Comparative Transcriptomics Analysis of Phytohormone-Related Genes and Alternative Splicing Events Related to Witches’ Broom in Paulownia


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Total RNAs Extraction and cDNA Library Construction
2.3. Obtaining Clean Reads, and Transcript Annotation
2.4. Differentially Expressed Genes among the Nine Libraries
2.5. Quantitative Real-time PCR (qRT-PCR) Analysis of DEGs
3. Results
3.1. Transcriptome Sequencing
3.2. Functional Annotation of DEGs
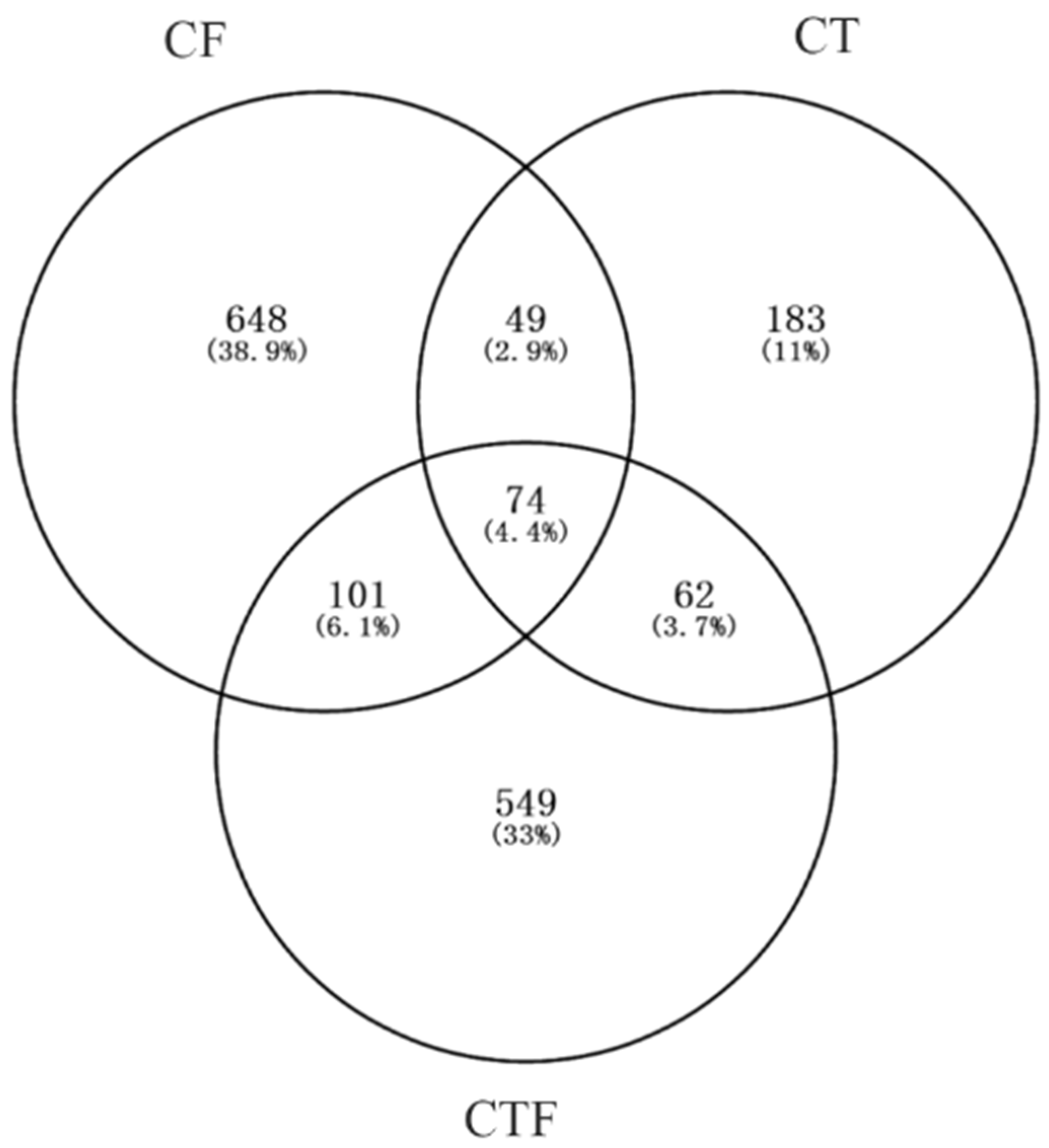
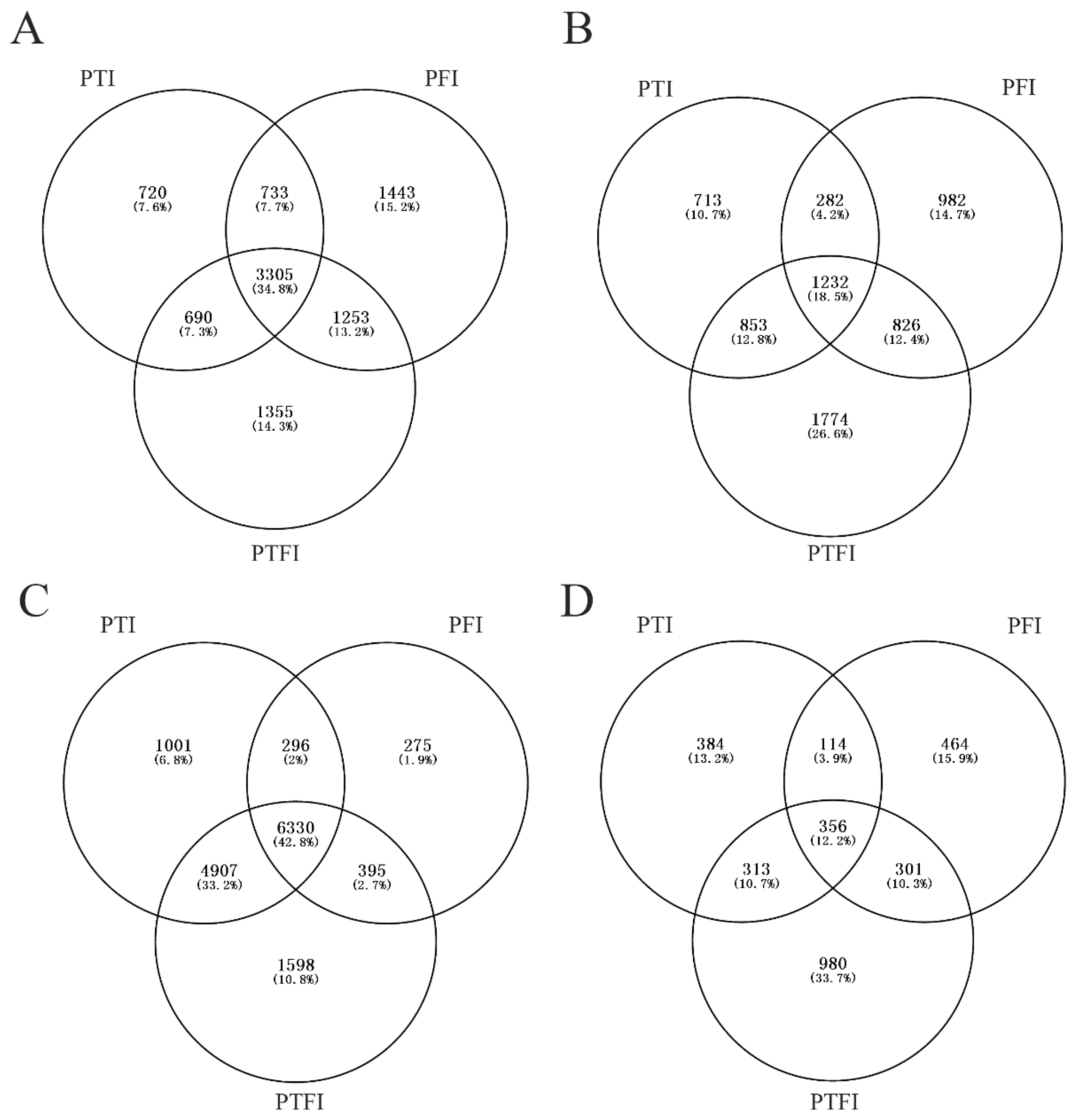
3.3. Analysis of the Common DEGs
3.4. Alternative Splicing Analysis between Healthy and PaWB-Infected Plants
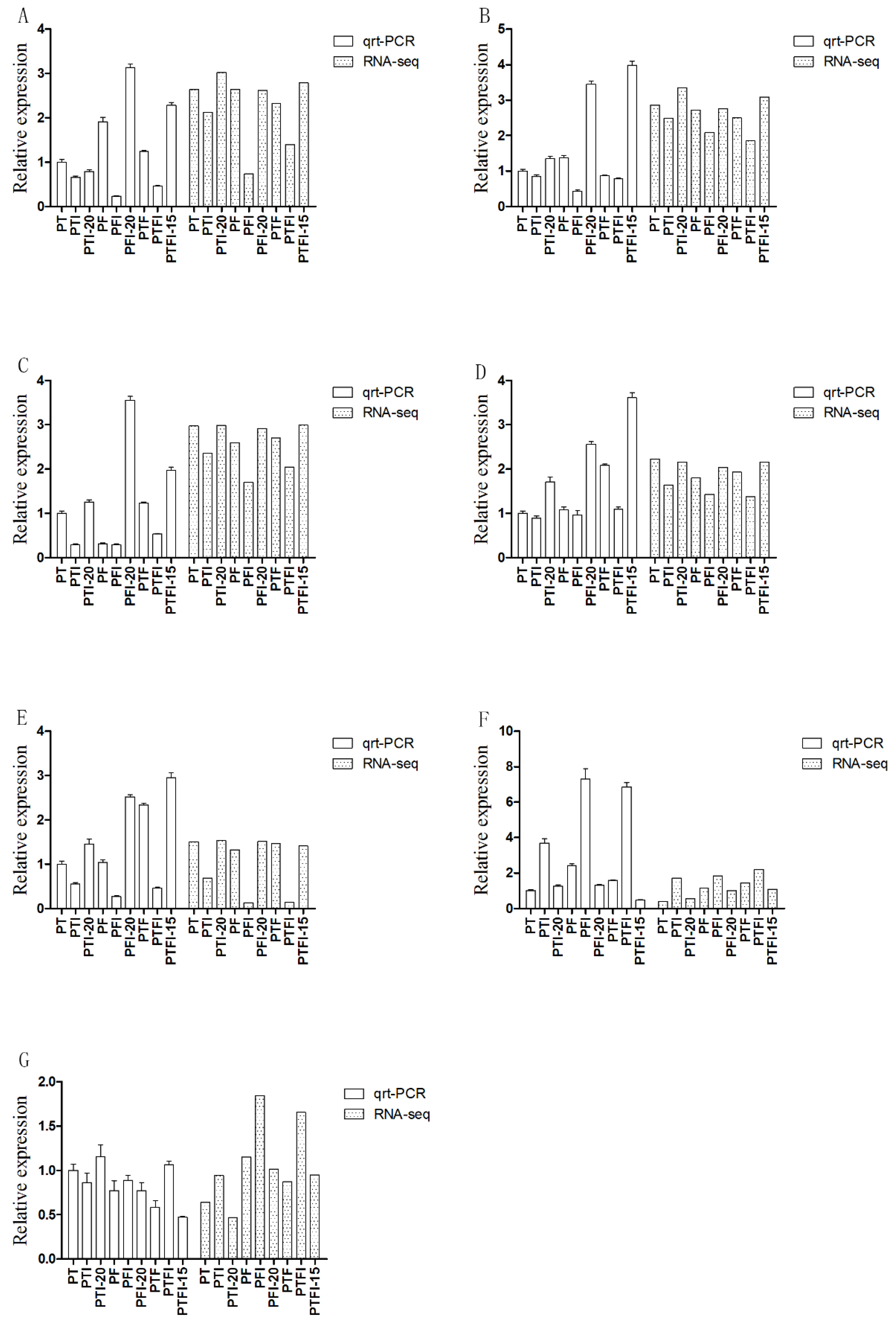
3.5. Verification of Candidate Genes by qRT-PCR
4. Discussion
4.1. Differences among the P. tomentosa × P. fortunei and Parental Responses to Phytoplasma
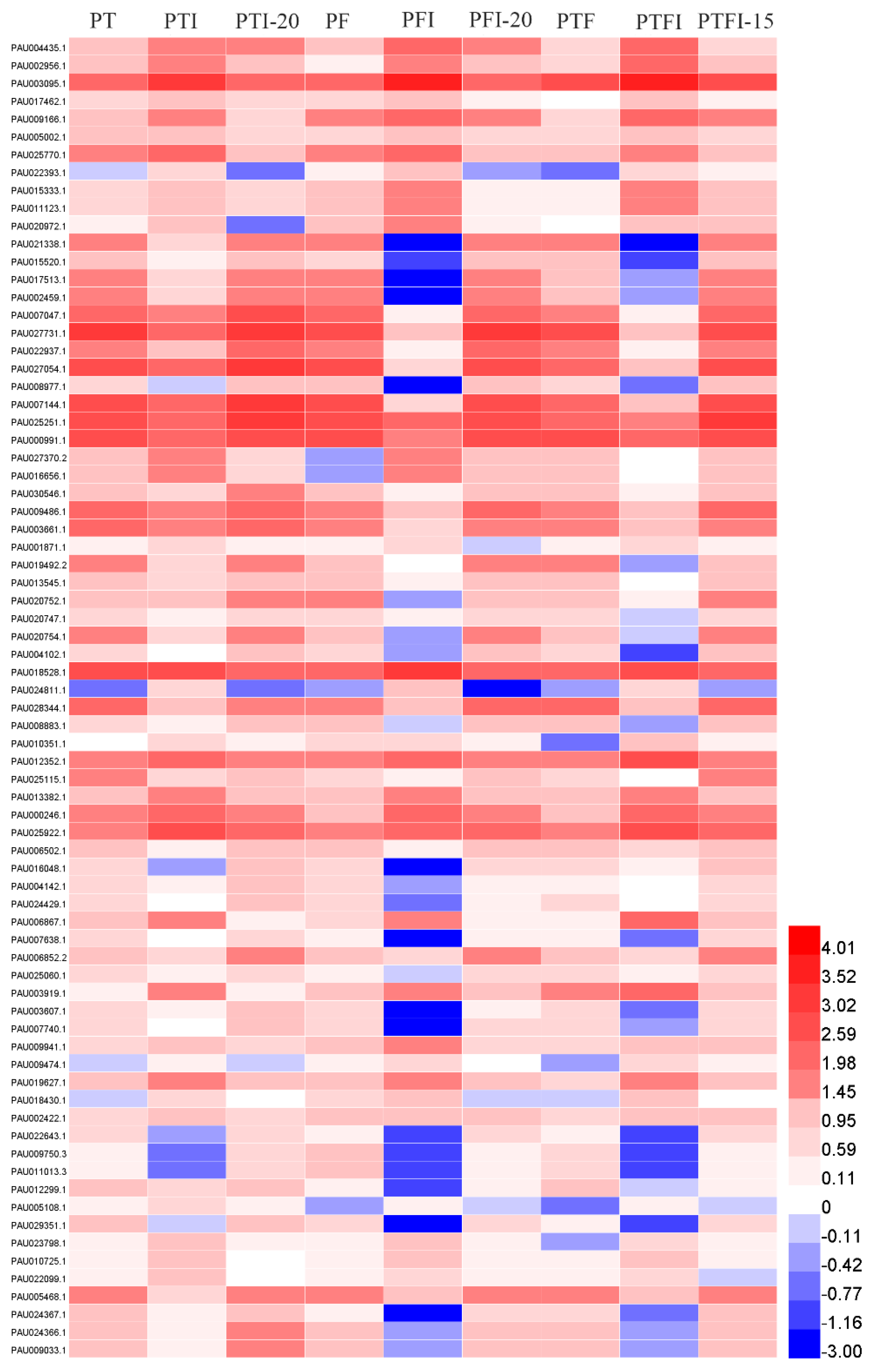
4.2. DEGs Related to Phytohormones
4.3. Relationship between AS and the Paulownia Plants Responses to PaWB
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kalaycioglu, H.; Deniz, I.; Hiziroglu, S. Some of the properties of particleboard made from paulownia. J. Wood Sci. 2005, 51, 410–414. [Google Scholar] [CrossRef]
- Wang, J. Genetic and serological analyses of elongation factor EF-Tu of paulownia witches’-broom phytoplasma (16SrI-D). Plant Pathol. 2010, 59, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.; Xu, E.; Deng, M.; Zhao, Z.; Niu, S. Phenylpropanoid metabolism, hormone biosynthesis and signal transduction-related genes play crucial roles in the resistance of Paulownia fortunei to paulownia witches’ broom phytoplasma infection. Genes Genom. 2015, 37, 913–929. [Google Scholar] [CrossRef]
- HY, J. ‘Candidatus Phytoplasma ziziphi’, a novel phytoplasma taxon associated with jujube witches’-broom disease. Int. J. Syst. Evol. Microbiol. 2002, 52, 1543–1549. [Google Scholar]
- Kawakita, H.; Saiki, T.; Wei, W.; Mitsuhashi, W.; Watanabe, K.; Sato, M. Identification of mulberry dwarf phytoplasmas in the genital organs and eggs of leafhopper hishimonoides sellatiformis. Phytopathology 2000, 90, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Tao, Z.; Li, H.F.; Fan, Z.F.; Yong, L.; Chungen, P.; Tian, G.Z. Molecular characterisation of two plasmids from paulownia witches’-broom phytoplasma and detection of a plasmid-encoded protein in infected plants. Eur. J. Plant Pathol. 2009, 123, 321–330. [Google Scholar] [CrossRef]
- Wang, Q.C.; Jpt, V. Efficient elimination of sweetpotato little leaf phytoplasma from sweetpotato by cryotherapy of shoot tips. Plant Pathol. 2008, 57, 338–347. [Google Scholar] [CrossRef] [Green Version]
- Bertaccini, A.; Duduk, B. Phytoplasma and phytoplasma diseases: A review of recent research. Phytopathol. Mediterr. 2009, 48, 355–378. [Google Scholar]
- Weintraub, P.; Beanland, L. Insect vectors of phytoplasmas. Annu. Rev. Entomol. 2006, 51, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Teranaka, M.; Yora, K.; Asuyama, H. Mycoplasma-or PLT group-like microorganisms found in the phloem elements of plants infected with mulberry dwarf, potato witches’ broom, aster yellows, or Paulownia witches’ broom. Jpn. J. Phytopathol. 1967, 33, 259–266. [Google Scholar] [CrossRef]
- Sugio, A.; Maclean, A.M.; Kingdom, H.N.; Grieve, V.M.; Manimekalai, R.; Hogenhout, S.A. Diverse targets of phytoplasma effectors: From plant development to defense against insects. Annu. Rev. Phytopathol. 2011, 49, 175. [Google Scholar] [CrossRef] [PubMed]
- Maclean, A.M. Phytoplasma effector SAP54 induces indeterminate leaf-like flower development in Arabidopsis plants. Plant Physiol. 2011, 157, 831. [Google Scholar] [CrossRef] [PubMed]
- Nault, L.R. Evolution of an insect pest: Maize and the corn leafhopper, a case study. Maydica 1990, 35, 165–175. [Google Scholar]
- Bai, L.J.; Ye, Y.T.; Chen, Q.; Tang, H.R. The complete chloroplast genome sequence of the white strawberry Fragaria pentaphylla. Conserv. Genet. Resour. 2017, 9, 1–3. [Google Scholar] [CrossRef]
- Jung, H.Y.; Miyata, S.; Oshima, K. First complete nucleotide sequence and heterologous gene organization of the two rRNA operons in the phytoplasma genome. DNA Cell Biol. 2003, 22, 209–215. [Google Scholar] [CrossRef] [PubMed]
- JBayliss, K.L.; Saqib, M.; Dell, B.; Jones, M.G.K.; Hardy, G.S.J. First record of ‘Candidatus phytoplasma australiense’ in Paulownia trees. Australas. Plant Pathol. 2005, 34, 123–124. [Google Scholar] [CrossRef]
- Hogenhout, S.A.; Music, M.Š.; Weintraub, P.G. Phytoplasma genomics, from sequencing to comparative and functional genomics—What have we learnt? In Phytoplasmas: Genomes, Plant Hosts and Vectors; CAB International: Wallingford, UK, 2009; Volume 41, pp. 19–36. [Google Scholar]
- Sugio, A.; Hogenhout, S.A. The genome biology of phytoplasma: Modulators of plants and insects. Curr. Opin. Microbiol. 2012, 15, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Cao, Y.; Deng, M.; Zhai, X.; Zhao, Z.; Niu, S.; Ren, Y. Identification and dynamic expression profiling of microRNAs and target genes of Paulownia tomentosa in response to Paulownia witches’ broom disease. Acta Physiol. Plant. 2017, 39, 28. [Google Scholar] [CrossRef]
- Liu, R.; Dong, Y.; Fan, G.; Zhao, Z.; Deng, M.; Cao, X.; Niu, S. Discovery of genes related to witches broom disease in Paulownia tomentosa × Paulownia fortunei by a De Novo assembled transcriptome. PLoS ONE 2013, 8, 80238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Quantitative proteome-level analysis of paulownia witches’ broom disease with methyl methane sulfonate assistance reveals diverse metabolic changes during the infection and recovery processes. PeerJ 2017, 5, e3495. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Fan, G.; Deng, M.; Zhao, Z.; Dong, Y. Identification of genes related to paulownia witches’ broom by AFLP and MSAP. Int. J. Mol. Sci. 2014, 15, 14669–14683. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Cao, X.; Fan, G. Growth of paulownia witches’ broom seedlings treated with methylmethane sulphonate and SSR analysis. Sci. Silvae Sin. 2010, 46, 176–181. [Google Scholar]
- Fan, G.; Cao, X.; Zhao, Z.; Deng, M. Transcriptome analysis of the genes related to the morphological changes of Paulownia tomentosa plantlets infected with phytoplasma. Acta Physiol. Plant. 2015, 37, 1–12. [Google Scholar] [CrossRef]
- Gu, S.; Fang, L.; Xu, X. Using SOAPaligner for short reads alignment. Curr. Protoc. Bioinform. 2013, 44, 1–17. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Götz, S. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Dalpiaz, D. Statistical Methods for Modeling RNA-Seq Short-Read Data. Ph.D. Thesis, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 2014. [Google Scholar]
- Chen, C. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Brian, A.W.; Kenneth, M.C.; Lorian, S.; Barbara, W. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Mastrangelo, A.M.; Marone, D.; Laidò, G.; De Leonardis, A.M.; De Vita, P. Alternative splicing: Enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 2012, 185–186, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Brendel, V. Genomewide comparative analysis of alternative splicing in Plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Xu, L.; Liang, D.; Wang, Y.; Zhang, W.; Zhu, X.; Zhu, Y.; Jiang, H.; Tang, M.; Liu, L. Comparative transcriptomics uncovers alternative splicing and molecular marker development in radish (Raphanus sativus L.). BMC Genom. 2017, 18, 505. [Google Scholar] [CrossRef] [PubMed]
- Bartish, I.V.; Jeppsson, N.; Nybom, H.; Swenson, U. Phylogeny of Hippophae (Elaeagnaceae) inferred from parsimony analysis of chloroplast DNA and morphology. Syst. Bot. 2007, 27, 41–54. [Google Scholar]
- Fahad, S.; Hussain, S.; Matloob, A.; Khan, F.A.; Khaliq, A.; Saud, S.; Hassan, S.; Shan, D.; Khan, F.; Ullah, N.; et al. Phytohormones and plant responses to salinity stress: A review. Plant Growth Regul. 2015, 75, 391–404. [Google Scholar] [CrossRef]
- Ahmad, P.; Bhardwaj, R.; Tuteja, N. Plant Signaling under Abiotic Stress Environment. In Environmental Adaptations and Stress Tolerance of Plants in the Era of Climate Change, 2012; Springer: New York, NY, USA, 2012; pp. 297–323. [Google Scholar]
- Browse, J.; Howe, G.A. New weapons and a rapid response against insect attack. Plant Physiol. 2008, 146, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Farmer, E.E.; Ryan, C.A. Interplant communication: Airborne methyl jasmonate induces synthesis of proteinase inhibitors in plant leaves. Proc. Natl. Acad. Sci. USA 1990, 87, 7713–7716. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Dong, Y.; Deng, M.; Zhao, Z.; Niu, S.; Xu, E. Plant-pathogen interaction, circadian rhythm, and hormone-related gene expression provide indicators of phytoplasma infection in Paulownia fortunei. Int. J. Mol. Sci. 2014, 15, 23141–23162. [Google Scholar] [CrossRef] [PubMed]
- Achard, P.; Genschik, P. Releasing the brakes of plant growth: How GAs shutdown DELLA proteins. J. Exp. Bot. 2009, 60, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.R.; Jones, J.D.G.; Hines, P.J.; Zahn, L.M. Hormone (dis)harmony moulds plant health and disease. Science 2009, 324, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Jin, X. Recent advances of MYB transcription factors involved in the regulation of flavonoid biosynthesis. Mol. Plant Breed. 2015, 13, 689–696. [Google Scholar]
- Ye, X.; Wang, H.; Chen, P.; Fu, B.; Zhang, M.; Li, J.; Zheng, X.; Tan, B.; Feng, J. Combination of iTRAQ proteomics and RNA-seq transcriptomics reveals multiple levels of regulation in phytoplasma-infected Ziziphus jujuba mill. Hortic. Res. 2017, 4, 17080. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jiang, W.; Dai, Y. A maize phytochrome-interacting factor 3 improves drought and salt stress tolerance in rice. Plant Mol. Biol. 2015, 87, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Carvalhais, L.C.; Schenk, P.M.; Dennis, P.G. Effects of jasmonic acid signalling on the wheat microbiome differ between body sites. Sci. Rep. 2017, 7, 41766. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adapter (%) | Low Quality (%) | |
|---|---|---|
| PF | 0 | 7.59 |
| PFI | 0 | 7.58 |
| PFI 20 | 0 | 8.41 |
| PT | 1.2 | 2.74 |
| PTI | 1.19 | 2.21 |
| PTI 20 | 0.7 | 4 |
| PTF | 1.55 | 3.22 |
| PTFI | 1.02 | 3.41 |
| PTFI 15 | 0.99 | 3.43 |
| qRT-PCR Gene ID | Species | Relative Expression | FPKM | NR |
|---|---|---|---|---|
| PAU007144.1 | PF | 1.906456 | 441.51 | Phylloplanin |
| PFI | 0.227004 | 5.45 | ||
| PFI-20 | 3.138247 | 420.54 | ||
| PT | 1 | 436.08 | ||
| PTI | 0.656896 | 135.57 | ||
| PTI-20 | 0.790961 | 1049.23 | ||
| PTF | 1.239057 | 213.39 | ||
| PTFI | 0.462863 | 24.65 | ||
| PTFI-15 | 2.287262 | 617.72 | ||
| PAU025251.1 | PF | 1.373163 | 528.61 | Pollen Ole e 1 allergen and extensin family protein |
| PFI | 0.431665 | 121.54 | ||
| PFI-20 | 3.452324 | 576.49 | ||
| PT | 1 | 733.94 | ||
| PTI | 0.851032 | 310.94 | ||
| PTI-20 | 1.350958 | 2268.65 | ||
| PTF | 0.875457 | 319.26 | ||
| PTFI | 0.786235 | 71.28 | ||
| PTFI-15 | 3.987706 | 1231.79 | ||
| PAU000991.1 | PF | 0.312058 | 394.01 | Lipid transfer protein 2 |
| PFI | 0.289438 | 50.47 | ||
| PFI-20 | 3.550041 | 826.48 | ||
| PT | 1 | 933.68 | ||
| PTI | 0.290733 | 228.72 | ||
| PTI-20 | 1.261053 | 972.13 | ||
| PTF | 1.238529 | 505.79 | ||
| PTFI | 0.533642 | 110.99 | ||
| PTFI-15 | 1.977384 | 979.78 | ||
| PAU009486.1 | PF | 1.084106 | 63.11 | Art v 3 allergen precursor |
| PFI | 0.968481 | 26.91 | ||
| PFI-20 | 2.564913 | 109.42 | ||
| PT | 1 | 170.46 | ||
| PTI | 0.890637 | 44.06 | ||
| PTI-20 | 1.710442 | 144.07 | ||
| PTF | 2.080034 | 85.19 | ||
| PTFI | 1.096989 | 24.08 | ||
| PTFI-15 | 3.611348 | 143.97 | ||
| PAU019492.2 | PF | 1.046466 | 21.08 | Putative prolyl 4-hydroxylase-like |
| PFI | 0.267094 | 1.03 | ||
| PFI-20 | 2.518455 | 32.82 | ||
| PT | 1 | 31.66 | ||
| PTI | 0.557728 | 4.88 | ||
| PTI-20 | 1.453677 | 33.94 | ||
| PTF | 2.3389 | 29.38 | ||
| PTFI | 0.467665 | 0.73 | ||
| PTFI-15 | 2.958268 | 26.26 | ||
| PAU003919.1 | PF | 2.409558 | 14.15 | RING finger and CHY zinc finger domain-containing protein 1 |
| PFI | 7.324452 | 70.03 | ||
| PFI-20 | 1.322104 | 10.39 | ||
| PT | 1 | 2.55 | ||
| PTI | 3.695298 | 51.49 | ||
| PTI-20 | 1.270922 | 3.65 | ||
| PTF | 1.575629 | 28.03 | ||
| PTFI | 6.850931 | 159.97 | ||
| PTFI-15 | 0.477221 | 12.32 | ||
| PAU010351.1 | PF | 1 | 4.37 | Allene oxide synthase, chloroplastic |
| PFI | 1.148782 | 8.77 | ||
| PFI-20 | 1.000575 | 2.95 | ||
| PT | 1.294857 | 1.07 | ||
| PTI | 1.115748 | 8.02 | ||
| PTI-20 | 1.501578 | 2.38 | ||
| PTF | 0.757176 | 7.46 | ||
| PTFI | 1.377922 | 45.4 | ||
| PTFI-15 | 0.610686 | 8.88 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Zhang, H.; Fan, G.; Zhai, X.; Wang, Z.; Cao, Y. Comparative Transcriptomics Analysis of Phytohormone-Related Genes and Alternative Splicing Events Related to Witches’ Broom in Paulownia. Forests 2018, 9, 318. https://0-doi-org.brum.beds.ac.uk/10.3390/f9060318
Dong Y, Zhang H, Fan G, Zhai X, Wang Z, Cao Y. Comparative Transcriptomics Analysis of Phytohormone-Related Genes and Alternative Splicing Events Related to Witches’ Broom in Paulownia. Forests. 2018; 9(6):318. https://0-doi-org.brum.beds.ac.uk/10.3390/f9060318
Chicago/Turabian StyleDong, Yanpeng, Huiyuan Zhang, Guoqiang Fan, Xiaoqiao Zhai, Zhe Wang, and Yabing Cao. 2018. "Comparative Transcriptomics Analysis of Phytohormone-Related Genes and Alternative Splicing Events Related to Witches’ Broom in Paulownia" Forests 9, no. 6: 318. https://0-doi-org.brum.beds.ac.uk/10.3390/f9060318