Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections



1
Department of Molecular Virology & Microbiology, Baylor College of Medicine, Houston, TX 77030, USA
2
Department of Regenerative Medicine & Cell Biology, Medical University of South Carolina, Charleston, SC 29425, USA
*
Author to whom correspondence should be addressed.
Gastroenterol. Insights 2021, 12(1), 41-55; https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12010005
Submission received: 2 December 2020
/
Revised: 28 December 2020
/
Accepted: 24 January 2021
/
Published: 1 February 2021
(This article belongs to the Section Gastrointestinal Disease)
{kind=link}
Abstract
:The lumen of the gastrointestinal tract harbors a diverse community of microbes, fungi, archaea, and viruses. In addition to occupying the same enteric niche, recent evidence suggests that microbes and viruses can act synergistically and, in some cases, promote disease. In this review, we focus on the disease-promoting interactions of the gut microbiota and rotavirus, norovirus, poliovirus, reovirus, and astrovirus. Microbes and microbial compounds can directly interact with viruses, promote viral fitness, alter the glycan structure of viral adhesion sites, and influence the immune system, among other mechanisms. These interactions can directly and indirectly affect viral infection. By focusing on microbe–virus interplay, we hope to identify potential strategies for targeting offending microbes and minimizing viral infection.
Keywords:
enteric viruses; rotavirus; norovirus; poliovirus; reovirus; astrovirus; microbes; microbiota1. Introduction
Enteric viruses enter the host via the fecal-oral route. In the gastrointestinal tract, viruses encounter a dynamic community of resident microorganisms, known as the gut microbiota. Intestinal microbes have three potential interactions with enteric viruses: (1) microbes may promote and aid viral infection, (2) microbes can inhibit viral infection, or (3) microbes have no effect on viral infection. Recent works by several groups have identified that select intestinal microbes can promote replication, transmission, and pathogenesis of several enteric viruses [1,2,3,4,5,6,7,8,9,10,11]. Microbes that augment viral infection are speculated to act through both direct and indirect mechanisms [12]. These mechanisms include the following: (1) aiding the delivery of virus to host cells via direct attachment, (2) removing decoy host glycans, (3) enhancing virion stability, or (4) dampening innate immune responses (Figure 1). The gut microbiota has been implicated in the pathogenesis of rotavirus, norovirus, poliovirus, reovirus, and astrovirus. This review focuses on bacteria that promote viral infections and attempts to cover known direct and indirect mechanisms by which microbes influence enteric viral infections.
2. Rotavirus (Family: Reoviridae, Genus: Rotavirus)
Rotavirus is a double-stranded, non-enveloped RNA virus that infects small intestinal epithelial cells. Currently, rotavirus is a leading cause of diarrheal diseases in children, with an estimated 258 million cases and 128,500 deaths per year [13]. To define the role of the gut microbiota in rotavirus infection, Uchiyama et al. treated adult C57BL/6 mice with the antibiotics ampicillin and neomycin, which reduced intestinal bacterial loads by 99% [8]. Infection of these microbiota-depleted mice with mouse EC rotavirus resulted in a 1-day delay in the appearance of fecal rotavirus antigen, a ~40% decrease in total rotavirus shedding by ELISA (enzyme-linked immunosorbent assay), and a 10-fold reduction in rotavirus genomes by RT-qPCR (reverse transcription polymerase chain reaction) [8]. Analysis of the ratio of positive sense to negative sense rotavirus strands, which reflects the extent of active rotavirus replication, revealed no differences in control or antibiotic treated mice, indicating that rotavirus replication was not affected by the microbiota. These findings suggest that microbiota ablation reduced viral entry rather than rotavirus replication. Consistent with these findings, germ-free C57BL/6 mice also exhibited a 1-day delay in initial appearance of infection [8]. In neonatal C57BL/6 mice, antibiotic treatment also resulted in lower daily rates of diarrhea and a ~34% reduction in total diarrhea incidence [8]. These findings indicate that gut microbiota influences rotavirus infectivity.
Rotavirus attaches to host cells via specific glycans [14,15,16], particularly those terminated with sialic acid and galactose residues [17]. The structure of these cell surface glycans is mirrored in mucins, which are secreted by goblet cells and overlay the intestinal epithelium. Similar to adhesion to cell surface glycans, rotavirus also adheres to mucin glycans [15,16,18,19]. As a result, mucus has been postulated to serve as a rotavirus decoy to prevent or delay attachment to host epithelial cells. In a neonatal Balb/c model, rhesus rotavirus infection correlated with mucin MUC2 expulsion from goblet cells 1-day post-infection [9]. This finding was also observed by Boshuizen and colleagues, who found that EDIM rotavirus in BALB/c mice induced loss of mucin-filled goblet cells [20]. Using mucin-producing cell lines and human jejunal enteroids (also known as organoids), rhesus rotavirus infection was shown to directly stimulate mucus release [9]. Consistent with the potential for mucus to act as a decoy, the addition of purified germ-free Muc2 to rhesus rotavirus significantly delayed infection of MA104 cells compared with rotavirus alone [9]. Work by other groups has also identified that crude and purified intestinal mucins from suckling and adult mice can inhibit rhesus rotavirus infection of MA104 cells [21]. In this study, the removal of mucus sialic acid by neuraminidase suppressed the ability of mucin to prevent rotavirus infection of MA104 [21], highlighting the importance of glycan structures to bind rotavirus. Boshuizen et al. also found that mouse colonic mucus was also able to neutralize infection by the human Wa rotavirus in Caco-2 cells [20]. These studies suggest that mucins possess molecular mimicry and can prevent virus–cell attachment.
Analysis of the gut microbiota in neonatal BALB/c mice during rhesus rotavirus infection revealed a time-dependent increase in ileal Bacteroides (phylum Bacteroidetes) and Akkermansia (phylum Verrucomicrobia) and a decrease in Lactobacillus (phylum Firmicutes) [9]. Bacteroides and Akkermansia species are known for their mucin-degrading activities and could potentially participate in rotavirus infection during the acute phase. Other groups have shown large-scale changes in Bacteroidetes in response to rotavirus infection. In neonatal gnotobiotic pigs, rotavirus infection correlated with increased Bacteroidetes levels [22,23]. Increased Bacteroidetes were observed in children with rotavirus induced gastroenteritis compared with healthy control children [24]. In another study, Zhang et al. identified three Bacteroides operational taxonomic units that could discriminate healthy children from rotavirus-infected children [25]. In addition to Bacteroides, Akkermansia has also been associated with rotavirus. Shi et al. demonstrated that an intestinal microbiota possessing high levels of Akkermansia supported mouse EC rotavirus infection in C57BL/6 mice [26]. In adults, Akkermanisa levels were found to correlate with increased susceptibility to rotavirus infection [27]. The potential for mucin-degrading microbes to influence rotavirus infection was recently demonstrated by Engevik et al. [9]. The authors found that pre-incubation of Muc2 with Bacteroides thetaiotaomicron or Akkermansia muciniphila, which degraded mucin glycans, reduced the ability of mucus to prevent rotavirus infection of MA104 cells [9]. These findings indicate that mucin-degrading microbes may modify mucin glycan structures and sialic acid content, thereby circumventing its decoy function and promoting access of rotavirus to the underlying epithelial cells. Dietary nutrients in the intestine are speculated to decrease during rotavirus infection owing to diarrhea and reduced dietary intake [28], and in the absence of luminal nutrients, mucin-degrading microbes may bloom in response to rotavirus-stimulated mucus release. Thus, mucin-degrading microbes may inadvertently enhance rotavirus infection. Based on these studies, we speculate that the release of mucus from goblet cells is a short-term defense mechanism of the host against the virus, but once the goblet cells are depleted, rotavirus can gain access to the host. The release of mucus also promotes mucin-degrading microbes, which likely then degrade any subsequently released mucus, thereby providing rotavirus with an additional advantage.
In addition to indirect changes in mucin composition, intestinal microbes may also directly influence rotavirus infection. Many enteric viruses can bind to bacterial surface polysaccharides [1] and adhesion of viruses to Gram-negative bacteria has been proposed as a mechanism to promote attachment to target cells. Rotavirus can bind to Gram-negative E. coli Nissile [29], and it is possible that rotavirus may bind to other intestinal microbes. As a result, intestinal bacteria could deliver rotavirus to intestinal cells, thereby directly promoting infection.
The immune system is also considered to be integral to rotavirus infection. Rotavirus initiates a robust adaptive immune response that resolves the infection and provides lasting protection [30]. This infection-induced immunity is the basis of the rotavirus vaccine and correlates with fecal immunoglobulin A (IgA) and serum immunoglobulin G (IgG) [30,31]. Uchiyama et al. demonstrated that antibiotic treatment (ampicillin and neomycin) of adult C57BL/6J mice for 1 week prior to inoculation and up to 11 weeks after inoculation enhanced levels of rotavirus-specific antibodies 9 weeks after inoculation, particularly serum and fecal IgA [8]. In neonatal mice, the authors also found that antibiotic treatment enhanced serum anti-rotavirus IgA production [8]. In addition to IgG and IgA, rotavirus also elicits interferon (IFN) responses, which are powerful antiviral effectors [32,33,34]. Murine rotavirus infection robustly activates type I IFN from immune cells in the intestine [35]. In separate studies, administration of antibiotics (ampicillin, gentamicin, metronidazole, neomycin, and vancomycin) to adult C57BL/6 mice impaired immune cell type I IFN responses to lymphocytic choriomeningitis virus (LCMV) infection [36]. It has been speculated that type I IFNs responses are mediated through plasmacytoid dendritic cells, which are substantially depleted in the gut in the absence of the microbiota [37]. Although no studies have linked the gut microbiota in IFN responses to rotavirus, we speculate that the microbiome may also be responsible for dampening these signalling cascades in the setting of rotavirus infection. These studies suggest that the microbiota dampens systemic and mucosal immune responses to rotavirus infection.
3. Norovirus (Family: Caliciviridae, Genus: Norovirus)
Norovirus is a positive stranded RNA virus that has become infamous for causing outbreaks of gastroenteritis in nursing homes and cruise ships [38]. Human noroviruses (HuNoVs) cause the majority of global gastroenteritis outbreaks and are a leading cause of severe childhood diarrhea [39,40]. In developing countries, HuNoVs are estimated to cause over 1 million clinic visits and 200,000 deaths in children each year [41]. Jones et al. identified that enteric microbes promote norovirus infection of B cells [3]. Using HuNov (GII.4-Sydney)-positive stool samples, the authors found that, in the presence of microbes, HuNov had enhanced infection in the BJAB human B cell line. However, genome replication was not observed when the stool sample was ultraviolet (UV)-inactivated or sterile filtered. Jones et al. also found that GII.4-Sydney HuNoV-positive stool was able to infect colon HT29 cells cultured with BJAB B cells [3]. These studies demonstrate that intestinal microbes promote norovirus infection of B cells. Interestingly, bacteria are not required for HuNoV GII.3 and GII.4 strains to infect human intestinal enteroid (HIE) cultures, also known as organoids [42,43]. Treatment of stool filtrates with polymyxin B, which reduces LPS levels, did not promote replication of HuNoV in HIEs [43]. These data suggest that bacteria are necessary for HuNoV to infect B cells, but not the intestinal epithelium.
To examine the role of the gut microbiota in norovirus infection in vivo, Baldridge et al. treated adult C57BL/6J mice for 2 weeks with broad-spectrum oral antibiotics (vancomycin, neomycin, ampicillin, and metronidazole), and then infected mice with murine norovirus (MNoV) CR6 [2], a strain that causes a persistent infection in mice. Antibiotics prevented persistent norovirus infection as measured by fecal viral shedding and decreased the levels of virus in intestinal tissues. Norovirus could still replicate in cultured cells, indicating that norovirus had decreased cell access or entry and not defects in replication [2]. To confirm the importance of bacteria in norovirus infection, Baldridge et al. performed an additional experiment where mice were treated with antibiotics, but the antibiotics were replaced with drinking water 3 days prior to norovirus infection to allow for drug clearance. In this setting, mice still exhibited an altered decreased microbiota, but antibiotics could not directly interact with MNoV CR6 strain [2]. Mice were still resistant to MNoV CR6 infection in this experiment, indicating that the gut microbiota, not direct antibiotic interaction, were responsible for the observed phenotype. Finally, the authors performed fecal transplantation from control C57BL/6 mice to replace the intestinal microbiota [2]. In antibiotic-treated mice who received untreated (control) feces, MNoV CR6 was able to persistently infect mice and establish infection in intestinal tissues. As a control, a separate cohort of mice received fecal transplants from antibiotic-treated mice. As expected, fecal transplants from antibiotic-treated mice did not restore MNoV CR6 infection [2].
In another study, C57BL/6 mice were pretreated with antibiotics (ampicillin, neomycin, metronidazole, and vancomycin) or PBS (phosphate buffered saline), and infected with 105 median tissue culture infectious dose (TCID50) units MNoV MNV-1 [44]. Analysis along the length of the gastrointestinal tract revealed that antibiotic treatment increased the viral load in the proximal portion of the small intestine, but had no effect on the mid-section of the small intestine. Interestingly, antibiotic treated mice had lower viral titers in the distal portion of the small intestine and colon [44], indicating regional specific microbiota effects on infection. Consistent with the results of antibiotic-treated mice, germ-free mice also exhibited a 25-fold increase in MNV-1 titers in the proximal small intestine, no changes in mid small intestine, and a 10-fold decrease in MNV-1 titers in the distal small intestine compared with conventional (complete microbiota) mice [44]. When susceptible adult type I IFN receptor-deficient (Ifnar−/−) C57BL/6 mice were pretreated with antibiotics or PBS, and infected with MNV-1, antibiotic-treated mice were equally susceptible to MNV-1 infection as PBS-treated mice and no statistical difference was observed in MNV-1 titers along the length of the gastrointestinal tract [44]. These data indicate that type I IFN is required for the antibiotic-mediated effects on MNV-1 infection. Interestingly, the same antibiotic regimen had a minimal effect on conventional mice with a persistent strain MNV-3 [44], indicating virus strain-specific differences in the role of the intestinal microbiota on persistent MNV infection.
In addition to mouse models, the role of commensal microbes in HuNoV infection has recently been demonstrated in a gnotobiotic pig model [45]. In the past, HuNoV infections have not been successful in conventional pigs, presumably owing to the porcine gut microbiota. To address the unique contributions of the human microbiota that facilitate HuNoV infection, Lei et al. transplanted infant feces into germ-free pigs, generating human gut microbiota gnotobiotic animals [45]. Compared with germ-free pigs, HuNoV GII.4/2006b inoculation of human gut microbiota pigs resulted in increased HuNoV shedding, as denoted by higher shedding titers on post inoculation day (PID) 3, 4, 6, 8, and 9; a significantly longer duration of virus shedding; and higher virus titers in the duodenum and ileum [45]. Microbiota analysis by 16S rRNA sequencing also identified that HuNoV infection dramatically altered intestinal microbiota in human gut microbiota pigs. Increased Bacteroidetes (Bacteroides) and decreased Proteobacteria (Enterobacteriaceae) and Firmicutes (Clostridium, Ruminococcus, Anaerococcus, Enterococcus) levels were observed in HuNoV infected pigs compared with non-infected pigs. Similar to rotavirus, norovirus has also been shown to bind to porcine gastric mucin and competitively inhibits norovirus binding to histo-blood group antigens and Caco-2 cells [46,47,48]. Although it has not been directly examined, it is possible that mucin degradation by Bacteroides may also promote norovirus infection.
Changes in the gut microbiota have also been observed in HuNoV-infected patients. In general, ‘healthy’ gut microbes such as Bifidobacterium spp. (Actinobacteria) and Lactobacillus spp. (Firmicutes) are decreased in children with HuNoV diarrhea as compared with healthy controls [49,50]. In another study, Chen et al. observed increased Proteobacteria in HuNoV infected children compared with healthy controls [24]. Microbiome analysis of symptomatic and asymptomatic HuNoV-infected patients by 16S rRNA sequencing revealed enrichment in Clostridia (Firmicutes) and depletion of Bacteroidia (Bacteroidetes) in symptomatic individuals compared with asymptomatic individuals [51]. These studies point to alterations in the gut microbiota in response to HuNoV.
Immune cell signaling, particularly IFN-λ signaling, is known to play a key role in controlling persistent MNoV infection [52]. Antibiotic treatment of Ifnlr1−/− mice (lacking IFN-λ receptor 1), Stat1−/− mice (lacking a key transcription factor that conveys IFN signals), and Irf3−/− mice (lacking the transcription factor that induces IFN expression) had no effect on the persistence of MNoV CR6 infection [2]. Another intriguing interaction between the gut microbiota, MNoV, and the immune system involves IgA production. Turula et al. found that Pigr−/− (polymeric immunoglobulin receptor) null mice, which lack intestinal IgA, have decreased MNV-1 infectivity compared with WT mice [53]. In a complementary experiment, no difference in susceptibility to MNoV was observed in germ-free mice, indicating a role for the gut microbiota in IgA-enhancement of MNoV [53]. Pigr−/− mice were found to have an altered gut microbiota relative to WT mice, which induced higher levels of IFN-γ and iNOS, thereby controlling MNoV infection [53]. These findings suggest that IgA maintains a normal gut microbiota structure, which decreases IFN-γ and iNOS levels and thus creates a permissive environment for NoV infection. These data suggest that gut microbes limit the efficacy of IFN-λ-dependent innate immunity that indirectly contributes to norovirus infection.
Norovirus also binds to host cells via specific glycans [3,12,54,55]. Several groups have now identified that human norovirus can bind to enteric bacteria that harbor similar glycans (histo-blood group antigens). Almand et al. found that human norovirus strains (GII.4 New Orleans 2009 and Sydney 2012, GI.6) adhered to Klebsiella spp., Citrobacter spp., Bacillus spp., Enterococcus faecium, Hafnia alvei, Staphylococcus aureus, and Enterobacter cloacae by microscopy [56]. Binding was observed on outer cell surfaces and pili structures of microbes. Jones et al. identified that Enterobacter cloacae, which expresses H type histo-blood group antigens, could bind the GII.4-Sydney HuNoV strain [3]. Li et al. found that HuNoV virus-like particles NoV VLPs GI.1 and GII.4 bound to E. coli LMG8223, which had high expression of type A histo-blood group antigen and E. coli LFMFP861, which had high expression of type B histo-blood group antigen [54]. With a similar histo-blood group antigen focus, Miura et al. isolated an enteric bacterium strain (SENG-6), closely related to Enterobacter cloacae, bearing histo-blood group antigen-like structures. This Enterobacter sp. SENG-6 bacteria was found to bind human NoV virus-like particles of a GI.1 wild-type strain (8fIIa) and a GII.6 strain, but not an NoVLP GI.1 mutant strain (W375A) that has lost the ability to bind to A antigen. It has been hypothesized that adhesion of norovirus to bacteria may enhance access of the virus to the host epithelium or transmit through the M cells and delivery to immune cells, such as B cells. It has also been speculated that adhesion to microbes may also facilitate retention of norovirus in the intestine and counteract the movement of particles via peristalsis [57].
Another component of the intestinal milieu that can affect viral infection is bile acids. Bile acids are produced in the liver, concentrated in the gallbladder, and released into the intestinal lumen in response to food as primary bile acids [58]. The gut microbiota then chemically modifies the bile acids to secondary bile acids, such as deoxycholic acid and lithocholic acid. Grau et al. identified that antibiotic administration reduced the overall levels of unconjugated primary bile acids and all secondary bile acids in the proximal small intestine compared with PBS-treated mice [44], suggesting that bile acids biotransformed by commensal bacteria participate in MNV infection. Bile acids have been found to engage the viral capsid of MNoV with its proteinaceous receptor CD300lf in vitro [59]. Bile acids have also been shown to directly bind the HuNoV capsid [58] and supplementation of bile, specifically the non-proteinaceous component, to human intestinal enteroids (HIEs) significantly increases HuNoV replication [43,60]. In the epithelium, bile acids appear to modify the host rather than the virus. Bile acids also facilitates HuNoV GII.2 interactions with diverse histo-blood group antigens [61]. As a result, microbial modulation of bile acids can serve as a cofactor for HuNoV infection.
A final mechanism by which the gut microbiota can influence norovirus infection is through the modulation of cell fate. Intestinal stem cells are highly influenced by microbial metabolites and these signals can influence the number and function of epithelial cell types [62,63,64,65]. Antibiotic treatment of mice, which reduces the number of microbes and metabolites, also reduces the number of colonic tuft cells, thereby reducing the number of cells available for persistent MNoV CR6 infection [66]. The role of tuft cells in MNoV infection was confirmed by supplementing antibiotic-treated mice with recombinant IL-4 or IL-25, which artificially increased tuft cell numbers and the CD300lf receptor and restored infection [66]. In hematopoietic cells, LPS stimulation has been shown to increase CD300lf receptor levels [67]. Thus, bacterial LPS may also be influencing the level of viral receptor in the intestine. Together, these studies indicate that intestinal microbes directly and indirectly enhance norovirus infection.
4. Poliovirus (Family: Picornaviridae, Genus: Poliovirus)
Poliovirus is a single-stranded, non-enveloped RNA virus in the Picornaviridae family. Poliovirus is responsible for the infectious disease polio [68]. In the early 20th century, polio outbreaks increased in frequency and size, disabling more than 35,000 people each year [69]. Although the number of worldwide polio cases has fallen in the 21st century, the virus remains in circulation in two countries: Afghanistan and Pakistan [69,70]. Poliovirus undergoes a primary replication cycle in the gastrointestinal tract. In rare cases, poliovirus disseminates from the gut, spreads through the bloodstream, and infects the central nervous system. In the central nervous system, poliovirus replicates in neurons and causes cell death, a phenomenon known as paralytic poliomyelitis. Poliomyelitis with irreversible paralysis only occurs in 1/200 infants infected with poliovirus and, among those paralyzed, only 5–10% die when their breathing muscles become immobilized [69,70].
To investigate the effects of the intestinal microbiota on poliovirus infection, Kuss et al. used a susceptible mouse model (C57BL/6 PVRtg-Ifnar1−/−), which is transgenic for the human poliovirus receptor (PVR) and immunodeficient (lacking the interferon-α/β receptor gene) [6]. PVRtg-Ifnar1−/− mice were treated with antibiotics (ampicillin, neomycin, metronidazole, and vancomycin) prior to oral inoculation with type 1 Mahoney poliovirus. Antibiotic treatment reduced mortality, viral titers, and viral replication compared with untreated mice. To confirm the importance of the microbiota, the authors also reintroduced microbes to the antibiotic-treated mice by fecal transplant. Fecal transplants enhanced poliovirus disease, suggesting that microbiota promote poliovirus pathogenesis.
To determine if diminished poliovirus replication and disease in antibiotic-treated mice was due to microbiota depletion rather than direct effects of antibiotic treatment, Kuss et al. also examined the infectivity and viability of poliovirus in response to bacteria [6]. First, the authors orally inoculated untreated, antibiotic-treated, or germ-free mice with poliovirus and harvested luminal contents from the lower small intestine at two hours post-infection to examine the infectivity of isolated poliovirus. Poliovirus isolated from untreated mice was found to be twice as infective as antibiotic-treated and germ-free intestinal virus, as determined by mouse embryonic fibroblasts titers and HeLa cell titers [6]. Moreover, incubation of poliovirus with untreated mouse stool increased poliovirus viability, as quantified by plaque assay. These findings were replicated with Gram-negative (Escherichia coli, Ochrobactrum intermedium) or Gram-positive (Bacillus cereus, Enterococcus faecalis) bacteria, which also significantly increased viability. Exposure to B. cereus was particularly potent, resulting in increased poliovirus cell adherence and infectivity over 500%. Interestingly, enhancement of poliovirus infectivity did not require live bacteria. Kuss et al. went on to identify that bacterial surface polysaccharides including lipopolysaccharide (LPS) and peptidoglycan specifically promoted poliovirus infectivity of HeLa cells [6].
Erickson et al. went on to screen 40 bacterial strains, representing both Gram-negative and Gram-positive microbes, for poliovirus binding and found that nearly all bacteria could bind the virus [1]. High levels of adhesion were observed between poliovirus and Lactobacillus johnsonii, Lactobacillus murinus, Bacteroides acidifaciens, Lactococcus garviaea, Bacillus cereus, Bacillus subtilus, Enterococcus durnas, Ochrobactrum intermedium, Escherichia coli, and Clostridium symbosium. In these adhesion assays, multiple virions bound to the surface of each bacterium [1], implicating that multiple viruses can be delivered simultaneously. Using a high-throughput flow cytometry-based assay with polioviruses encoding DsRed or GFP (green fluorescent protein) to quantify infection of HeLa cells, Erickson et al. found 32% (13/40) of the bacterial strains increased poliovirus infectivity [1]. Infection appeared to correlate with the ability of bacteria to adhere to host cells. Interestingly, exposure to select microbes also enhanced poliovirus genetic recombination. This was elegantly demonstrated using mutant polioviruses sensitive or resistant to either a specific drug (guanidine hydrochloride) or temperature (39.5 °C) [1]. Finally, the authors found that select bacteria could facilitate the co-infection of multiple parental viruses, with up to six different parental viruses observed in a single plaque [1].
Work from the same group further examined the thermal inactivation of poliovirus in the presence of glycans [7]. The authors found that exposure to GlcNAc-containing polysaccharides, including the bacterial surface polysaccharides LPS and peptidoglycan, enhanced the stability of type 1 Mahoney poliovirus [7]. Detoxified LPS lacking lipid A was also able to enhance poliovirus stability, suggesting that the polysaccharide component of LPS was sufficient for stabilization [7]. Interestingly, chitin, a long polysaccharide that also contains GlcNAc found on yeast/fungi, stabilized poliovirus. Based on lack of stabilization of poliovirus with a six-unit GlcNAc oligosaccharide (chitohexaose), monomeric GlcNAc, deacetylated chitin (chitosan), and a GlcNAc-containing polysaccharide with sulfate groups (heparan), the authors concluded that viral stabilization required an acetylated GlcNAc-containing polysaccharide longer than six units [7]. In addition to providing stability, bacterial polysaccharides including LPS also enhance poliovirus cell attachment to HeLa cells by increasing binding to the viral receptor, PVR [7]. By enhancing stability and cell attachment of poliovirus, enteric microbes are likely enhancing viral infectivity in the gastrointestinal tract.
5. Reovirus (Family: Reoviridae, Genus: Orthoreovirus)
Reovirus (nonfusogenic mammalian orthoreovirus) is a non-enveloped, segmented double stranded RNA virus of the Reoviridae family [71]. Reoviruses commonly infect children, but infection seldom results in disease [72,73,74]. Reovirus infection is known to influence the intestinal immune system and infection can affect tolerance to dietary antigens such as gluten [75]. Similar to other enteric pathogens, reovirus attaches to host glycans and infects the small intestinal epithelium. It can also be transported by intestinal M cells to underlying Peyer’s patches [76,77,78]. Berger et al. explored the interaction of microbes with reovirus [71], demonstrating that reovirus prototypical strains Type 1 Lang (T1L) and Type 3 Dearing (T3D) directly interacted with Gram-positive Bacillus subtilis and Gram-negative E. coli and enhanced infection of HeLa cells [71]. Enhanced virion thermostability and infection was conveyed by bacterial envelope components LPS and peptidoglycan [71]. These data suggest that enteric microbes, particularly their polysaccharide components, enhance reovirus infection by enhancing the thermal stability of the reovirus particle.
The role of the gut microbiota in reovirus infection was also demonstrated by Kuss et al. [6]. The authors infected untreated or antibiotic-treated (ampicillin, neomycin, metronidazole, and vancomycin) C57BL/6 PVRtg-Ifnar1−/− mice with reovirus T3SA+ [6]. Mice infected with reovirus T3SA+ exhibited yellow oily stool, increased Peyer’s patch diameter, and high viral titers, indications of disease. In contrast, antibiotic-treated mice had stool that appeared normal, no change in Peyer’s patch diameter, and decreased viral titers. These findings suggest that intestinal microbes promote reovirus disease.
6. Astrovirus (Family: Astroviridae, Genus: Astrovirus)
Astroviruses are single-stranded positive, non-enveloped RNA viruses from the Astroviridae family [79]. Astroviruses are responsible for an estimated 47.8 million infections worldwide and are considered a leading cause of pediatric diarrhea [80,81]. Similar to other enteric viral infections, astrovirus infections can range from subclinical manifestations to severe diarrhea [82]. Pérez-Rodriguez et al. recently investigated whether microbial components in the stool environment could affect human astrovirus (HAstV) stability and infectivity [83]. The authors heat-inactivated two different HAstV genotypes, HAstV-1 and HAstV-8, which resulted in ~75% loss of infectivity in human colon Caco-2 cells, as determined by immunofluorescence. Addition of filtered human stool samples enhanced heat-inactivated astrovirus infection of Caco-2 cells [83]. 16S rRNA gene sequencing revealed that the stool was dominated by Firmicutes and Bacteroidetes and, to a lesser extent, Actinobacteria and Proteobacteria. To further investigate the effect of fecal bacterial communities on HAstV infectivity, Pérez-Rodriguez et al. tested bacterial species isolated from human stools: Escherichia coli (Gram-negative) and Enterococcus faecalis (Gram-positive). Incubation of HAstVs with heat-inactivated bacteria strains preserved viral infectivity when the bacteria were present at high concentrations (2.5 × 108 CFU/mL) [83]. The authors also identified that LPS (from Escherichia coli O111:B4) and peptidoglycan (from Bacillus subtilis) showed a dose-dependent enhancement of viral infectivity [83]. These data indicate that the presence of bacteria and bacterial surface molecules enhances HAstV persistence after mild thermal stress.
Recently, Cortez et al. identified that murine astrovirus (MuAstV) infects goblet cells in the small intestines of adult C57BL/6 mice [84]. Similar findings were observed by Kolawole et al., who found that HAstV VAI could infect goblet cells and sucrose isomaltose positive enterocytes in HIEs [85]. In mice, infection by astrovirus was also found to alter the gut microbiota [86]. Infection of MuAstV strain SJ002 in adult C57BL/6 mice [87] resulted in decreased microbial diversity and increased abundance of Clostridium cluster XVIII compared with the mock-infected animals [86]. Altered microbiota composition has also been noted in children, poultry, and bats with astrovirus infection [88,89,90,91]. To identify the contribution of the gut microbiota to astrovirus, Cortez and colleagues treated mice with broad-spectrum antibiotics (vancomycin, ampicillin, neomycin, and metronidazole) and found that antibiotic treatment significantly reduced virus infection and shedding [84]. Although the exact mechanisms have not been elucidated, murine astrovirus STL5 also induces IFN-λ [92]. HAstV VAI elicits IFN-mediated innate immune responses in HIEs [85]. It is possible that, similar to norovirus, the microbiota may indirectly interfere with IFN-λ production and thus promote infection. An alternative hypothesis is that the microbiota provides signals that induce mucus secretion [63,93] and mucus may be necessary for the transmission of astrovirus. Despite the gaps in knowledge, it is clear that the microbiota contributes to the pathogenesis of astrovirus and likely contributes through direct and indirect mechanisms.
7. Conclusions
This review highlights common mechanisms by which certain microbes enhance infection. Rotavirus, norovirus, poliovirus, and reovirus can directly bind to bacteria, which can facilitate delivery to epithelial cells. In the case of poliovirus and norovirus, multiple virions can attach to a single bacterium, which may promote co-infection of different parental viruses in the same cell. Another common feature among the highlighted enteric viruses is the ability to bind to specific host glycans, structures that are mirrored in mucins. In the setting of norovirus and rotavirus, studies indicate that mucin glycans can serve as a potential decoy for viral attachment to the host and, in rotavirus infection, mucin-degrading microbes promote infection by cleaving these decoy structures. For astrovirus, the preference for mucin glycans is apparent by the fact that astrovirus infects mucin-secreting goblet cells. Adhesion of viruses to bacteria also stabilizes the virus and enhances infectivity. This has been elegantly demonstrated for poliovirus, reovirus, and astrovirus. Finally, the microbiota appears to dampen epithelial and immune signals, creating a permissive environment for infection. Although not all mechanisms have been demonstrated for all enteric viruses, we predict that bacterial adhesion, mucin-degradation, enhancing viral stability, and dampening the immune system are common pathways by which microbes augment viral infection.
Although we cover studies where the gut microbiota supports infection, there are also several studies that indicate that select members of the gut microbiota can inhibit enteric viruses. For example, segmented filamentous bacteria (SFB) impedes rotavirus infection via induction of IL-17 and IL-22 and by increasing epithelial cell turnover [26]. Additionally, Bifidobacteria and Lactobacilli have been found to modulate epithelial and immune signaling and decrease rotavirus infection in various models [29,94,95,96,97,98,99,100]. For norovirus infection, Bacillus spp. were found to produce poly-γ-glutamic acid (γ-PGA), which acts as a non-canonical TLR4 ligand to induce IFN-β, thereby inhibiting MNoV MNV-1 infection [101]. Another study identified that retinoic acid (Vitamin A) administration increased Lactobacillus spp., which induced cytokines such as IFN-β and IFN-γ, which in turn inhibited MNoV MNV-1 infection [102]. As proof of concept, retinoic acid administration to Mexican children in a randomized, placebo-controlled trial demonstrated that retinoic acid reduced the prevalence of HuNoV GII infections and decreased the prevalence of HuNoV-associated diarrhea [103]. These are just a few examples of how microbes can also inhibit viral infection. These studies support recent findings that microbial composition plays a large role in the infectivity, persistence, and duration of viral infections. Inter-personal variations in the intestinal microbiota help explain why some individuals develop more severe infections.
These studies beg the question of whether antibiotics should be used for the clinical treatment of enteric viral infections or as an adjuvant to vaccines. Unfortunately, early life antibiotic exposure has been shown to incite long-lasting microbiota perturbation [104,105,106]. As a healthy gut microbiota influences many attributes of human health, we do not recommend antibiotics for viral infections. As an antibiotic alternative, we believe that the gut microbiota can be positively manipulated to protect the host from prolonged infection. We speculate that delivery of a defined bacterial cocktail, designed to stimulate the immune system and shift the microbiota composition, could diminish enteric infection. Probiotic microbes such as Bifidobacteria and Lactobacilli coupled with prebiotics to promote their growth may be a viable treatment strategy as well. However, care must be taken in selecting Bifidobacteria and Lactobacilli species, as species effects have been documented. Diet has been shown to shift microbial composition within 24 h [107], and this approach may also provide benefits for enteric viruses. Finally, supplementation of compounds that mirror host glycans may provide a valuable resource for resolving viral infection. Addition of human milk oligosaccharides (HMOs), which mirror host glycan structures and support Bifidobacteria growth [108,109,110], serve as a decoy for rotavirus and norovirus [111,112,113,114], and enhance the intestinal epithelial barrier [115,116,117,118], may provide a benefit for viral infections. We believe these strategies merit further investigation.
A potential caveat to the existing work is that many studies use stool samples as a global assessment of the gut microbiota. Work by Engevik et al. [9] and Grau et al. [44] identified site-specific changes in the gut microbiota in response to rotavirus and norovirus infection. These studies suggest that analysis of the microbial composition of fecal or colon samples does not reflect the bacteria present in the small intestine, where viral infection occurs. Additionally, other studies have highlighted key differences between the luminal and mucosa-associated microbiota [119,120], considerations that should be taken into account when examining the microbiota changes in response to viral infection. This information should prompt more analysis of the microbiota in different regions of the small intestine (duodenum, jejunum, and ileum). Small intestinal microbes identified in these regions could then be selected for adhesion, viral stability, and host delivery properties. The findings presented herein emphasize the importance of considering trans-kingdom interactions in the pathogenesis of infectious diseases. Future research focused on microbiota–viral interactions will undoubtedly unveil new mechanisms and new opportunities for therapeutic interventions for viral diseases.
Author Contributions
Conceptualization, K.A.E. and M.A.E.; literature review, K.A.E. and M.A.E.; writing—original draft preparation, K.A.E., writing—review and editing, K.A.E. and M.A.E.; visualization, M.A.E.; supervision, M.A.E.; funding acquisition, M.A.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH NIDDK, grant number T32DK00766428 (K.A.E.) and K01K12319501 (M.A.E.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria Facilitate Enteric Virus Co-infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe 2018, 23, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, M.; Case, L.K.; Kopaskie, K.; Kozlova, A.; MacDearmid, C.; Chervonsky, A.V.; Golovkina, T.V. Successful transmission of a retrovirus depends on the commensal microbiota. Science 2011, 334, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuss, S.K.; Etheredge, C.A.; Pfeiffer, J.K. Multiple host barriers restrict poliovirus trafficking in mice. PLoS Pathog. 2008, 4, e1000082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, R.; Chassaing, B.; Zhang, B.; Gewirtz, A.T. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J. Infect. Dis. 2014, 210, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engevik, M.A.; Banks, L.D.; Engevik, K.A.; Chang-Graham, A.L.; Perry, J.L.; Hutchinson, D.S.; Ajami, N.J.; Petrosino, J.F.; Hyser, J.M. Rotavirus infection induces glycan availability to promote ileum-specific changes in the microbiome aiding rotavirus virulence. Gut Microbes 2020, 11, 1324–1347. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Virgin, H.W. Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 2016, 351. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, J.K.; Sonnenburg, J.L. The intestinal microbiota and viral susceptibility. Front. Microbiol. 2011, 2, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almand, E.A.; Moore, M.D.; Jaykus, L.A. Virus-Bacteria Interactions: An Emerging Topic in Human Infection. Viruses 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Bomsel, M.; Alfsen, A. Entry of viruses through the epithelial barrier: Pathogenic trickery. Nat. Rev. Mol. Cell Biol. 2003, 4, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, S.; Woodruff, A.L.; Xia, M.; Tan, M.; Kennedy, M.A.; Jiang, X. Structural basis of glycan specificity of P[19] VP8*: Implications for rotavirus zoonosis and evolution. PLoS Pathog. 2017, 13, e1006707. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.L.; Wang, M.X.; Sun, X.M.; Yuan, Y.; Qing, Y.; Xin, Y.; Zhang, J.Y.; Li, D.D.; Duan, Z.J. Glycan binding patterns of human rotavirus P[10] VP8* protein. Virol. J. 2018, 15, 161. [Google Scholar] [CrossRef]
- Jolly, C.L.; Beisner, B.M.; Holmes, I.H. Rotavirus infection of MA104 cells is inhibited by Ricinus lectin and separately expressed single binding domains. Virology 2000, 275, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin glycan foraging in the human gut microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, D.; Qi, J.; Chai, W.; Wang, L.; Wang, L.; Peng, R.; Wang, H.; Zhang, Q.; Pang, L.; et al. Glycan Binding Specificity and Mechanism of Human and Porcine P[6]/P[19] Rotavirus VP8*s. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Boshuizen, J.A.; Reimerink, J.H.; Korteland-Van Male, A.M.; Van Ham, V.J.; Bouma, J.; Gerwig, G.J.; Koopmans, M.P.; Buller, H.A.; Dekker, J.; Einerhand, A.W. Homeostasis and function of goblet cells during rotavirus infection in mice. Virology 2005, 337, 210–221. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Baylor, M.; Bass, D.M. Murine intestinal mucins inhibit rotavirus infection. Gastroenterology 1993, 105, 84–92. [Google Scholar] [CrossRef]
- Twitchell, E.L.; Tin, C.; Wen, K.; Zhang, H.; Becker-Dreps, S.; Azcarate-Peril, M.A.; Vilchez, S.; Li, G.; Ramesh, A.; Weiss, M.; et al. Modeling human enteric dysbiosis and rotavirus immunity in gnotobiotic pigs. Gut Pathog. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Vlasova, A.N.; Deblais, L.; Huang, H.C.; Wijeratne, A.; Kandasamy, S.; Fischer, D.D.; Langel, S.N.; Paim, F.C.; Alhamo, M.A.; et al. Impact of nutrition and rotavirus infection on the infant gut microbiota in a humanized pig model. BMC Gastroenterol. 2018, 18, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Tsai, C.N.; Lee, Y.S.; Lin, C.Y.; Huang, K.Y.; Chao, H.C.; Lai, M.W.; Chiu, C.H. Intestinal microbiome in children with severe and complicated acute viral gastroenteritis. Sci. Rep. 2017, 7, 46130. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.; Zhang, C.; Du, H.; Wei, G.; Pang, X.; Zhou, H.; Liu, B.; Zhao, L. Pattern extraction of structural responses of gut microbiota to rotavirus infection via multivariate statistical analysis of clone library data. FEMS Microbiol. Ecol. 2009, 70, 21–29. [Google Scholar] [CrossRef]
- Shi, Z.; Zou, J.; Zhang, Z.; Zhao, X.; Noriega, J.; Zhang, B.; Zhao, C.; Ingle, H.; Bittinger, K.; Mattei, L.M.; et al. Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 2019, 179, 644–685.e613. [Google Scholar] [CrossRef]
- Rodriguez-Diaz, J.; Garcia-Mantrana, I.; Vila-Vicent, S.; Gozalbo-Rovira, R.; Buesa, J.; Monedero, V.; Collado, M.C. Relevance of secretor status genotype and microbiota composition in susceptibility to rotavirus and norovirus infections in humans. Sci. Rep. 2017, 7, 45559. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.M.; Wahed, M.A. Direct Nutrient Loss and Diarrhea. In Diarrhea and Malnutrition: Interactions, Mechanisms, and Interventions; Chen, L.C., Scrimshaw, N.S., Eds.; Springer: Boston, MA, USA, 1983; pp. 155–160. [Google Scholar] [CrossRef]
- Kandasamy, S.; Vlasova, A.N.; Fischer, D.; Kumar, A.; Chattha, K.S.; Rauf, A.; Shao, L.; Langel, S.N.; Rajashekara, G.; Saif, L.J. Differential Effects of Escherichia coli Nissle and Lactobacillus rhamnosus Strain GG on Human Rotavirus Binding, Infection, and B Cell Immunity. J. Immunol. 2016, 196, 1780–1789. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, H.B.; Estes, M.K. Rotaviruses: From pathogenesis to vaccination. Gastroenterology 2009, 136, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Blutt, S.E.; Miller, A.D.; Salmon, S.L.; Metzger, D.W.; Conner, M.E. IgA is important for clearance and critical for protection from rotavirus infection. Mucosal Immunol. 2012, 5, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct Roles of Type I and Type III Interferons in Intestinal Immunity to Homologous and Heterologous Rotavirus Infections. PLoS Pathog. 2016, 12, e1005600. [Google Scholar] [CrossRef]
- Hernandez, P.P.; Mahlakoiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hoelscher, C.; Dumoutier, L.; et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, A.; Rothenberg, M.E.; Mukherjee, G.; Feng, N.; Kalisky, T.; Nair, N.; Johnstone, I.M.; Clarke, M.F.; Greenberg, H.B. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc. Natl. Acad. Sci. USA 2012, 109, 20667–20672. [Google Scholar] [CrossRef] [Green Version]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Swiecki, M.; Miller, H.L.; Sesti-Costa, R.; Cella, M.; Gilfillan, S.; Colonna, M. Microbiota induces tonic CCL2 systemic levels that control pDC trafficking in steady state. Mucosal Immunol. 2017, 10, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Vashist, S.; Bailey, D.; Putics, A.; Goodfellow, I. Model systems for the study of human norovirus Biology. Future Virol. 2009, 4, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.C.; Vinje, J.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Weinberg, G.A.; Hall, C.B.; Chappell, J.; Bernstein, D.I.; Curns, A.T.; et al. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 2013, 368, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Karst, S.M.; Wobus, C.E. A working model of how noroviruses infect the intestine. PLoS Pathog. 2015, 11, e1004626. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinje, J.; Parashar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef]
- Estes, M.K.; Ettayebi, K.; Tenge, V.R.; Murakami, K.; Karandikar, U.; Lin, S.C.; Ayyar, B.V.; Cortes-Penfield, N.W.; Haga, K.; Neill, F.H.; et al. Human Norovirus Cultivation in Nontransformed Stem Cell-Derived Human Intestinal Enteroid Cultures: Success and Challenges. Viruses 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Grau, K.R.; Zhu, S.; Peterson, S.T.; Helm, E.W.; Philip, D.; Phillips, M.; Hernandez, A.; Turula, H.; Frasse, P.; Graziano, V.R.; et al. The intestinal regionalization of acute norovirus infection is regulated by the microbiota via bile acid-mediated priming of type III interferon. Nat. Microbiol. 2020, 5, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Twitchell, E.L.; Ramesh, A.K.; Bui, T.; Majette, E.; Tin, C.M.; Avery, R.; Arango-Argoty, G.; Zhang, L.; Becker-Dreps, S.; et al. Enhanced GII.4 human norovirus infection in gnotobiotic pigs transplanted with a human gut microbiota. J. Gen. Virol. 2019, 100, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Brandl, M.; Mandrell, R. Porcine gastric mucin binds to recombinant norovirus particles and competitively inhibits their binding to histo-blood group antigens and Caco-2 cells. Lett. Appl. Microbiol. 2005, 41, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Yang, D.; Jiang, X.; Zhong, W.; Cannon, J.L.; Burkhardt, W., 3rd; Woods, J.W.; Hartman, G.; Lindesmith, L.; Baric, R.S.; et al. Specificity and kinetics of norovirus binding to magnetic bead-conjugated histo-blood group antigens. J. Appl. Microbiol. 2010, 109, 1753–1762. [Google Scholar] [CrossRef]
- Li, X.; Chen, H. Evaluation of the porcine gastric mucin binding assay for high-pressure-inactivation studies using murine norovirus and tulane virus. Appl. Environ. Microbiol. 2015, 81, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.M.; Walk, S.T.; Taube, S.; Taniuchi, M.; Houpt, E.R.; Wobus, C.E.; Young, V.B. Disruption of the human gut microbiota following Norovirus infection. PLoS ONE 2012, 7, e48224. [Google Scholar] [CrossRef] [Green Version]
- Walker, F.C.; Baldridge, M.T. Interactions between noroviruses, the host, and the microbiota. Curr. Opin. Virol. 2019, 37, 1–9. [Google Scholar] [CrossRef]
- Patin, N.V.; Pena-Gonzalez, A.; Hatt, J.K.; Moe, C.; Kirby, A.; Konstantinidis, K.T. The Role of the Gut Microbiome in Resisting Norovirus Infection as Revealed by a Human Challenge Study. mBio 2020, 11. [Google Scholar] [CrossRef]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turula, H.; Bragazzi Cunha, J.; Mainou, B.A.; Ramakrishnan, S.K.; Wilke, C.A.; Gonzalez-Hernandez, M.B.; Pry, A.; Fava, J.; Bassis, C.M.; Edelman, J.; et al. Natural Secretory Immunoglobulins Promote Enteric Viral Infections. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Breiman, A.; Le Pendu, J.; Uyttendaele, M. Binding to histo-blood group antigen-expressing bacteria protects human norovirus from acute heat stress. Front. Microbiol. 2015, 6, 659. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Sano, D.; Suenaga, A.; Yoshimura, T.; Fuzawa, M.; Nakagomi, T.; Nakagomi, O.; Okabe, S. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J. Virol. 2013, 87, 9441–9451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almand, E.A.; Moore, M.D.; Outlaw, J.; Jaykus, L.A. Human norovirus binding to select bacteria representative of the human gut microbiota. PLoS ONE 2017, 12, e0173124. [Google Scholar] [CrossRef]
- Karst, S.M. The influence of commensal bacteria on infection with enteric viruses. Nat. Rev. Microbiol. 2016, 14, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Kilic, T.; Koromyslova, A.; Hansman, G.S. Structural Basis for Human Norovirus Capsid Binding to Bile Acids. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.N.; Orchard, R.C.; Kim, A.S.; Stegeman, R.A.; Hsieh, L.L.; Smith, T.J.; Virgin, H.W.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E9201–E9210. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Tenge, V.R.; Karandikar, U.C.; Lin, S.C.; Ramani, S.; Ettayebi, K.; Crawford, S.E.; Zeng, X.L.; Neill, F.H.; Ayyar, B.V.; et al. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 1700–1710. [Google Scholar] [CrossRef]
- Mallory, M.L.; Lindesmith, L.C.; Brewer-Jensen, P.D.; Graham, R.L.; Baric, R.S. Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains. Viruses 2020, 12, 989. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe 2018, 24, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engevik, M.A.; Luk, B.; Chang-Graham, A.L.; Hall, A.; Herrmann, B.; Ruan, W.; Endres, B.T.; Shi, Z.; Garey, K.W.; Hyser, J.M.; et al. Bifidobacterium dentium Fortifies the Intestinal Mucus Layer via Autophagy and Calcium Signaling Pathways. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troll, J.V.; Hamilton, M.K.; Abel, M.L.; Ganz, J.; Bates, J.M.; Stephens, W.Z.; Melancon, E.; Van der Vaart, M.; Meijer, A.H.; Distel, M.; et al. Microbiota promote secretory cell determination in the intestinal epithelium by modulating host Notch signaling. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, B.C.E.; Shanahan, M.T.; Singh, A.P.; Sethupathy, P. Gut Microbial Influences on the Mammalian Intestinal Stem Cell Niche. Stem Cells Int. 2017, 2017, 5604727. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Lee, S.; Hsieh, L.L.; Orchard, R.C.; Desai, C.; Hykes, B.L., Jr.; McAllaster, M.R.; Balce, D.R.; Feehley, T.; Brestoff, J.R.; et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 2018, 360, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, K.; Kitaura, J.; Yamanishi, Y.; Matsuoka, T.; Oki, T.; Shibata, F.; Kumagai, H.; Nakajima, H.; Maeda-Yamamoto, M.; Hauchins, J.P.; et al. Functional analysis of activating receptor LMIR4 as a counterpart of inhibitory receptor LMIR3. J. Biol. Chem. 2007, 282, 17997–18008. [Google Scholar] [CrossRef] [Green Version]
- Hogle, J.M. Poliovirus cell entry: Common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef] [Green Version]
- Zarocostas, J. WHO keeps polio on the international health emergency list. Lancet 2018, 392, 2425. [Google Scholar] [CrossRef]
- Stower, H. WHO declares Africa polio-free. Nat. Med. 2020, 26, 1805. [Google Scholar] [CrossRef]
- Berger, A.K.; Yi, H.; Kearns, D.B.; Mainou, B.A. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog. 2017, 13, e1006768. [Google Scholar] [CrossRef] [Green Version]
- Ouattara, L.A.; Barin, F.; Barthez, M.A.; Bonnaud, B.; Roingeard, P.; Goudeau, A.; Castelnau, P.; Vernet, G.; Paranhos-Baccala, G.; Komurian-Pradel, F. Novel human reovirus isolated from children with acute necrotizing encephalopathy. Emerg. Infect. Dis. 2011, 17, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.H.; Williams, J.V.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr.; Dermody, T.S. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J. Infect. Dis. 2005, 191, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L. Serologic grouping of reoviruses by hemagglutination-inhibition. Am. J. Hyg. 1960, 71, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antar, A.A.; Konopka, J.L.; Campbell, J.A.; Henry, R.A.; Perdigoto, A.L.; Carter, B.D.; Pozzi, A.; Abel, T.W.; Dermody, T.S. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 2009, 5, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, D.H.; Eaton, M.A.; Anderson, A.O. Reovirus infection in adult mice: The virus hemagglutinin determines the site of intestinal disease. Microb. Pathog. 1986, 1, 79–87. [Google Scholar] [CrossRef]
- Amerongen, H.M.; Wilson, G.A.; Fields, B.N.; Neutra, M.R. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J. Virol. 1994, 68, 8428–8432. [Google Scholar] [CrossRef] [Green Version]
- Vu, D.L.; Bosch, A.; Pinto, R.M.; Guix, S. Epidemiology of Classic and Novel Human Astrovirus: Gastroenteritis and Beyond. Viruses 2017, 9, 33. [Google Scholar] [CrossRef]
- Olortegui, M.P.; Rouhani, S.; Yori, P.P.; Salas, M.S.; Trigoso, D.R.; Mondal, D.; Bodhidatta, L.; Platts-Mills, J.; Samie, A.; Kabir, F.; et al. Astrovirus Infection and Diarrhea in 8 Countries. Pediatrics 2018, 141. [Google Scholar] [CrossRef] [Green Version]
- GBD Diarrhoeal Diseases Collaborators. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 2017, 17, 909–948. [Google Scholar] [CrossRef] [Green Version]
- Cortez, V.; Meliopoulos, V.A.; Karlsson, E.A.; Hargest, V.; Johnson, C.; Schultz-Cherry, S. Astrovirus Biology and Pathogenesis. Annu. Rev. Virol. 2017, 4, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rodriguez, F.J.; Vieille, G.; Turin, L.; Yildiz, S.; Tapparel, C.; Kaiser, L. Fecal Components Modulate Human Astrovirus Infectivity in Cells and Reconstituted Intestinal Tissues. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, V.; Boyd, D.F.; Crawford, J.C.; Sharp, B.; Livingston, B.; Rowe, H.M.; Davis, A.; Alsallaq, R.; Robinson, C.G.; Vogel, P.; et al. Astrovirus infects actively secreting goblet cells and alters the gut mucus barrier. Nat. Commun. 2020, 11, 2097. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, A.O.; Mirabelli, C.; Hill, D.R.; Svoboda, S.A.; Janowski, A.B.; Passalacqua, K.D.; Rodriguez, B.N.; Dame, M.K.; Freiden, P.; Berger, R.P.; et al. Astrovirus replication in human intestinal enteroids reveals multi-cellular tropism and an intricate host innate immune landscape. PLoS Pathog. 2019, 15, e1008057. [Google Scholar] [CrossRef] [Green Version]
- Cortez, V.; Margolis, E.; Schultz-Cherry, S. Astrovirus and the microbiome. Curr. Opin. Virol. 2019, 37, 10–15. [Google Scholar] [CrossRef]
- Cortez, V.; Sharp, B.; Yao, J.; Livingston, B.; Vogel, P.; Schultz-Cherry, S. Characterizing a Murine Model for Astrovirus Using Viral Isolates from Persistently Infected Immunocompromised Mice. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Wu, X.; Nawaz, M.; Li, J.; Yu, P.; Moore, J.E.; Xu, J. Molecular characterization of fecal microbiota in patients with viral diarrhea. Curr. Microbiol. 2011, 63, 259–266. [Google Scholar] [CrossRef]
- Qureshi, M.A.; Saif, Y.M.; Heggen-Peay, C.L.; Edens, F.W.; Havenstein, G.B. Induction of functional defects in macrophages by a poult enteritis and mortality syndrome-associated turkey astrovirus. Avian Dis. 2001, 45, 853–861. [Google Scholar] [CrossRef]
- Edens, F.W.; Parkhurst, C.R.; Qureshi, M.A.; Casas, I.A.; Havenstein, G.B. Atypical Escherichia coli strains and their association with poult enteritis and mortality syndrome. Poult. Sci. 1997, 76, 952–960. [Google Scholar] [CrossRef]
- Wasimuddin; Brandel, S.D.; Tschapka, M.; Page, R.; Rasche, A.; Corman, V.M.; Drosten, C.; Sommer, S. Astrovirus infections induce age-dependent dysbiosis in gut microbiomes of bats. ISME J. 2018, 12, 2883–2893. [Google Scholar] [CrossRef]
- Ingle, H.; Lee, S.; Ai, T.; Orvedahl, A.; Rodgers, R.; Zhao, G.; Sullender, M.; Peterson, S.T.; Locke, M.; Liu, T.C.; et al. Viral complementation of immunodeficiency confers protection against enteric pathogens via interferon-lambda. Nat. Microbiol. 2019, 4, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Willing, B.; Keeney, K.M.; Menendez, A.; Bergstrom, K.S.; Gill, N.; Russell, S.L.; Vallance, B.A.; Finlay, B.B. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect. Immun. 2011, 79, 1536–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, S.; Chattha, K.S.; Vlasova, A.N.; Rajashekara, G.; Saif, L.J. Lactobacilli and Bifidobacteria enhance mucosal B cell responses and differentially modulate systemic antibody responses to an oral human rotavirus vaccine in a neonatal gnotobiotic pig disease model. Gut Microbes 2014, 5, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.; Vimont, A.; Darveau, A.; Fliss, I.; Jean, J. Study of the Ability of Bifidobacteria of Human Origin to Prevent and Treat Rotavirus Infection Using Colonic Cell and Mouse Models. PLoS ONE 2016, 11, e0164512. [Google Scholar] [CrossRef]
- Ishizuka, T.; Kanmani, P.; Kobayashi, H.; Miyazaki, A.; Soma, J.; Suda, Y.; Aso, H.; Nochi, T.; Iwabuchi, N.; Xiao, J.Z.; et al. Immunobiotic Bifidobacteria Strains Modulate Rotavirus Immune Response in Porcine Intestinal Epitheliocytes via Pattern Recognition Receptor Signaling. PLoS ONE 2016, 11, e0152416. [Google Scholar] [CrossRef]
- Phuapradit, P.; Varavithya, W.; Vathanophas, K.; Sangchai, R.; Podhipak, A.; Suthutvoravut, U.; Nopchinda, S.; Chantraruksa, V.; Haschke, F. Reduction of rotavirus infection in children receiving bifidobacteria-supplemented formula. J. Med. Assoc. Thail. 1999, 82, S43–S48. [Google Scholar]
- Azagra-Boronat, I.; Massot-Cladera, M.; Knipping, K.; Garssen, J.; Ben Amor, K.; Knol, J.; Franch, A.; Castell, M.; Rodriguez-Lagunas, M.J.; Perez-Cano, F.J. Strain-Specific Probiotic Properties of Bifidobacteria and Lactobacilli for the Prevention of Diarrhea Caused by Rotavirus in a Preclinical Model. Nutrients 2020, 12, 498. [Google Scholar] [CrossRef] [Green Version]
- Vlasova, A.N.; Kandasamy, S.; Chattha, K.S.; Rajashekara, G.; Saif, L.J. Comparison of probiotic lactobacilli and bifidobacteria effects, immune responses and rotavirus vaccines and infection in different host species. Vet. Immunol. Immunopathol. 2016, 172, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Duffy, L.C.; Griffiths, E.; Dryja, D.; Leavens, A.; Rossman, J.; Rich, G.; Riepenhoff-Talty, M.; Locniskar, M. Immune responses in rhesus rotavirus-challenged BALB/c mice treated with bifidobacteria and prebiotic supplements. Pediatr. Res. 2002, 51, 750–755. [Google Scholar] [CrossRef]
- Lee, W.; Kim, M.; Lee, S.H.; Jung, H.G.; Oh, J.W. Prophylactic efficacy of orally administered Bacillus poly-gamma-glutamic acid, a non-LPS TLR4 ligand, against norovirus infection in mice. Sci. Rep. 2018, 8, 8667. [Google Scholar] [CrossRef]
- Lee, H.; Ko, G. Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome. Sci. Rep. 2016, 6, 25835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, K.Z.; Garcia, C.; Santos, J.I.; Rosado, J.L.; Hertzmark, E.; Dupont, H.L.; Ko, G. Vitamin A supplementation has divergent effects on norovirus infections and clinical symptoms among Mexican children. J. Infect. Dis. 2007, 196, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roubaud-Baudron, C.; Ruiz, V.E.; Swan, A.M., Jr.; Vallance, B.A.; Ozkul, C.; Pei, Z.; Li, J.; Battaglia, T.W.; Perez-Perez, G.I.; Blaser, M.J. Long-Term Effects of Early-Life Antibiotic Exposure on Resistance to Subsequent Bacterial Infection. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparrini, A.J.; Wang, B.; Sun, X.; Kennedy, E.A.; Hernandez-Leyva, A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Dantas, G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019, 4, 2285–2297. [Google Scholar] [CrossRef]
- Sbihi, H.; Boutin, R.C.; Cutler, C.; Suen, M.; Finlay, B.B.; Turvey, S.E. Thinking bigger: How early-life environmental exposures shape the gut microbiome and influence the development of asthma and allergic disease. Allergy 2019, 74, 2103–2115. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Lawson, M.A.E.; O’Neill, I.J.; Kujawska, M.; Gowrinadh Javvadi, S.; Wijeyesekera, A.; Flegg, Z.; Chalklen, L.; Hall, L.J. Breast milk-derived human milk oligosaccharides promote Bifidobacterium interactions within a single ecosystem. ISME J. 2020, 14, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Thongaram, T.; Hoeflinger, J.L.; Chow, J.; Miller, M.J. Human milk oligosaccharide consumption by probiotic and human-associated bifidobacteria and lactobacilli. J. Dairy Sci. 2017, 100, 7825–7833. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, A.; Katoh, T.; Sakanaka, M.; Ling, Y.; Yamada, C.; Asakuma, S.; Urashima, T.; Tomabechi, Y.; Katayama-Ikegami, A.; Kurihara, S.; et al. Sharing of human milk oligosaccharides degradants within bifidobacterial communities in faecal cultures supplemented with Bifidobacterium bifidum. Sci. Rep. 2018, 8, 13958. [Google Scholar] [CrossRef]
- Krammer, E.M.; Bouckaert, J.M.J. Norovirus devours human milk oligosaccharides rich in alpha-fucose. J. Biol. Chem. 2018, 293, 11966–11967. [Google Scholar] [CrossRef] [Green Version]
- Koromyslova, A.; Tripathi, S.; Morozov, V.; Schroten, H.; Hansman, G.S. Human norovirus inhibition by a human milk oligosaccharide. Virology 2017, 508, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Laucirica, D.R.; Triantis, V.; Schoemaker, R.; Estes, M.K.; Ramani, S. Milk Oligosaccharides Inhibit Human Rotavirus Infectivity in MA104 Cells. J. Nutr. 2017, 147, 1709–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, S.; Stewart, C.J.; Laucirica, D.R.; Ajami, N.J.; Robertson, B.; Autran, C.A.; Shinge, D.; Rani, S.; Anandan, S.; Hu, L.; et al. Human milk oligosaccharides, milk microbiome and infant gut microbiome modulate neonatal rotavirus infection. Nat. Commun. 2018, 9, 5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chichlowski, M.; De Lartigue, G.; German, J.B.; Raybould, H.E.; Mills, D.A. Bifidobacteria isolated from infants and cultured on human milk oligosaccharides affect intestinal epithelial function. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.Y.; Li, B.; Koike, Y.; Maattanen, P.; Miyake, H.; Cadete, M.; Johnson-Henry, K.C.; Botts, S.R.; Lee, C.; Abrahamsson, T.R.; et al. Human Milk Oligosaccharides Increase Mucin Expression in Experimental Necrotizing Enterocolitis. Mol. Nutr. Food Res. 2019, 63, e1800658. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Kong, C.; Walvoort, M.T.C.; Faas, M.M.; De Vos, P. Human Milk Oligosaccharides Differently Modulate Goblet Cells Under Homeostatic, Proinflammatory Conditions and ER Stress. Mol. Nutr. Food Res. 2020, 64, e1900976. [Google Scholar] [CrossRef] [Green Version]
- Natividad, J.M.; Rytz, A.; Keddani, S.; Bergonzelli, G.; Garcia-Rodenas, C.L. Blends of Human Milk Oligosaccharides Confer Intestinal Epithelial Barrier Protection in Vitro. Nutrients 2020, 12, 3047. [Google Scholar] [CrossRef]
- Engevik, M.A.; Aihara, E.; Montrose, M.H.; Shull, G.E.; Hassett, D.J.; Worrell, R.T. Loss of NHE3 alters gut microbiota composition and influences Bacteroides thetaiotaomicron growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G697–G711. [Google Scholar] [CrossRef] [Green Version]
- Engevik, M.A.; Hickerson, A.; Shull, G.E.; Worrell, R.T. Acidic conditions in the NHE2(−/−) mouse intestine result in an altered mucosa-associated bacterial population with changes in mucus oligosaccharides. Cell Physiol. Biochem. 2013, 32, 111–128. [Google Scholar] [CrossRef]
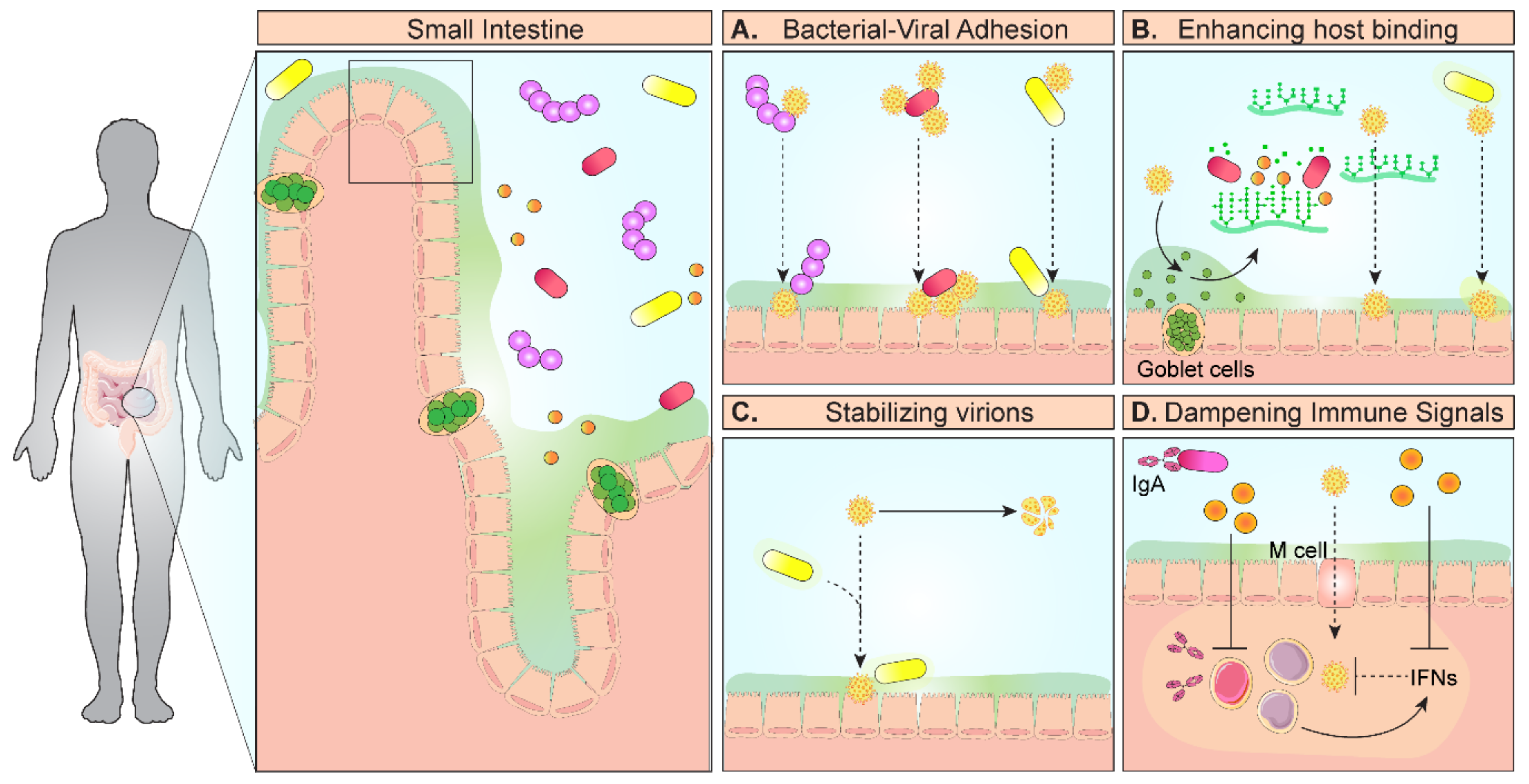
Figure 1.
Model diagram of mechanisms of microbe–virus interactions that promote infection. In the gastrointestinal tract, enteric viruses encounter the gut microbiota and select microbes facilitate infection. (A) Microbes in the small intestine can directly bind to viruses via histo-blood group antigens and deliver multiple viruses to the host epithelium. (B) Microbes can also degrade mucin glycans, which normally act as decoys to prevent viral adhesion to cell surface glycans, thereby promoting viral access to epithelial cells. (C) Microbes and their cell surface components, particularly lipopolysaccharide (LPS) and peptidoglycan, can increase the thermostability of enteric viruses. In addition to stability, bacterial polysaccharides can enhance viral engagement of cellular receptors, leading to increased infectivity. (D) Finally, intestinal microbes can dampen host immune signals designed to target viruses, such as interferons (IFN-λ, γ, β) and IgA production. Through these direct and indirect interactions, certain gut microbes can enhance viral infection.
Figure 1.
Model diagram of mechanisms of microbe–virus interactions that promote infection. In the gastrointestinal tract, enteric viruses encounter the gut microbiota and select microbes facilitate infection. (A) Microbes in the small intestine can directly bind to viruses via histo-blood group antigens and deliver multiple viruses to the host epithelium. (B) Microbes can also degrade mucin glycans, which normally act as decoys to prevent viral adhesion to cell surface glycans, thereby promoting viral access to epithelial cells. (C) Microbes and their cell surface components, particularly lipopolysaccharide (LPS) and peptidoglycan, can increase the thermostability of enteric viruses. In addition to stability, bacterial polysaccharides can enhance viral engagement of cellular receptors, leading to increased infectivity. (D) Finally, intestinal microbes can dampen host immune signals designed to target viruses, such as interferons (IFN-λ, γ, β) and IgA production. Through these direct and indirect interactions, certain gut microbes can enhance viral infection.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Engevik, K.A.; Engevik, M.A. Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections. Gastroenterol. Insights 2021, 12, 41-55. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12010005
AMA Style
Engevik KA, Engevik MA. Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections. Gastroenterology Insights. 2021; 12(1):41-55. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12010005
Chicago/Turabian StyleEngevik, Kristen A., and Melinda A. Engevik. 2021. "Partners in Infectious Disease: When Microbes Facilitate Enteric Viral Infections" Gastroenterology Insights 12, no. 1: 41-55. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12010005