoprecipitation experiments [40]. Most recently, the CRAPome was published, which highlights the major problems with affinity purification-mass spectrometry data sets [41]. Indeed, they showed that 94% of all FLAG purifications data sets detect PRMT5 peptides. Thus, the misassignment of PRMT5 in many FLAG-tagged protein complex purifications has led many researchers astray, and these artifacts have found their way deep into the published literature.
2.5. Mouse Models Reveal a Number of Biological Roles for the Methylosome
It is very likely that loss of PRMT5 and MEP50 in mice will phenocopy each other, as they are codependent on each other for protein stability. Indeed, the interdependence and essentiality of MEP50 and PRMT5 complexing is supported by the fact that the mouse knockouts of both PRMT5 and MEP50 result in early lethal developmental defects. MEP50 knockout mice display an early embryonic lethal phenotype with no null embryos detected at E8.5 [42]. The PRMT5 knockout mice also display a very early embryonic lethal phenotype [35].
The early lethality of these total knockouts has made it necessary to generate conditional alleles for both PRMT5 and MEP50, to help elucidate the biological roles of this protein complex in vivo. Importantly, conditional knockouts of PRMT5 have provided additional insights into its roles in T and B cell development, limb development and neural development [43,44,45,46,47]. A conditional allele for the study of MEP50 loss in adult mice is also available, but has only been used in two studies related to prostate development [48] and lung development [49], although the conditional knockout was performed ex vivo in the latter study.
The first conditional PRMT5 knockout mice were generated by crossing PRMT5fl/fl and Nestincre mice, which resulted in postnatal lethality in all homozygous null mice, and implicated PRMT5 in neuronal development [43]. Further exploration determined that this mortality was linked to splicing variations of Mdm4, which induces a p53 response, leading to severe cranial abnormalities. Subsequently, PRMT5 was conditionally knocked out in oligodendrocytes, using Olig1cre, and identified as a key factor for myelination [50]. Myelin basic protein has long been known to be a robust substrate for PRMT5 in in vitro studies, and the myelination defect in the conditional knockout mice provides in vivo evidence for the functional importance of this PTM [51].
A number of additional conditional knockouts of PRMT5 have been performed in adult mice. PRMT5 is also essential for the initiation and maintenance of hematopoiesis [44,52]. Methyl-transferase localization appears to impact the modification of splicing machinery, whereas loss of PRMT5 results in alternative splicing defects via intron retention and exon skipping, which is critical for hematopoietic stem cell quiescence and viability [44,53]. Both conditional knockout and small-molecule inhibitor studies reveal that loss of PRMT5 has anti-tumor activity against MLL-rearranged acute myeloid leukemia (AML) likely due to hypomethylation of essential splicing factor like SRSF1 [54,55], and further vulnerability of cancer to PRMT5 loss is bestowed on the tumors that harbor driver mutation in splicing factors [56]. Using a CD4cre, it was recently shown that PRMT5 is dispensable for late T cell development, and is required for peripheral T cell expansion and survival [46]. The removal of PRMT5 activity from pancreatic beta cells, using the Paxcre, reveals its role on regulation of insulin expression in vivo [57]. PRMT5 has also been shown to play a role in muscle stem cell expansion in adult mice (using Pax7cre), but does not seem important for the proliferation and differentiation of myogenic progenitor cells during embryonic development [58].
In a mouse embryo developmental biology setting, PRMT5 has been identified as a key for certain differentiated chondrocytes, and in this case Prxcre was used to remove PRMT5 from developing limb buds [59]. Conditional deletion of PRMT5 in hind limbs of mice led to severe phenotypes of atrophied long bone and knee. While essential for some chondrocyte lineages, PRMT5 is dispensable for general chondrocyte maintenance in adult mice. Inactivation of PRMT5 in germ cells (using Tnapcre) results in defects in spermatogenesis [60], and loss of PRMT5 in the developing lung epithelial cells (using Shhcre) causes defects in branching morphogenesis [61].
Although PRMT5 biology has been studied extensively through conditional knockouts in both adult mice and embryos, far fewer mouse genetic studies have been performed with the other key component of the methylosome—namely MEP50. Importantly, a conditional allele for mouse MEP50 has been generated [48]. However, it has only been used in one study, and that was to investigate the role of MEP50 in the prostate (which we mentioned earlier). Using the Probasincre mouse, MEP50 was conditionally removed from all lobes of the developing mouse prostate. This inactivation of PRMT5 had a severe inhibitory effect on prostate development during embryogenesis, which is likely mediated by the deregulation of androgen receptor (AR) target genes due to the ability of MEP50 (and likely PRMT5) to function as an AR cofactor.
While PRMT5 and MEP50 knockouts have been shown to be essential for many key developmental pathways, PRMT5 also harbors many oncogenic characteristics through its ability to repress the expression of the tumor suppressors ST7 and NM23 [62]. Likewise, loss of E-cadherin, a characteristic of epithelial to mesenchymal transition (EMT) which is key for metastasis, is actively repressed through the binding of PRMT5 and AJUBA to SNAIL [63]. Overexpression of PRMT5 further induces hyperproliferation of cell lines in culture [64,65]. In addition, PRMT5 has been shown to be overexpressed in many different cancers including gastric [66], colorectal [67], lung [68,69], lymphoma [64], ovarian [70], melanoma [71], and glioblastoma [72,73]. The focus of this review is on the overexpression of PRMT5 in HCC and there are numerous reports of elevated PRMT5 levels in liver cancer [12,74,75,76,77,78]. Most of these published studies demonstrate that PRMT5 is overexpressed in many different cancer types, and PRMT5 overexpression correlates with aggressive tumors and poor prognosis. However, it is not clear whether PRMT5 is an oncogenic driver, or whether the elevated PRMT5 levels are an aftereffect of a transformed state. In other words, it is still unknown whether high PRMT5 expression is a cause or a consequence of cellular transformation.
3. Biological Roles of SND1—A Major Reader of SDMA Marks
SND1 (also known as TSN, p100, or TDRD11) is a ubiquitously expressed Tudor domain-containing protein [79]. Unique characteristics of SND1 include four tandem SN domains which convey nucleic acid binding and nuclease activity [79,80] (Figure 2). The SN domains are followed by a single Tudor domain that exclusively recognizes SDMA marks, which is fused to a fifth split SN domain [79,81]. This dual ability to simultaneously interface with nucleic and amino acids allows SND1 to impinge on a wide range of different signaling pathways. Some have claimed that this multifacility endows SND1 with the ability to “positively impact all hallmarks of cancer” [82]. Despite its preferential SDMA reading specificity, SND1 has been called a promiscuous binder given its affinity for RNA and DNA, and it regulates multiple pathways that control various aspects of gene expression [83,84].
3.1. The Tudor Domain of SND1 Interacts Selectively with SDMA Marks
Methylated lysine motifs are bound by at least eight different domain types—Chromo, PHD, MBT, Tudor, PWWP, Ank, BAH and WD40 domains. In the case of methylated arginine motifs, only members of the Tudor family are known effectors, with a handful of Tudor domain-containing proteins either binding SDMA or ADMA marks [8]. Importantly, there are a few individual PHD and WD40 domains whose binding affinity is also impacted by arginine methylation. Tudor domains were identified simultaneously by two research groups, which both realized that the Drosophila melanogaster Tudor protein contains previously unrecognized repeating domains, which were found in a number of other proteins in many different species [9,85]. Interestingly, SND1 was one of the first proteins to be identified as a Tudor domain-containing protein [85]. Initial structural studies involving the Tudor domain of SND1 identified an aromatic cage that suggested it might recognize methylated peptide ligands [86]. Subsequent crystal and NMR structural studies found that the extended Tudor domain of fly SND1 bound a short GAR motif from SmB, only when this motif was symmetrically dimethylated [87]. Finally, work involving the human SND1 protein revealed that it bound PIWIL1 in an arginine methylation-dependent manner, with a strong preference for SDMA motifs over ADMA motifs [88]. Thus, SND1 reads marks that PRMT5 deposits.
3.2. SND1 as a Transcriptional Coactivator
SND1 was originally identified as a transcriptional coactivator of EBNA2 (Epstein–Barr nuclear antigen-2) [89], which interacts with many general transcription factors and coactivators, and positions SND1 as a central player in transcriptional regulation. Indeed, SND1 has been found to directly engage with a number of transcription factors. Signal transducer and activator of transcription 6 (STAT6) is a key player in transcriptional activation upon IL-4 stimulation. SND1 acts as a transcriptional coactivator by binding the C-terminus of STAT6 along with RNA Polymerase II. In this way, SND1 acts as a bridge between STAT6 nuclear localization and transcriptional activation [90]. A second STAT protein, STAT5, induces transcriptional activation in response to lactogenic hormones, which is facilitated in a similar fashion by SND1 binding the C-terminal transcriptional activation domain [91]. SND1 also functions as a coactivator for the c-Myb transcription factor [92], and in this context it is regulated by phosphorylation. Using a protein domain microarray approach, we identified the Tudor domain of SND1 as a reader of a PRMT5 deposited SDMA motif within the E2F1 transcription factor [81]. E2F proteins are widely known for their central role in transcriptional activity and their close association to proliferation and cancer. Follow-up mechanistic studies by the La Thangue group revealed that the recruitment of SND1 to arginine methylated E2F1, results in cross-talk between transcriptional regulation and altered splicing regulation of a subset of E2F1 transcriptional target genes [93]. Independent luciferase-based assays have validated the ability of SND1 to coactivate E2F1 transcriptional activity [94]. SND1 is also an interactor and coactivator of the transcription factor peroxisome proliferator-activated receptor γ (PPARγ), and regulates adipogenesis [95]. Thus, SND1 interacts directly with a number of transcription factors (STAT5/6, c-Myb, E2F1 and PPARγ) and promotes their transcriptional activity.
3.3. SND1 Is a Splicing Factor
The splicing of precursor mRNA is a highly ordered process that is orchestrated by the spliceosome. The spliceosome is composed of five small nuclear ribonucleoprotein particles, which by definition harbor a mix of small nuclear RNAs (snRNAs U1, 2, 4/6 and 5) and proteins (Sm proteins plus additional splicing factors). SND1 interacts with both the RNA and protein components of the spliceosome. Indeed, due to its unique structure, with a Tudor domain for protein binding and SN domains for RNA binding, it plays a role in spliceosome complex formation. The Tudor domain of SND1 interacts directly with the arginine methylated forms of SmB/B’ and SmD1/D3 [96], and also the splicing factor Sam68 [97]. The interaction of SND1 with the U RNAs is likely mediated through the Sm core proteins, which bind directly to U1, U2, U4, U5, and U6 snRNAs, as the Tudor domain of SND1 can pulldown U RNAs, but the SN domains cannot [98].
As mentioned above, SND1 directly associates with a number of transcription factors. It has been proposed that the recruitment of SND1 by transcription factors to enhancer/promoter elements can directly impact the alternative splicing of the transcripts that are being activated by that particular transcription factor, at least in the case of E2F1 [93]. In summary, PRMT5 is a key regulator of RNA splicing [55], and as an effector molecule for SDMA marks deposited by PRMT5, it is not surprising that SND1 is also integral to the maintenance of normal splicing programs that can go awry in a cancer setting.
3.4. SND1 Regulates RNA Stability
Apart from splicing, SND1 is involved in many other aspects of RNA biogenesis. SND1 is not only a component of the spliceosome, but also the RNA-induced silencing complex (RISC), which are both ribonucleoprotein particles. The RISC is a “carrier” of miRNA and siRNA, which when loaded with Argonaute can target mRNA for cleavage to regulate gene expression. A biochemical purification of RISC in Drosophila identified SND1 along with Argonaute 2 and VIG-1 [99], and SND1 is enriched in size fractioned extracts that also contain the 250 kDa miRNA complex. SND1 was also confirmed to be a component of the mammalian RISC enzyme. SND1 was shown to have ribonuclease activity that is specific for inosine-containing dsRNAs [100], and it was subsequently found that SND1 selectively degrades a highly edited pri-miR-142 that is not processed by Drosha [101]. SND1 has four intact SN domains (the 5th SSN domain is split by the Tudor domain), and structural studies have revealed that a minimum of two tandem SN domains are necessary and sufficient for RNA binding [102]. Recent studies have found that SND1 is involved in regulating the turnover of a sub-set of mature miRNAs with a common CA/UA dinucleotide sequence signature [103]. This process is known as Tudor-staphylococcal/micrococcal-like nuclease (TSN)-mediated miRNA decay (TumiD), and it is promoted by the UPF1 helicase [104]. Thus, as a component of RISC, SND1 is actively involved in processing miRNAs.
3.5. SND1 as a Component of Exosome Cargo
Extracellular vesicles (EVs), including exosomes and microvesicles, carry high levels of SND1 as part of their secreted cargo. Exosomes are critical carriers of molecular and signaling information in the extracellular environment of organ systems, where they transfer molecules from one cell to another via membrane vesicle trafficking. Exosomes are approximately 100 nm in size, and are produced in the endosomal compartment (the Golgi network) of most eukaryotic cells. With the development of cancer, exosomes become important messenger packages that “speak to” the tumor microenvironment. The first hint that SND1 was sorted into vesicles came from a study showing the presence of SND1 protein in lipid droplets that are generated by milk secreting cells [105]. Subsequent work has revealed that EVs are enriched for miRNAs, mRNAs and Ago2, which a key protein component of RISC [106,107,108]. SND1 is also an integral component of RISC [99], and it is thus not surprising that it is also part of these miRNA/Ago-enriched EVs. An analysis of the changes in the protein composition of exosomes, after ionizing radiation, reveals an increase in SND1 [109]. In addition, patient urinary EVs, which are secreted by bladder cancer cells also contain high levels of SND1 [110]. Exosome-mediated intracellular communication within the tumor microenvironment seems to play an important role in the development and progression of HCC [111].
3.6. SND1 Expression Patterns at the RNA and Protein Levels
Detailed profiling of SND1 expression has found it to be ubiquitously expressed, with the highest expression in proliferating cells and active secretory organs such as exocrine pancreas, lactating mammary glands and the liver [112]. Western analysis of SND1 reveals that it is most highly expressed in the pancreas and the liver [88] (see Figure S1 in Liu et. al., 2010). We have reproduced this data and also see a very similar expression pattern (Figure 3). RNA-seq analysis, curated by the NCBI, was performed on 24 different human tissues from 95 individuals, and also reveals fairly ubiquitous RNA expression of SND1, with approximately a two-fold variable between tissues (Figure 3A). We performed Western analysis on protein extracted from a panel of cell lines, and we observed ubiquitous expression of SND1, albeit at varying levels (Figure 3B). When comparing the protein and RNA expression of SND1, there seems to be a disconnect between the high protein levels of SND1 in pancreas and the liver, and the equal and ubiquitous RNA expression of SND1 in different human tissues. This observation could be explained by the post-transcriptional regulation of SND1 possibly by the proteasome. Indeed, mass spectrometric analysis of SND1 reveals at least 10 different lysine residues that can be ubiquitinated (see the CST—PhosphoSite database). However, no studies have yet been performed to evaluate the protein stability of SND1 in different tissue settings. Alternatively, certain organs such as the liver and pancreas harbor levels of exosome activity, and SND1 may by sorted and secreted in the tissues from these organs, resulting in extracellular accumulation and retention.
3.7. Mouse Models of SND1 Overexpression Support Its Potential Oncogenic Functions
The SND1 knockout mouse has yet to be described. However, it does seem that this mouse has been generated [95], but the knockout phenotype was never presented, and these mice were only ever used to generate SND1 knockout mouse embryonic fibroblasts (MEFs) for further analysis [94,113]. Phenotyping performed by the mouse genome informatics (MIP) project at the Jackson Labs and the international mouse phenotyping consortium (IMPC), which both perform high-throughput phenotyping of spontaneous and trapping mutant mice, suggests that the SND1 knockout is partially viable, with knockout mice appearing at lower than expected Mendelian rations (approximately 50% of the expected ration). This data suggests that adult SND1 knockout mice will be available for detailed analysis (albeit at low numbers). However, very little information has yet been gleaned from the systemic knockout of SND1. It would be very informative to compare an SND1 knockout phenotype to a SND1 Tudor-dead knockin phenotype, as this would reveal the importance of the methylarginine reader abilities of SND1, and reveal what signaling pathways are dependent of SND1’s ability to read PRMT5 deposited marks, and what SND1 functions are independent of PRMT5 activity.
An overexpression model has revealed that SND1 is a driver for HCC when the induction of expression is focused on the liver, using an Albumin promoter (see Section 4.2) [14]. Similar overexpression models for PRMT5 would be extremely valuable to investigate whether this mouse would phenocopy the SND1 as a driver of HCC. Although we have generated other PRMT overexpression transgenic mouse models (including PRMT1, CARM1 and PRMT6) [114], the PRMT5 overexpression transgenic mouse has not yet been developed.
3.8. Mouse Syngeneic Tumor Models Reveal a Role for SND1 (and PRMT5) in Antitumor Immunity
The melanoma B16F10 cell line has been used to investigate the role of SND1 in facilitating immune evasion of tumor cells [115]. B16F10-SND1-KO cells were transferred into the flank of syngeneic mice and monitored over a number of days. The resulting tumor size and weight were smaller for growths seeded with SND1-KO cells than in the control parental cell. Furthermore, it was found that SND1-null tumors elicited a robust immune response, when compared to the parental cells. This suggests that the loss of SND1 may sensitize tumors to immune checkpoint inhibitors. Importantly, very similar experiments involving siRNA-mediated PRMT5 knockdown or PRMT5 inhibition by small-molecule treatment in B16F10 cells also showed that the presence of PRMT5 activity attenuates immune checkpoint therapy [116]. Thus, the loss of either PRMT5 or SND1 will convert an immunologically “cold” microenvironment into a “hot” one, further supporting a mechanistic link between this writer–reader pair. HCC may be sensitized to respond to immune checkpoint inhibitors (which block PD1, PD-L1 and CTLA-4 activity) by prior treatment with PRMT5 inhibitors.
3.9. SND1 Is Likely an Oncogene
Like PRMT5 [16], SND1 is upregulated in many different cancer types [117,118]. There is a particular interest in HCC, stemming from the observation made ten years ago, that SND1 protein levels are elevated in HCC and that its expression increases with the stage of the disease [119]. Interestingly, the analysis of RNA expression databases (TCGA and GEO) does not support a role for SND1 expression in the clinical progression of liver cancer [117], but the analysis of protein expression by immunohistochemistry does [119]. This suggests that SND1 may be post-transcriptionally regulated in certain tissues, as we have eluded to above (Figure 3). We will next summarize the reported roles of PRMT5 and SND1 in HCC, which can serve as a pre-clinical (mouse) and clinical (human) model system for understanding the link between this enzyme and its effector.
4. Hallmarks of HCC
Liver cancer comes in a variety of types and frequencies, from the common HCC and cholangiocarcinoma to the rare liver angiosarcoma and pediatric hepatoblastoma. HCC is cited as constituting approximately 75% of all liver cancers according to the Cancer Treatment Centers of America. Among all cancers, HCC is the fourth leading cause of cancer mortality worldwide [13]. Traditionally, HCC has affected more males than females, though recent studies have begun to suggest occurrence may have less gender disparities than previously thought [120,121]. Induction of HCC is often preceded by other hepatic ailments which ultimately develop into HCC. Non-alcoholic fatty liver disease (NAFLD) is one such malady that ultimately leads to HCC and is associated with obesity and metabolic syndrome [122,123]. Progression towards HCC from NAFLD often occurs in successive stages from NAFLD which develops into non-alcoholic steatohepatitis (NASH), increasing to fibrosis and cirrhosis, concluding with HCC and metastasis [124].
Atypical lipid accumulation is characteristic of hepatic damage and malignancies and HCC is no exception [125,126]. Hallmark metabolic dysregulation in hepatic tissues include increased de novo synthesis of lipids over extracellular lipid uptake to fulfill the lipid requirements needed for excessive cell division in the transformed state [126,127,128]. Upswing in lipogenesis arises in part from the liver being the center of lipid synthesis allowing a microenvironment permissible for de novo lipid synthesis. Sterol regulatory element-binding protein (SREBP) is a transcription factor directly upregulated in hepatocytes with active de novo lipogenesis. Accordingly, in HCC and premalignant hepatocytes, lipid accumulation is a tell-tale indicator of increased HCC risk. Lipid accumulation can be grossly visualized as lipid droplets within cells. Additional markers of HCC used in the clinic include measurement of alpha-fetoprotein (AFP), Alanine transaminase (ALT), and Aspartate transaminase (AST). Each of these enzymes, when elevated in the blood stream, can suggest malignancy and dysregulation of hepatocytes as they are typically seen in low abundance extra-hepatically. HCC is highly vascularized allowing excessive signaling and growth factor secretion for rapid expansion of cancer cells and is subsequently supported by sustained nutrient availability [129]. Hepatitis B/C (HBV and HCV, respectively) is the most common risk factor for developing HCC. Many reviews have explored HBV and HBC as they relate to HCC. Interestingly, PRMT5 can methylate the HBV core protein [130], and regulates it nuclear accumulation. Thus, there may be cross-talk between PRMT5–SND1 axis and hepatitis, but this issue will not be further addressed here.
4.1. PRMT5 and HCC
PRMT5 has, in recent years, become an increasingly prominent character in HCC research. Multiple studies have reported a worse prognosis of HCC patients with increased PRMT5 expression [12,74,75,76,131]. PRMT5 combined with the lysine methyltransferase SET8 have been identified as predictors of overall survival and recurrence in HCC patients [131].
EMT and invasion are key hallmarks of metastasis and PRMT5 has been identified as important in both. In vitro knockdown of PRMT5 in HCC and colon cancer cells decreases matrix metalloproteinase-2 expression [75]. This impaired expression decreases invasiveness of these lines along with decreasing proliferation which is supported by another study confirming increased HCC proliferation in PRMT5 competent lines [78]. A current model of E-cadherin depletion, characteristic of EMT, proposes the zinc finger domains of SNAIL directly bind the E-cadherin enhancer. AJUBA links PRMT5–MEP50 complex to SNAIL thereby permitting PRMT5 to mediate the SNAIL-dependent gene repression of E-cadherin [63].
A cellular defense against EMT in hepatocytes is the transcription of hepatocyte nuclear factor 4α (HNF4α). HNF4α has been shown to re-differentiate HCC cells towards hepatocytes and repress EMT, thereby blocking hepatocarcinogenesis [121,132,133]. While HNF4α works to drive differentiation towards hepatic cellularity, PRMT5 antagonizes HNF4α expression assisting in liver cancer stem cell maintenance. In HCC cells, PRMT5 binds H4R3 generating H4R3me2s at the HNF4α promoter. This methylation of H4R3 represses HNF4α transcription, while inhibition of PRMT5 activity restores HNF4α transcription and differentiation activity [78].
Another tumor suppressor, BTG2, is known to suppress proliferation, and is typically inhibited in cancers. While tumor suppressors are commonly inactivated by genetic mutations, deleterious mutations in BTG2 have not to date been identified, suggesting its downregulation may be a result of epigenetic reprogramming. PRMT5 has recently been linked to repressing BTG2 expression (again, in the context of HCC) through ERK signaling, though the mechanism of repression remains unknown [74].
As previously mentioned, lipid accumulation is concurrent with hepatic damage [125]. Lipid accumulation is driven by de novo lipogenesis over extracellular uptake, implying that drivers of lipid synthesis harbor oncogenic potential in hepatocytes. Sterol regulatory element-binding protein 1 (SREBP1) is a transcription factor that regulates the expression of genes involved in the synthesis of fatty acids, triglycerides and phospholipids, and has recently been shown to directly interact with PRMT5 [134]. Furthermore, PRMT5 methylates SREBP1, which stabilizes this transcription factor and helps promote the expression of its target genes and consequently also de novo lipogenesis. The overexpression of PRMT5 in HepG2 cells increases the levels of intracellular triglyceride levels, and conversely, the knockdown of PRMT5 results in a decrease in Oil red-O staining (a marker for intracellular lipid droplet accumulation). Furthermore, the overexpression of SREBP1 causes an increase in Oil red-O staining, which is not observed when the mutant form of SREBP1 (that cannot be methylated by PRMT5) is overexpressed. Thus, PRMT5 promotes de novo lipogenesis by methylating a single site on SREBP1 [134].
Long non-coding RNAs (lncRNAs) have emerged as key regulators of normal physiology as well as pathogenesis. Long intergenic non-coding RNA 1q21.2 (LINC01138) has been shown to correlate with PRMT5 expression as well as HCC tumor size, AFP levels, and hepatitis B surface antigen levels. PRMT5 and LINC01138 were shown to interact in HCC, which allowed PRMT5 to evade proteasomal degradation [77]. Increased PRMT5 stability may explain why we see increased protein expression of PRMT5, but not an increase in PRMT5 mRNA levels in some patient samples.
Recent findings reveal that PRMT5, in combination with SND1, promotes the dynamic regulation of E2F1 target genes. PRMT5 has been shown to promote cell growth, which contrasts to PRMT1 asymmetric dimethylation of E2F1 which promotes apoptosis [67,81]. PRMT5 methylation of E2F1, and the subsequent recognition of this methylated site by SND1, expands traditional E2F1 transcriptional control of genes to an extended set of targets that are traditionally poorly regulated by E2F1. Extended targeting is accomplished by E2F1-dependent alternative splicing of targets. This alternative splicing activity is dependent on both PRMT5 activity as well as SND1 recognition of SDMA marks on E2F1 [93]. While PRMT5 driven proliferation via E2F1 methylation has been identified, it remains unclear whether this signaling pathway is active or important for the development of HCC. However, given the emerging role of E2F1 in HCC [135], the activation of this regulatory node could be a critical consequence of elevated PRMT5 and/or SND1 protein levels.
4.2. SND1 and HCC
While the role that PRMT5 plays in the development of HCC is largely circumstantial—elevated PRMT5 levels clearly correlate with the promotion of HCC and poor prognosis of these cancer patients—its effector molecule, SND1, is more solidly implicated as a driver of HCC. Indeed, a landmark study of SND1 in HCC emerged from a mouse model with SND1 overexpression [14]. Transgenic mice carrying SND1 under the control of an albumin promoter/enhancer element, selectively overexpress SND1 in the liver, and this is sufficient to drive spontaneous HCC formation with partial penetrance. While half of the overexpressing mice develop HCC spontaneously, all overexpressing mice showed more aggressive tumors in HCC that is chemically induced by diethylnitrosamine (DEN). Hepatocytes from SND1 overexpressing mice have higher levels of spheroid-generating tumor-initiating cells. Furthermore, SND1 overexpression resulted in a steady proinflammatory state [14], similar to what is seen in chronic inflammation, a central hallmark of HCC progression.
SND1 contributes to alterations in the signaling cascades within HCC that control both transcriptional and post-transcriptional regulation. Angiotensin 2 receptor 1 (AT1R) mRNA stability is augmented through overexpression of SND1 [136]. Importantly, upregulation of AT1R is associated with both the progression of HCC, as well as unfavorable outcomes with respect to overall survival of cancer patients [137]. This increased stability of AT1R mRNA, and subsequent elevation of its protein levels, activates the ERK and SMAD signaling pathways, leading to a downstream increase in TGFβ signaling in HCC cells [138]. TGFβ signaling is known to drive proliferation and EMT progression [139]. Furthermore, TGFβ signaling also induces SND1 transcriptional activation in a feed forward loop [140]. This feed forward activity can be seen in other SND1-regulated pathways including NF-κB and SREBPs [141]. SND1 further promotes proliferative signals by degradation of miRNA via its nuclease domains. Elbarbary et. al. used transcriptome profiling to identify miRNAs that increase after knockdown or knockout of SND1, and show that these upregulated miRNAs in turn downregulate a cohort of mRNAs that are needed for G1/S transition [103]. We can speculate that the opposite is also true, when SND1 is upregulated, the G1/S transition may be shortened, and this would help explain one of the characteristics of liver cancer, which is a deregulated cell cycle [142].
NF-κB is a transcription factor that regulates innate immunity, and activation of this pathway promotes an inflammatory response and cellular growth. Chronic inflammation has been linked to many cancers, including HCC [143,144,145]. Indeed, chronic inflammation and hepatic injury serve as malignant drivers and precede 90% of HCC occurrences [146]. SND1 interacts with AEG-1 (also called metadherin) [119], which regulates multiple signaling pathways including NF-κB, PI3K/Akt and Wnt [147]. The activation of the NF-κB pathway by SND1 overexpression is reported to increase oncogenic miRNAs (oncomiRs) such as miR-221, that target and degrade tumor suppressor RNAs [80,148]. SND1 overexpression functions to increase inflammatory driving cytokines to promote HCC formation, as well as factors such as CXCL16 and angiogenin which promote angiogenesis. The inhibition of NF-κB blocks SND1-induced angiogenesis [80]. The benefits, to liver cancer cells, of having elevated SND1 can thus be explained, in part, by its pro-inflammatory and pro-angiogenic roles.
In order for cancer cells to persist and multiply, they evolve aggressive survival responses to stress signals to evade cell death pathways, which represents one of the major hallmarks of cancer. Stress granules are membraneless organelles that collect in the cytoplasm of cells, and serve as a means of stalling cellular machinery while the cell responds to the strain [149]. This suspended state can buy time sufficient for cells to respond with survival signaling, thereby evading apoptosis. SND1 has been shown to be enriched in stress granules induced from oxidative stress [150,151,152]. Whether SND1′s role is primarily as a nuclease or as a recruiting/scaffolding protein remains unclear. It has been noted, however, that phosphorylation of SND1 promotes its binding to G3BP [152], which in turn stimulates stress granule formation, suggesting that SND1 works in a recruiting role in stress granule formation. The ability of SND1 to help liver cancer cells evade cell death is not limited to its role in stress granule formation and function. SND1 can also promote the expression of UCA1 expression in HepG2 and SMMC-7721 cells [153]. UCA1 is an oncogenic lncRNA that has anti-apoptotic activity, and is itself a predictor of poor overall survival for patients with HCC [154]. Thus, high levels of SND1 helps tumor cells evade cell death.
A third survival pathway for cancer cells is DNA damage response. Using a laser microirradiation approach, SND1 is clearly localized to the laser-induced DNA damaged stripes [113]. Mechanistically, SND1 is recruited by PARP1 to damaged DNA, where it serves as a scaffold for chromatin remodeling proteins that then help facilitate DNA repair, such as SMARCA5 and GCN5 [113]. These two proteins are an ATP-dependent remodeler and a histone acetyltransferase, respectively, and they both act at sites of DNA damage to help open up chromatin so that the repair machinery can access the damaged DNA. SND1 overexpression thereby potentially provides a survival advantage in DNA damaged cells [82]. The combination of the liver as the body’s detoxification center, along with the ability of elevated SND1 levels to promote survival advantages under DNA damaging conditions, may partially explain why HCC is often chemoresistant and radioresistant. Targeting SND1 may reverse this resistance.
Like PRMT5, SND1 has been noted to have a variety of functions in lipogenesis. SND1 facilitates lipid droplet formation in mammary cells and hepatocytes. This association is lost in milk globules suggesting that SND1 association is specific to formation, but not maintenance of fat droplets [105,155]. SND1 overexpressing cells show a significantly altered lipoprotein secretion content and are saturated with phospholipids over other metabolically common lipids [156]. SREBPs appear to be regulated by SND1, although the difference in activity between normal and diseased states remains unknown [156]. More recently, it was found that hepatoma cells overexpressing SND1 display low triglyceride synthesis and accelerated cholesterol ester synthesis, likely because fatty acids are preferentially used for cholesterol esterification [157]. While profiling the target genes of SND1s transcriptional coactivator activity, using human hepatoma HepG2 cells, it was found that cohort of glycerolipid genes (such as CHPT1, LPGAT1, PTDSS1 and LPIN1) are regulated by SND1, in response to proinflammatory TNFα signaling [83]. SND1 is thus key for sustaining glycerophospholipid homeostasis in human HCC cells. The roles of SND1 in lipid metabolism has recently been reviewed in detail [158].
5. Targeting Elevated SND1 Levels with PRMT5 Inhibitors
As highlighted above, PRMT5 is overexpressed in a large number of different tumor types, and the inhibition of PRMT5 has been linked to tumor regression in mouse models [2,16,54]. These findings indicate that PRMT5 might be a promising therapeutic target for both solid and liquid tumors. Indeed, PRMT5 is currently a very popular target for the development of small-molecule inhibitors by both pharma and biomedical startup companies. A search of the published and patent literature reveals the development of at least 13 distinct PRMT5 inhibitors (Table 1). These inhibitors have six different mechanisms of action (MOA) [159,160], namely (1) inhibitors that compete with SAM (but not the peptide substrate); (2) inhibitors that compete with peptide substrate (but not SAM-competitive); (3) inhibitors that block both SAM and the substrate peptide from binding; (4) covalent inhibitors that form a stable bond with Cys449 in the active site and prevent SAM binding; (5) the development of a PROTAC probe that is based on the GSK3326595 compound, and targets PRMT5 for proteasomal degradation; (6) an allosteric inhibitor which causes the formation of an 11 amino acid acidic loop that blocks both SAM binding and peptide substrate binding.
In addition to the panel of 13 different PRMT5 inhibitors that have been developed, it may be possible to target elevated PRMT5 levels using inhibitors for PRMT1 and perhaps even CARM1 (PRMT4). This is because there is clear evidence of redundancy between different PRMT family members. Indeed, we have shown that PRMT1 and PRMT5 share many substrates [7]. Furthermore, GSK and the Guccione lab have shown that PRMT5 and PRMT1 inhibitors function synergistically to target MTAP-null cancer cells [161] and tumors that are driven by splicing mutations [56]. Finally, using a CRISPR-screening approach, we have found that in the presence of PRMT5 inhibitors cells are sensitized to PRMT1 and CARM1 loss [162]. Thus, it may be important to evaluate the effects of a CARM1 inhibitor (GSK3359088) [163] and a Type I PRMT inhibitor (GSK3368715) [161] for their ability to retard the growth of tumors with elevated PRMT5–SND1 signaling.
6. Conclusion and Future Direction
HCC is a major health concern worldwide. It has a 20% five-year survival rate and 1% of the global population are expected to develop HCC in their life time. This disease poses a significant health burden which urges additional study. The majority of therapeutic options are hepatic resection and transplant, though transplant needs far outweigh available organs [13]. Accordingly, a more comprehensive molecular understanding of HCC development is needed to approach the HCC epidemic. The PRMT5–SND1 axis has emerged recently as a key point of inquiry, though we are far from understanding its intricacies in HCC. A lot of the interest in PRMT5 is driven by the fact that there are now very good inhibitors available that target this enzyme, raising therapeutic hope for diseases that are driven by PRMT5 overexpression, or by increased effector molecule activity (such as SND1 overexpression). SND1 has been shown to be a potent hepatic-oncogene, though many important questions still need to be addressed. Clearly, there remains a lot of low-hanging fruit to be picked; for example: (1) In the context of HHC, is SND1 the primary effector molecule for SDMA marks that are deposited by PRMT5? (2) Does SND1 compete with other Tudor domain-containing proteins such as SMN for binding to PRMT5 deposited marks? (3) Will PRMT5 inhibitors block the oncogenic effects of SND1 overexpression in the liver? (4) How important is the Tudor domain of SND1 for its oncogenic function? Many of these questions will require the development of new genetically engineered mouse models that will facilitate pre-clinical studies. A detailed mechanistic elucidation of the PRMT5–SND1 axis in HCC promises to illuminate novel and rational approaches towards treating this terrible disease.
Author Contributions
T.W. wrote the first draft of this review. M.T.B. then revised the review. Y.W. edited the review and contributed to Figure 3. All authors have read and agreed to the published version of the manuscript.
Funding
M.T.B. is supported by an NIH grant—GM126421.
Conflicts of Interest
M.T.B. is a co-founder of EpiCypher.
References
- Chen, J.; Sagum, C.; Bedford, M.T. Protein domain microarrays as a platform to decipher signaling pathways and the histone code. Methods 2019, 184, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF7 methylates arginine 85 in the NDUFS2 subunit of human complex I. J. Biol. Chem. 2013, 288, 33016–33026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Hadjikyriacou, A.; Xia, Z.; Gayatri, S.; Kim, D.; Zurita-Lopez, C.; Kelly, R.; Guo, A.; Li, W.; Clarke, S.G.; et al. PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun. 2015, 6, 6428. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M. Epsilon-N-methylated lysine and guanidine-N-methylated arginine of proteins. 3. Presence and distribution in nature and mammals. Seikagaku 1972, 44, 364–370. [Google Scholar]
- Paik, W.K.; Kim, S. Natural Occurrence of Various Methylated Amino Acid Derivatives; John Wiley & Sons: New York, NY, USA, 1980; pp. 8–25. [Google Scholar]
- Dhar, S.; Vemulapalli, V.; Patananan, A.N.; Huang, G.L.; Di Lorenzo, A.; Richard, S.; Comb, M.J.; Guo, A.; Clarke, S.G.; Bedford, M.T. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 2013, 3, 1311. [Google Scholar] [CrossRef]
- Gayatri, S.; Bedford, M.T. Readers of histone methylarginine marks. Biochim. Biophys. Acta 2014, 1839, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P. Tudor domains in proteins that interact with RNA. Trends Biochem. Sci. 1997, 22, 51–52. [Google Scholar] [CrossRef]
- Friesen, W.J.; Massenet, S.; Paushkin, S.; Wyce, A.; Dreyfuss, G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Selenko, P.; Sprangers, R.; Stier, G.; Buhler, D.; Fischer, U.; Sattler, M. SMN tudor domain structure and its interaction with the Sm proteins. Nat. Struct. Biol. 2001, 8, 27–31. [Google Scholar]
- Shimizu, D.; Kanda, M.; Sugimoto, H.; Shibata, M.; Tanaka, H.; Takami, H.; Iwata, N.; Hayashi, M.; Tanaka, C.; Kobayashi, D.; et al. The protein arginine methyltransferase 5 promotes malignant phenotype of hepatocellular carcinoma cells and is associated with adverse patient outcomes after curative hepatectomy. Int. J. Oncol. 2017, 50, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, N.; Rajasekaran, D.; Mendoza, R.G.; Shen, X.N.; Siddiq, A.; Akiel, M.A.; Robertson, C.L.; Subler, M.A.; Windle, J.J.; Fisher, P.B.; et al. Oncogenic Role of SND1 in Development and Progression of Hepatocellular Carcinoma. Cancer Res. 2017, 77, 3306–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Xu, R.M.; Thompson, P.R. Substrate specificity, processivity, and kinetic mechanism of protein arginine methyltransferase 5. Biochemistry 2013, 52, 5430–5440. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef]
- Friesen, W.J.; Wyce, A.; Paushkin, S.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 2002, 277, 8243–8247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Dhar, S.; Bedford, M.T. PRMT5 regulates IRES-dependent translation via methylation of hnRNP A1. Nucleic Acids Res. 2017, 45, 4359–4369. [Google Scholar] [CrossRef] [Green Version]
- Timm, D.E.; Bowman, V.; Madsen, R.; Rauch, C. Cryo-electron microscopy structure of a human PRMT5:MEP50 complex. PLoS ONE 2018, 13, e0193205. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.C.; Wilczek, C.; Bonanno, J.B.; Xing, L.; Seznec, J.; Matsui, T.; Carter, L.G.; Onikubo, T.; Kumar, P.R.; Chan, M.K.; et al. Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS ONE 2013, 8, e57008. [Google Scholar] [CrossRef]
- Antonysamy, S.; Bonday, Z.; Campbell, R.M.; Doyle, B.; Druzina, Z.; Gheyi, T.; Han, B.; Jungheim, L.N.; Qian, Y.; Rauch, C.; et al. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. USA 2012, 109, 17960–17965. [Google Scholar] [CrossRef] [Green Version]
- Hosohata, K.; Li, P.; Hosohata, Y.; Qin, J.; Roeder, R.G.; Wang, Z. Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol. Cell. Biol. 2003, 23, 7019–7029. [Google Scholar] [CrossRef] [Green Version]
- Wilczek, C.; Chitta, R.; Woo, E.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Shechter, D. Protein arginine methyltransferase Prmt5-Mep50 methylates histones H2A and H4 and the histone chaperone nucleoplasmin in Xenopus laevis eggs. J. Biol. Chem. 2011, 286, 42221–42231. [Google Scholar] [CrossRef] [Green Version]
- Meister, G.; Eggert, C.; Buhler, D.; Brahms, H.; Kambach, C.; Fischer, U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–1994. [Google Scholar] [CrossRef] [Green Version]
- Pesiridis, G.S.; Diamond, E.; Van Duyne, G.D. Role of pICLn in methylation of Sm proteins by PRMT5. J. Biol. Chem. 2009, 284, 21347–21359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paknia, E.; Chari, A.; Stark, H.; Fischer, U. The Ribosome Cooperates with the Assembly Chaperone pICln to Initiate Formation of snRNPs. Cell Rep. 2016, 16, 3103–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guderian, G.; Peter, C.; Wiesner, J.; Sickmann, A.; Schulze-Osthoff, K.; Fischer, U.; Grimmler, M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011, 286, 1976–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, M.; Messaoudi, S.E.; Rodier, G.; Le Cam, A.; Sardet, C.; Fabbrizio, E. The histone-binding protein COPR5 is required for nuclear functions of the protein arginine methyltransferase PRMT5. EMBO Rep. 2008, 9, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Tamiya, H.; Kim, H.; Klymenko, O.; Kim, H.; Feng, Y.; Zhang, T.; Han, J.Y.; Murao, A.; Snipas, S.J.; Jilaveanu, L.; et al. SHARPIN-mediated regulation of protein arginine methyltransferase 5 controls melanoma growth. J. Clin. Investig. 2018, 128, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Lin, X.; Segers, F.; Suganthan, R.; Hildrestrand, G.A.; Rinholm, J.E.; Aas, P.A.; Sousa, M.M.L.; Holm, S.; Bolstad, N.; et al. OXR1A, a Coactivator of PRMT5 Regulating Histone Arginine Methylation. Cell Rep. 2020, 30, 4165–4178. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef] [Green Version]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrakis, K.J.; McDonald, E.R., 3rd; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; deWeck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Pollack, B.P.; Kotenko, S.V.; He, W.; Izotova, L.S.; Barnoski, B.L.; Pestka, S. The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J. Biol. Chem. 1999, 274, 31531–31542. [Google Scholar] [CrossRef] [Green Version]
- Tee, W.W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, F.M.; Cote, J.; Boulanger, M.C.; Richard, S. A Proteomic Analysis of Arginine-methylated Protein Complexes. Mol. Cell. Proteom. 2003, 2, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Musiani, D.; Bok, J.; Massignani, E.; Wu, L.; Tabaglio, T.; Ippolito, M.R.; Cuomo, A.; Ozbek, U.; Zorgati, H.; Ghoshdastider, U.; et al. Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Nishioka, K.; Reinberg, D. Methods and tips for the purification of human histone methyltransferases. Methods 2003, 31, 49–58. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Chen, G.I.; Gingras, A.C. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods 2007, 42, 298–305. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wu, H.; Lee, P.; Wang, Z. Roles of the androgen receptor cofactor p44 in the growth of prostate epithelial cells. J. Mol. Endocrinol. 2006, 37, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Cheng, G.; Hamard, P.J.; Greenblatt, S.; Wang, L.; Man, N.; Perna, F.; Xu, H.; Tadi, M.; Luciani, L.; et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Investig. 2015, 125, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Litzler, L.C.; Zahn, A.; Meli, A.P.; Hebert, S.; Patenaude, A.M.; Methot, S.P.; Sprumont, A.; Bois, T.; Kitamura, D.; Costantino, S.; et al. PRMT5 is essential for B cell development and germinal center dynamics. Nat. Commun. 2019, 10, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Nagai, Y.; Okumura, M.; Greene, M.I.; Kambayashi, T. PRMT5 Is Required for T Cell Survival and Proliferation by Maintaining Cytokine Signaling. Front. Immunol. 2020, 11, 621. [Google Scholar] [CrossRef]
- Norrie, J.L.; Li, Q.; Co, S.; Huang, B.L.; Ding, D.; Uy, J.C.; Ji, Z.; Mackem, S.; Bedford, M.T.; Galli, A.; et al. PRMT5 is essential for the maintenance of chondrogenic progenitor cells in the limb bud. Development 2016, 143, 4608–4619. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Wu, H.; Wang, F.; Wang, Z. Altered differentiation and proliferation of prostate epithelium in mice lacking the androgen receptor cofactor p44/WDR77. Endocrinology 2010, 151, 3941–3953. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Zhang, F.; Wang, Z.Q.; Ma, W.; Davis, R.E.; Wang, Z. The p44/wdr77-dependent cellular proliferation process during lung development is reactivated in lung cancer. Oncogene 2013, 32, 1888–1900. [Google Scholar] [CrossRef] [Green Version]
- Scaglione, A.; Patzig, J.; Liang, J.; Frawley, R.; Bok, J.; Mela, A.; Yattah, C.; Zhang, J.; Teo, S.X.; Zhou, T.; et al. PRMT5-mediated regulation of developmental myelination. Nat. Commun. 2018, 9, 2840. [Google Scholar] [CrossRef]
- Rho, J.; Choi, S.; Seong, Y.R.; Cho, W.K.; Kim, S.H.; Im, D.S. Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J. Biol. Chem. 2001, 276, 11393–11401. [Google Scholar] [CrossRef] [Green Version]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.Q.; Li, Y.; Yang, C.; Li, J.; Tan, S.H.; Chin, D.W.L.; Nakamura-Ishizu, A.; Yang, H.; Suda, T. PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep. 2019, 26, 2316–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Liu, F.; Veazey, K.J.; Gao, G.; Das, P.; Neves, L.F.; Lin, K.; Zhong, Y.; Lu, Y.; Giuliani, V.; et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Radzisheuskaya, A.; Shliaha, P.V.; Grinev, V.; Lorenzini, E.; Kovalchuk, S.; Shlyueva, D.; Gorshkov, V.; Hendrickson, R.C.; Jensen, O.N.; Helin, K. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat. Struct. Mol. Biol. 2019, 26, 999–1012. [Google Scholar] [CrossRef]
- Fong, J.Y.; Pignata, L.; Goy, P.A.; Kawabata, K.C.; Lee, S.C.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; He, X.; Cao, Y.; O’Dwyer, K.; Szigety, K.M.; Wu, Y.; Gurung, B.; Feng, Z.; Katona, B.W.; Hua, X. Islet-specific Prmt5 excision leads to reduced insulin expression and glucose intolerance in mice. J. Endocrinol. 2020, 244, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gunther, S.; Looso, M.; Kunne, C.; Kruger, M.; Kim, J.; Zhou, Y.; Braun, T. Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat. Commun. 2015, 6, 7140. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, J.; Liu, Z.; Gray, R.S.; Vokes, S.A. PRMT5 is necessary to form distinct cartilage identities in the knee and long bone. Dev. Biol. 2019, 456, 154–163. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, T.; Li, Q.; Liu, C.; Han, F.; Chen, M.; Zhang, L.; Cui, X.; Qin, Y.; Bao, S.; et al. Prmt5 is required for germ cell survival during spermatogenesis in mice. Sci. Rep. 2015, 5, 11031. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Jiao, J.; Li, H.; Wan, H.; Zheng, C.; Cai, J.; Bao, S. Histone arginine methylation by Prmt5 is required for lung branching morphogenesis through repression of BMP signaling. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24, 9630–9645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.; Peng, H.; Ayyanathan, K.; Yan, K.P.; Langer, E.M.; Longmore, G.D.; Rauscher, F.J., 3rd. The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 2008, 28, 3198–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.; Baiocchi, R.A.; Byrd, J.C.; Grever, M.R.; Jacob, S.T.; Sif, S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pal, S.; Sif, S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 2008, 28, 6262–6277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Sohn, H.Y.; Yoon, S.Y.; Oh, J.H.; Yang, J.O.; Kim, J.H.; Song, K.S.; Rho, S.M.; Yoo, H.S.; Kim, Y.S.; et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin. Cancer Res. 2005, 11, 473–482. [Google Scholar] [PubMed]
- Cho, E.C.; Zheng, S.; Munro, S.; Liu, G.; Carr, S.M.; Moehlenbrink, J.; Lu, Y.C.; Stimson, L.; Khan, O.; Konietzny, R.; et al. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012, 31, 1785–1797. [Google Scholar] [CrossRef]
- Jing, P.; Xie, N.; Zhu, X.; Dang, H.; Gu, Z. The methylation induced by protein arginine methyltransferase 5 promotes tumorigenesis and progression of lung cancer. J. Thorac. Dis. 2018, 10, 7014–7019. [Google Scholar] [CrossRef]
- Wei, T.Y.; Juan, C.C.; Hisa, J.Y.; Su, L.J.; Lee, Y.C.; Chou, H.Y.; Chen, J.M.; Wu, Y.C.; Chiu, S.C.; Hsu, C.P.; et al. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer Sci. 2012, 103, 1640–1650. [Google Scholar] [CrossRef]
- Bao, X.; Zhao, S.; Liu, T.; Liu, Y.; Liu, Y.; Yang, X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 2013, 61, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, C.; Yang, J.; Peters, S.B.; Bill, M.A.; Baiocchi, R.A.; Yan, F.; Sif, S.; Tae, S.; Gaudio, E.; Wu, X.; et al. PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1.). PLoS ONE 2013, 8, e74710. [Google Scholar] [CrossRef]
- Han, X.; Li, R.; Zhang, W.; Yang, X.; Wheeler, C.G.; Friedman, G.K.; Province, P.; Ding, Q.; You, Z.; Fathallah-Shaykh, H.M.; et al. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J. Neurooncol. 2014, 118, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–1765. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhu, Y.; Zhou, Z.; Xu, J.; Jin, S.; Xu, K.; Zhang, H.; Sun, Q.; Wang, J.; Xu, J. PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 2018, 7, 869–882. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Lee, J.S.; Park, E.R.; Shen, Y.N.; Kim, M.Y.; Shin, H.J.; Joo, H.Y.; Cho, E.H.; Moon, S.M.; Shin, U.S.; et al. Protein arginine methyltransferase 5 is implicated in the aggressiveness of human hepatocellular carcinoma and controls the invasive activity of cancer cells. Oncol. Rep. 2018, 40, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Dong, S.; Li, Z.; Lu, L.; Zhang, S.; Chen, X.; Cen, X.; Wu, Y. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta-catenin. J. Transl. Med. 2015, 13, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, J.; Liu, X.; Li, S.; Wang, Q.; Di, C.; Hu, Z.; Yu, T.; Ding, J.; Li, J.; et al. The LINC01138 drives malignancies via activating arginine methyltransferase 5 in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1572. [Google Scholar] [CrossRef]
- Zheng, B.N.; Ding, C.H.; Chen, S.J.; Zhu, K.; Shao, J.; Feng, J.; Xu, W.P.; Cai, L.Y.; Zhu, C.P.; Duan, W.; et al. Targeting PRMT5 Activity Inhibits the Malignancy of Hepatocellular Carcinoma by Promoting the Transcription of HNF4alpha. Theranostics 2019, 9, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ki, B.S.; Hong, K.; Park, S.P.; Ko, J.J.; Choi, Y. Tudor Domain Containing Protein TDRD12 Expresses at the Acrosome of Spermatids in Mouse Testis. Asian-Australas J. Anim. Sci. 2016, 29, 944–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhekadur, P.K.; Das, S.K.; Gredler, R.; Chen, D.; Srivastava, J.; Robertson, C.; Baldwin, A.S., Jr.; Fisher, P.B.; Sarkar, D. Multifunction protein staphylococcal nuclease domain containing 1 (SND1) promotes tumor angiogenesis in human hepatocellular carcinoma through novel pathway that involves nuclear factor kappaB and miR-221. J. Biol. Chem. 2012, 287, 13952–13958. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Moehlenbrink, J.; Lu, Y.C.; Zalmas, L.P.; Sagum, C.A.; Carr, S.; McGouran, J.F.; Alexander, L.; Fedorov, O.; Munro, S.; et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell 2013, 52, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Chidambaranathan-Reghupaty, S.; Mendoza, R.; Fisher, P.B.; Sarkar, D. The multifaceted oncogene SND1 in cancer: Focus on hepatocellular carcinoma. Hepatoma Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arretxe, E.; Armengol, S.; Mula, S.; Chico, Y.; Ochoa, B.; Martinez, M.J. Profiling of promoter occupancy by the SND1 transcriptional coactivator identifies downstream glycerolipid metabolic genes involved in TNFalpha response in human hepatoma cells. Nucleic Acids Res. 2015, 43, 10673–10688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Beltran, E.; Denisenko, T.V.; Zhivotovsky, B.; Bozhkov, P.V. Tudor staphylococcal nuclease: Biochemistry and functions. Cell Death Differ. 2016, 23, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Mornon, J.P. The human EBNA-2 coactivator p100: Multidomain organization and relationship to the staphylococcal nuclease fold and to the tudor protein involved in Drosophila melanogaster development. Biochem. J. 1997, 321 Pt 1, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Shaw, N.; Zhao, M.; Cheng, C.; Xu, H.; Saarikettu, J.; Li, Y.; Da, Y.; Yao, Z.; Silvennoinen, O.; Yang, J.; et al. The multifunctional human p100 protein ‘hooks’ methylated ligands. Nat. Struct. Mol. Biol. 2007, 14, 779–784. [Google Scholar] [CrossRef]
- Friberg, A.; Corsini, L.; Mourao, A.; Sattler, M. Structure and ligand binding of the extended Tudor domain of D. melanogaster Tudor-SN. J. Mol. Biol. 2009, 387, 921–934. [Google Scholar] [CrossRef]
- Liu, K.; Chen, C.; Guo, Y.; Lam, R.; Bian, C.; Xu, C.; Zhao, D.Y.; Jin, J.; MacKenzie, F.; Pawson, T.; et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc. Natl. Acad. Sci. USA 2010, 107, 18398–18403. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.; Drapkin, R.; Yalamanchili, R.; Mosialos, G.; Kieff, E. The Epstein-Barr virus nuclear protein 2 acidic domain forms a complex with a novel cellular coactivator that can interact with TFIIE. Mol. Cell. Biol. 1995, 15, 4735–4744. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Aittomaki, S.; Pesu, M.; Carter, K.; Saarinen, J.; Kalkkinen, N.; Kieff, E.; Silvennoinen, O. Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. EMBO J. 2002, 21, 4950–4958. [Google Scholar] [CrossRef] [Green Version]
- Paukku, K.; Yang, J.; Silvennoinen, O. Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Mol. Endocrinol. 2003, 17, 1805–1814. [Google Scholar] [CrossRef]
- Leverson, J.D.; Koskinen, P.J.; Orrico, F.C.; Rainio, E.M.; Jalkanen, K.J.; Dash, A.B.; Eisenman, R.N.; Ness, S.A. Pim-1 kinase and p100 cooperate to enhance c-Myb activity. Mol. Cell 1998, 2, 417–425. [Google Scholar] [CrossRef]
- Roworth, A.P.; Carr, S.M.; Liu, G.; Barczak, W.; Miller, R.L.; Munro, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. Arginine methylation expands the regulatory mechanisms and extends the genomic landscape under E2F control. Sci. Adv. 2019, 5, eaaw4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.; Zhang, C.; Tecle, A.; Fu, X.; He, J.; Song, J.; Zhang, W.; Sun, X.; Ren, Y.; Silvennoinen, O.; et al. Tudor staphylococcal nuclease (Tudor-SN), a novel regulator facilitating G1/S phase transition, acting as a co-activator of E2F-1 in cell cycle regulation. J. Biol. Chem. 2015, 290, 7208–7220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Z.; Zhao, X.; Fu, X.; Su, C.; Xin, L.; Saarikettu, J.; Yang, X.; Yao, Z.; Silvennoinen, O.; Wei, M.; et al. Tudor-SN, a novel coactivator of peroxisome proliferator-activated receptor gamma protein, is essential for adipogenesis. J. Biol. Chem. 2014, 289, 8364–8374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Zhao, X.; Zhu, Y.; He, J.; Shao, J.; Su, C.; Zhang, Y.; Zhang, W.; Saarikettu, J.; Silvennoinen, O.; et al. Tudor staphylococcal nuclease (Tudor-SN) participates in small ribonucleoprotein (snRNP) assembly via interacting with symmetrically dimethylated Sm proteins. J. Biol. Chem. 2012, 287, 18130–18141. [Google Scholar] [CrossRef] [Green Version]
- Cappellari, M.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Saarikettu, J.; Silvennoinen, O.; Sette, C. The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 2014, 33, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Valineva, T.; Hong, J.; Bu, T.; Yao, Z.; Jensen, O.N.; Frilander, M.J.; Silvennoinen, O. Transcriptional co-activator protein p100 interacts with snRNP proteins and facilitates the assembly of the spliceosome. Nucleic Acids Res. 2007, 35, 4485–4494. [Google Scholar] [CrossRef] [Green Version]
- Caudy, A.A.; Ketting, R.F.; Hammond, S.M.; Denli, A.M.; Bathoorn, A.M.; Tops, B.B.; Silva, J.M.; Myers, M.M.; Hannon, G.J.; Plasterk, R.H. A micrococcal nuclease homologue in RNAi effector complexes. Nature 2003, 425, 411–414. [Google Scholar] [CrossRef]
- Scadden, A.D. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat. Struct. Mol. Biol. 2005, 12, 489–496. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Yang, W.Z.; Chen, Y.P.; Yuan, H.S. Structural and functional insights into human Tudor-SN, a key component linking RNA interference and editing. Nucleic Acids Res. 2008, 36, 3579–3589. [Google Scholar] [CrossRef] [Green Version]
- Elbarbary, R.A.; Miyoshi, K.; Myers, J.R.; Du, P.; Ashton, J.M.; Tian, B.; Maquat, L.E. Tudor-SN-mediated endonucleolytic decay of human cell microRNAs promotes G1/S phase transition. Science 2017, 356, 859–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbarbary, R.A.; Miyoshi, K.; Hedaya, O.; Myers, J.R.; Maquat, L.E. UPF1 helicase promotes TSN-mediated miRNA decay. Genes Dev. 2017, 31, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.W.; Winter, S.; Rackwitz, H.R.; Heid, H.W. Nuclear coactivator protein p100 is present in endoplasmic reticulum and lipid droplets of milk secreting cells. Biochim. Biophys. Acta 2000, 1523, 84–90. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Makarova, J.A.; Shkurnikov, M.U.; Wicklein, D.; Lange, T.; Samatov, T.R.; Turchinovich, A.A.; Tonevitsky, A.G. Intracellular and extracellular microRNA: An update on localization and biological role. Prog. Histochem. Cytochem. 2016, 51, 33–49. [Google Scholar] [CrossRef]
- Nakamura, K.; Sawada, K.; Yoshimura, A.; Kinose, Y.; Nakatsuka, E.; Kimura, T. Clinical relevance of circulating cell-free microRNAs in ovarian cancer. Mol. Cancer 2016, 15, 48. [Google Scholar] [CrossRef] [Green Version]
- Jelonek, K.; Wojakowska, A.; Marczak, L.; Muer, A.; Tinhofer-Keilholz, I.; Lysek-Gladysinska, M.; Widlak, P.; Pietrowska, M. Ionizing radiation affects protein composition of exosomes secreted in vitro from head and neck squamous cell carcinoma. Acta Biochim. Pol. 2015, 62, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Silvers, C.R.; Miyamoto, H.; Messing, E.M.; Netto, G.J.; Lee, Y.F. Characterization of urinary extracellular vesicle proteins in muscle-invasive bladder cancer. Oncotarget 2017, 8, 91199–91208. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, L.; Lv, D.; Zhu, X.; Tang, H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J. Hematol. Oncol. 2019, 12, 53. [Google Scholar] [CrossRef]
- Fashe, T.; Saarikettu, J.; Isomaki, P.; Yang, J.; Silvennoinen, O. Expression analysis of Tudor-SN protein in mouse tissues. Tissue Cell 2013, 45, 21–31. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, C.; Meng, H.; Zhang, K.; Shi, L.; Cao, C.; Wang, Y.; Su, C.; Xin, L.; Ren, Y.; et al. Oncoprotein Tudor-SN is a key determinant providing survival advantage under DNA damaging stress. Cell Death Differ. 2018, 25, 1625–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Di Lorenzo, A.; Lin, K.; Lu, Y.; Zhong, Y.; Sebastian, M.M.; Muller, W.J.; Yang, Y.; Bedford, M.T. Mouse Models of Overexpression Reveal Distinct Oncogenic Roles for Different Type I Protein Arginine Methyltransferases. Cancer Res. 2019, 79, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, X.; Cui, X.; Zhuo, Y.; Li, H.; Ha, C.; Xin, L.; Ren, Y.; Zhang, W.; Sun, X.; et al. Oncoprotein SND1 hijacks nascent MHC-I heavy chain to ER-associated degradation, leading to impaired CD8+ T cell response in tumor. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, X.; Liu, M.; Zhao, C.; Zhang, N.; Ren, Y.; Su, C.; Zhang, W.; Sun, X.; He, J.; et al. A pan-cancer analysis of the oncogenic role of staphylococcal nuclease domain-containing protein 1 (SND1) in human tumors. Genomics 2020. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Ochiai, M.; Nakashima, K.; Ubagai, T.; Sugimura, T.; Nakagama, H. SND1, a component of RNA-induced silencing complex, is up-regulated in human colon cancers and implicated in early stage colon carcinogenesis. Cancer Res. 2007, 67, 9568–9576. [Google Scholar] [CrossRef] [Green Version]
- Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Chen, D.; Emdad, L.; Bhutia, S.; Pannell, L.; Fisher, P.B.; Sarkar, D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology 2011, 53, 1538–1548. [Google Scholar] [CrossRef] [Green Version]
- Petrick, J.L.; Kelly, S.P.; Altekruse, S.F.; McGlynn, K.A.; Rosenberg, P.S. Future of Hepatocellular Carcinoma Incidence in the United States Forecast Through 2030. J. Clin. Oncol. 2016, 34, 1787–1794. [Google Scholar] [CrossRef]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Mohamed, A.M.T.; Kolonin, M.G.; Sladek, F.M.; Younes, M.; Eckel-Mahan, K.L. HNF4alpha-Deficient Fatty Liver Provides a Permissive Environment for Sex-Independent Hepatocellular Carcinoma. Cancer Res. 2019, 79, 5860–5873. [Google Scholar] [CrossRef] [Green Version]
- Seyda Seydel, G.; Kucukoglu, O.; Altinbasv, A.; Demir, O.O.; Yilmaz, S.; Akkiz, H.; Otan, E.; Sowa, J.P.; Canbay, A. Economic growth leads to increase of obesity and associated hepatocellular carcinoma in developing countries. Ann. Hepatol. 2016, 15, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Hasanin, M.; Kaif, M.; Wiesner, R.; Kuo, Y.F. Nonalcoholic Steatohepatitis is the Most Rapidly Growing Indication for Simultaneous Liver Kidney Transplantation in the United States. Transplantation 2016, 100, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Tarao, K.; Tanaka, K.; Nozaki, A.; Sato, A.; Ishii, T.; Komatsu, H.; Ikeda, T.; Komatsu, T.; Matsushima, S.; Oshige, K. Efficacy and safety of dual therapy with daclatasvir and asunaprevir in elderly patients. World J. Hepatol. 2017, 9, 544–550. [Google Scholar] [CrossRef]
- Gu, J.; Yao, M.; Yao, D.; Wang, L.; Yang, X.; Yao, D. Nonalcoholic Lipid Accumulation and Hepatocyte Malignant Transformation. J. Clin. Transl. Hepatol. 2016, 4, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Twaddel, W.; Goloubeva, O.G.; Wong, K.K.; Saxena, N.K.; Biswal, S.; Girnun, G.D. Metformin prevents liver tumorigenesis by inhibiting pathways driving hepatic lipogenesis. Cancer Prev. Res. 2012, 5, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, R.J.; Voorneveld, P.W.; Kodach, L.L.; Hardwick, J.C. Cholesterol metabolism and colorectal cancers. Curr. Opin. Pharmacol. 2012, 12, 690–695. [Google Scholar] [CrossRef]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef]
- Lin, Z.; Jia, H.; Hong, L.; Zheng, Y.; Shao, W.; Ren, X.; Zhu, W.; Lu, L.; Lu, M.; Zhang, J.; et al. Prognostic impact of SET domain-containing protein 8 and protein arginine methyltransferase 5 in patients with hepatocellular carcinoma following curative resection. Oncol. Lett. 2018, 16, 3665–3673. [Google Scholar] [CrossRef]
- Yin, C.; Lin, Y.; Zhang, X.; Chen, Y.X.; Zeng, X.; Yue, H.Y.; Hou, J.L.; Deng, X.; Zhang, J.P.; Han, Z.G.; et al. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology 2008, 48, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Ning, B.F.; Ding, J.; Yin, C.; Zhong, W.; Wu, K.; Zeng, X.; Yang, W.; Chen, Y.X.; Zhang, J.P.; Zhang, X.; et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res. 2010, 70, 7640–7651. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhao, X.; Zhao, L.; Li, J.; Yang, H.; Zhu, Z.; Liu, J.; Huang, G. Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Farra, R.; Grassi, G.; Tonon, F.; Abrami, M.; Grassi, M.; Pozzato, G.; Fiotti, N.; Forte, G.; Dapas, B. The Role of the Transcription Factor E2F1 in Hepatocellular Carcinoma. Curr. Drug Deliv. 2017, 14, 272–281. [Google Scholar] [CrossRef]
- Paukku, K.; Kalkkinen, N.; Silvennoinen, O.; Kontula, K.K.; Lehtonen, J.Y. p100 increases AT1R expression through interaction with AT1R 3’-UTR. Nucleic Acids Res. 2008, 36, 4474–4487. [Google Scholar] [CrossRef]
- Ji, Y.; Chen, H.; Gow, W.; Ma, L.; Jin, Y.; Hui, B.; Yang, Z.; Wang, Z. Potential biomarkers Ang II/AT1R and S1P/S1PR1 predict the prognosis of hepatocellular carcinoma. Oncol. Lett. 2020, 20, 208. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Akiel, M.; Emdad, L.; Gredler, R.; Srivastava, J.; Rajasekaran, D.; Robertson, C.L.; Mukhopadhyay, N.D.; Fisher, P.B.; Sarkar, D. Staphylococcal nuclease domain containing-1 (SND1) promotes migration and invasion via angiotensin II type 1 receptor (AT1R) and TGFbeta signaling. FEBS Open Bio 2014, 4, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Yu, L.; Liu, X.; Cui, K.; Di, Y.; Xin, L.; Sun, X.; Zhang, W.; Yang, X.; Wei, M.; Yao, Z.; et al. SND1 Acts Downstream of TGFbeta1 and Upstream of Smurf1 to Promote Breast Cancer Metastasis. Cancer Res. 2015, 75, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, B.; Chico, Y.; Martinez, M.J. Insights into SND1 Oncogene Promoter Regulation. Front. Oncol. 2018, 8, 606. [Google Scholar] [CrossRef]
- Bisteau, X.; Caldez, M.J.; Kaldis, P. The Complex Relationship between Liver Cancer and the Cell Cycle: A Story of Multiple Regulations. Cancers 2014, 6, 79–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caselmann, W.H. Pathogenesis of hepatocellular carcinoma. Digestion 1998, 59 (Suppl. 2), 60–63. [Google Scholar] [CrossRef]
- Mauad, T.H.; van Nieuwkerk, C.M.; Dingemans, K.P.; Smit, J.J.; Schinkel, A.H.; Notenboom, R.G.; van den Bergh Weerman, M.A.; Verkruisen, R.P.; Groen, A.K.; Oude Elferink, R.P.; et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 1994, 145, 1237–1245. [Google Scholar] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, G.; Srivastava, N.; Goyal, M.; Rakha, E.; Lothion-Roy, J.; Mongan, N.P.; Miftakhova, R.R.; Khaiboullina, S.F.; Rizvanov, A.A.; Baranwal, M. Metadherin: A Therapeutic Target in Multiple Cancers. Front. Oncol. 2019, 9, 349. [Google Scholar] [CrossRef] [PubMed]
- Pineau, P.; Volinia, S.; McJunkin, K.; Marchio, A.; Battiston, C.; Terris, B.; Mazzaferro, V.; Lowe, S.W.; Croce, C.M.; Dejean, A. miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Grabocka, E. Stress Granules in Cancer. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Gao, X.; Ge, L.; Shao, J.; Su, C.; Zhao, H.; Saarikettu, J.; Yao, X.; Yao, Z.; Silvennoinen, O.; Yang, J. Tudor-SN interacts with and co-localizes with G3BP in stress granules under stress conditions. FEBS Lett. 2010, 584, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Weissbach, R.; Scadden, A.D. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA 2012, 18, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Gao, X.; Yang, W.; Zhao, Y.; Fu, X.; Cui, X.; Zhang, C.; Xin, L.; Ren, Y.; Li, L.; et al. Phosphorylation of Tudor-SN, a novel substrate of JNK, is involved in the efficient recruitment of Tudor-SN into stress granules. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhao, C.; Yao, X.; Qian, B.; Su, C.; Ren, Y.; Yao, Z.; Gao, X.; Yang, J. SND1 acts as an anti-apoptotic factor via regulating the expression of lncRNA UCA1 in hepatocellular carcinoma. RNA Biol. 2018, 15, 1364–1375. [Google Scholar] [CrossRef] [Green Version]
- Yao, F.; Wang, Q.; Wu, Q. The prognostic value and mechanisms of lncRNA UCA1 in human cancer. Cancer Manag. Res. 2019, 11, 7685–7696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arcos, I.; Rueda, Y.; Gonzalez-Kother, P.; Palacios, L.; Ochoa, B.; Fresnedo, O. Association of SND1 protein to low density lipid droplets in liver steatosis. J. Physiol. Biochem. 2010, 66, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Palacios, L.; Ochoa, B.; Gomez-Lechon, M.J.; Castell, J.V.; Fresnedo, O. Overexpression of SND p102, a rat homologue of p100 coactivator, promotes the secretion of lipoprotein phospholipids in primary hepatocytes. Biochim. Biophys. Acta 2006, 1761, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Imaz, H.; Chico, Y.; Rueda, Y.; Fresnedo, O. Channeling of newly synthesized fatty acids to cholesterol esterification limits triglyceride synthesis in SND1-overexpressing hepatoma cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 137–146. [Google Scholar] [CrossRef]
- Navarro-Imaz, H.; Ochoa, B.; Garcia-Arcos, I.; Martinez, M.J.; Chico, Y.; Fresnedo, O.; Rueda, Y. Molecular and cellular insights into the role of SND1 in lipid metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158589. [Google Scholar] [CrossRef]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Palte, R.L.; Schneider, S.E.; Altman, M.D.; Hayes, R.P.; Kawamura, S.; Lacey, B.M.; Mansueto, M.S.; Reutershan, M.; Siliphaivanh, P.; Sondey, C.; et al. Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5). ACS Med. Chem. Lett 2020, 11, 1688–1693. [Google Scholar] [CrossRef]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.P.; Montes de Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 9711. [Google Scholar] [CrossRef]
- Metz, P.J.; Ching, K.A.; Xie, T.; Delgado Cuenca, P.; Niessen, S.; Tatlock, J.H.; Jensen-Pergakes, K.; Murray, B.W. Symmetric Arginine Dimethylation Is Selectively Required for mRNA Splicing and the Initiation of Type I and Type III Interferon Signaling. Cell Rep. 2020, 30, 1935–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, K.J.; Zitzer, N.C.; Gao, Y.; Choe, H.K.; Sell, N.E.; Neidemire-Colley, L.; Ignaci, A.; Kale, C.; Devine, R.D.; Abad, M.G.; et al. PRMT5 regulates T cell interferon response and is a target for acute graft-versus-host disease. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Wang, M.; Zhang, Y.W.; Tong, S.; Leal, R.A.; Shetty, R.; Vaddi, K.; Luengo, J.I. Discovery of Potent and Selective Covalent Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617. [Google Scholar] [CrossRef]
- Barczak, W.; Jin, L.; Carr, S.M.; Munro, S.; Ward, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. PRMT5 promotes cancer cell migration and invasion through the E2F pathway. Cell Death Dis. 2020, 11, 572. [Google Scholar] [CrossRef]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, L.; Villarreal, O.D.; He, W.; Su, D.; Bedford, E.; Moh, P.; Shen, J.; Shi, X.; Bedford, M.T.; et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 2019, 47, 5038–5048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, A.E.; Moradei, O.; Jacques, S.L.; Rioux, N.; Boriack-Sjodin, A.P.; Allain, C.; Scott, M.P.; Jin, L.; Raimondi, A.; Handler, J.L.; et al. Identification of a CARM1 Inhibitor with Potent In Vitro and In Vivo Activity in Preclinical Models of Multiple Myeloma. Sci. Rep. 2017, 7, 17993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The PRMTs, the marks they deposit, and the effectors of those marks. PRMT5 and PRMT9 deposit the SDMA mark, in “green”. PRMT1-4, 6 and 8 all deposit the ADMA, in “blue”. Methyl groups are highlighted in “red”. The effectors, or readers, of the methyl marks are Tudor domain-containing proteins.
Figure 1.
The PRMTs, the marks they deposit, and the effectors of those marks. PRMT5 and PRMT9 deposit the SDMA mark, in “green”. PRMT1-4, 6 and 8 all deposit the ADMA, in “blue”. Methyl groups are highlighted in “red”. The effectors, or readers, of the methyl marks are Tudor domain-containing proteins.

Figure 2.
PRMT5 and SND1 structural domains. PRMT5 contains three distinct regions. A TIM barrel, which contains eight consecutive alpha helices, followed by a Rossmann fold, which contains the amino acid residues that bind S-adenosylmethionine. The C-terminus of PRMT5 contains a beta barrel in which the dimerization domain has been mapped. In hetero-octamerization complexing with MEP50, PRMT5 dimerizes head to toe with other PRMT5 proteins. SND1 contains four intact SN-like domains capable of binding nucleic acids and have nuclease activity. A fifth truncated SN-like domain is split by the Tudor.
Figure 2.
PRMT5 and SND1 structural domains. PRMT5 contains three distinct regions. A TIM barrel, which contains eight consecutive alpha helices, followed by a Rossmann fold, which contains the amino acid residues that bind S-adenosylmethionine. The C-terminus of PRMT5 contains a beta barrel in which the dimerization domain has been mapped. In hetero-octamerization complexing with MEP50, PRMT5 dimerizes head to toe with other PRMT5 proteins. SND1 contains four intact SN-like domains capable of binding nucleic acids and have nuclease activity. A fifth truncated SN-like domain is split by the Tudor.

Figure 3.
SND1 RNA and protein expression. (A) SND1 RNA expression in different human tissues. Data was obtained from PRJEB4337 (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/bioproject/PRJEB4337/). The expression values of SND1 in 24 human tissues were obtained from RNA-seq RPKM (Reads Per Kilobase per Million mapped reads) values and analyzed by Graphpad. (B) SND1 protein expression in different cell lines and mice tissues. MEF were generated from E13.5 mouse embryo following a standard 3T3-MEF generation protocol. MEF cells and other cell lines were lysed. Total cell lysates were analyzed by Western blotting. Different tissues from 8-week-old FVB mice were homogenized and lysed, total tissue lysates were analyzed by Western blotting. The antibodies used were anti-SND1 (Bethyl, #A302-883A) and anti-ACTIN (Sigma, #A1978).
Figure 3.
SND1 RNA and protein expression. (A) SND1 RNA expression in different human tissues. Data was obtained from PRJEB4337 (https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/bioproject/PRJEB4337/). The expression values of SND1 in 24 human tissues were obtained from RNA-seq RPKM (Reads Per Kilobase per Million mapped reads) values and analyzed by Graphpad. (B) SND1 protein expression in different cell lines and mice tissues. MEF were generated from E13.5 mouse embryo following a standard 3T3-MEF generation protocol. MEF cells and other cell lines were lysed. Total cell lysates were analyzed by Western blotting. Different tissues from 8-week-old FVB mice were homogenized and lysed, total tissue lysates were analyzed by Western blotting. The antibodies used were anti-SND1 (Bethyl, #A302-883A) and anti-ACTIN (Sigma, #A1978).


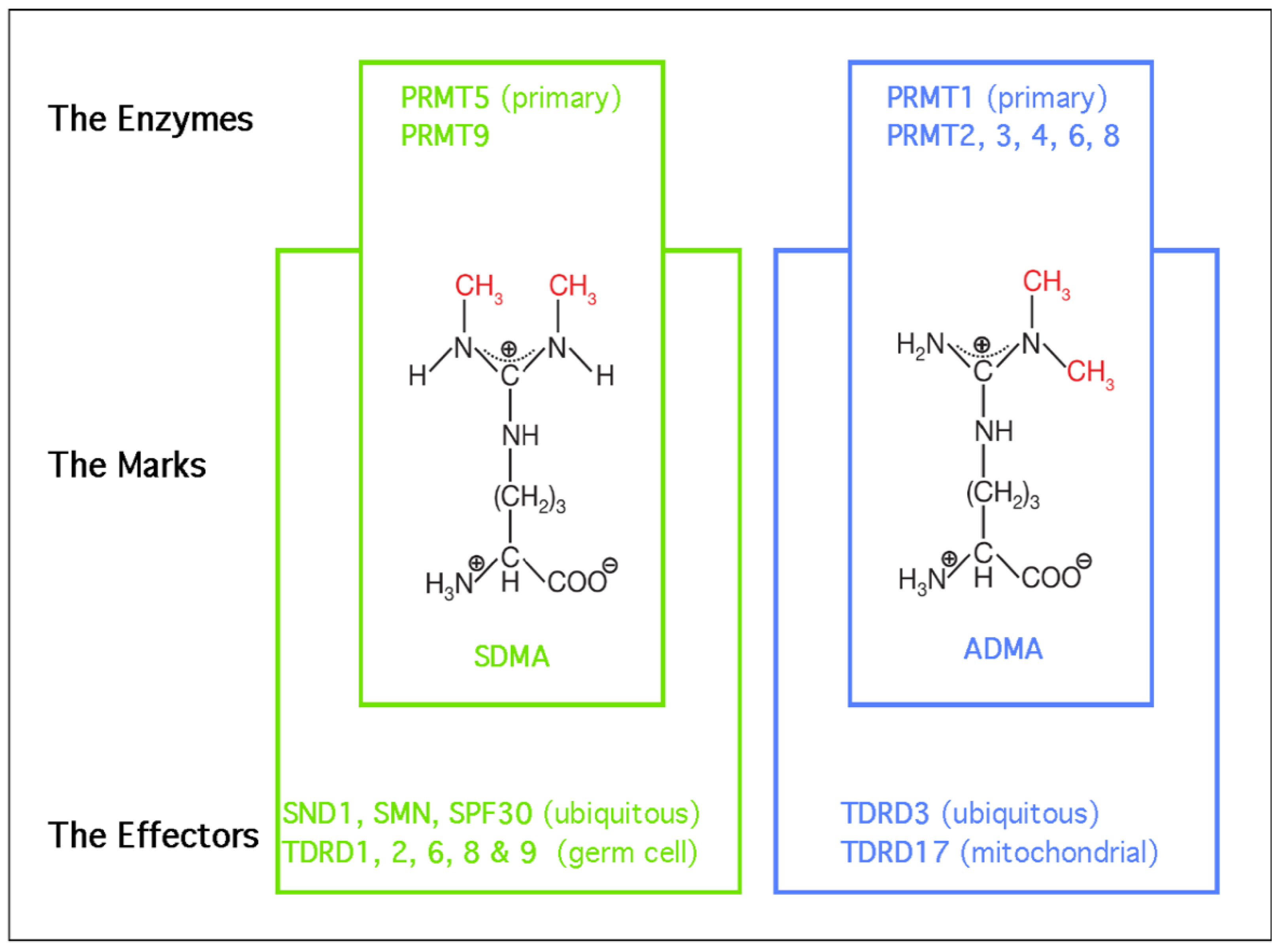{kind=link}
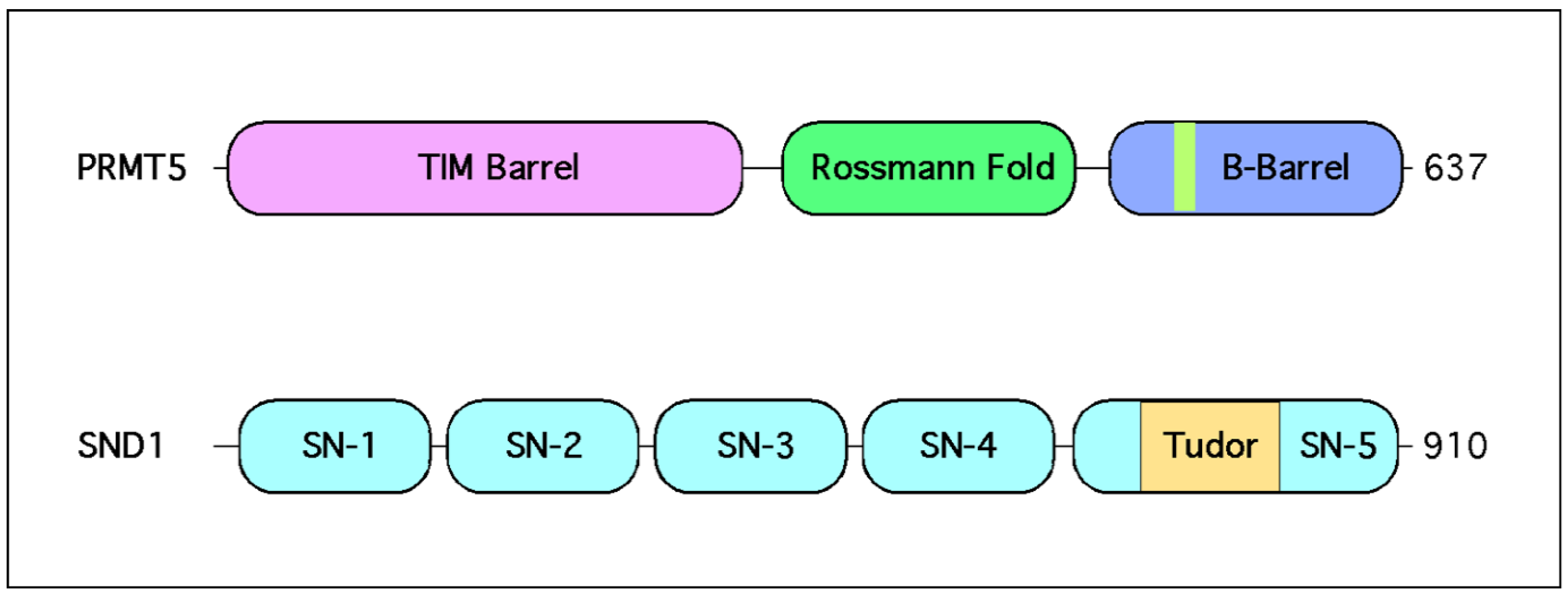{kind=link}
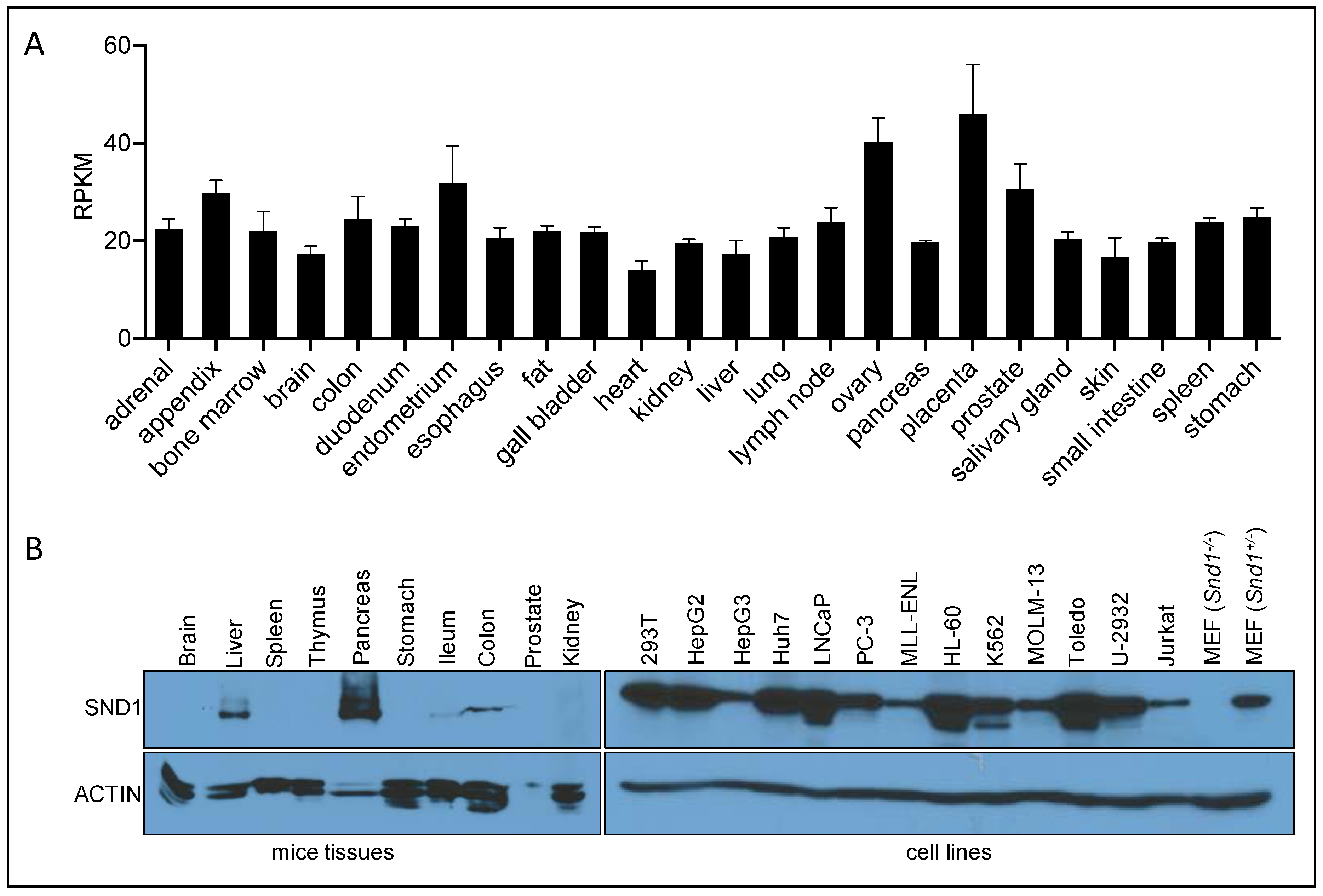{kind=link}
Table 1.
PRMT5 inhibitor and their different mechanisms of action.
| Company | Compound | Trials | MOA | Ref. or Patent |
|---|---|---|---|---|
| GSK/Epizyme | GSK3326595 | Ph 1 NCT02783300Ph 2 NCT03614728 | b | [164] |
| Pfizer | PF-06939999 | Ph 1 NCT03854227 | a | [165] |
| Janssen | JNJ64619178 | Ph 1 NCT03573310 | c | AACR |
| Prelude | PRT543 | Ph 1 NCT03886831 | a | [166] |
| Prelude | PRT811 | Ph 1 NCT04089449 | a | [166] |
| Prelude | C449 | Pre-clinical | f | [167] |
| Eli Lilly | LLY-283 | Pre-clinical | a | [168] |
| Argonaut | T1-44 | Pre-clinical | b | [169] |
| Jian Jin | MS4322 | Pre-clinical | d | [159] |
| Merck | Comp 1A | Pre-clinical | e | [160] |
| Aurigene | AU-14755 | Pre-clinical | b | WO2019-180628 |
| Angex | Not named | Pre-clinical | a | WO2019-112719 |
| Jubilant | JPRMT5i | Pre-clinical | b | WO2019-102494 |
Mechanism of action (MOA): (a) SAM competitive; (b) SAM cooperative and peptide substrate competitive; (c) SAM and peptide substrate competitive; (d) PROTAC degrader; (e) allosteric modulator; and (f) covalent inhibitor. The “WO2019” numbers refer to patent submissions.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wright, T.; Wang, Y.; Bedford, M.T. The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma. Epigenomes 2021, 5, 2. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010002
AMA Style
Wright T, Wang Y, Bedford MT. The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma. Epigenomes. 2021; 5(1):2. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010002
Chicago/Turabian StyleWright, Tanner, Yalong Wang, and Mark T. Bedford. 2021. "The Role of the PRMT5–SND1 Axis in Hepatocellular Carcinoma" Epigenomes 5, no. 1: 2. https://0-doi-org.brum.beds.ac.uk/10.3390/epigenomes5010002