The Response of the Soil Microbiota to Long-Term Mineral and Organic Nitrogen Fertilization is Stronger in the Bulk Soil than in the Rhizosphere

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Site, Experimental Design and Sample Collection
2.2. DNA Extraction and IonTorrent Sequencing
2.3. IonTorrent Sequence Analysis
2.4. Co-Occurrence Network Analysis
2.5. Soil Chemical Analyses
2.6. Quantitative PCR
3. Results
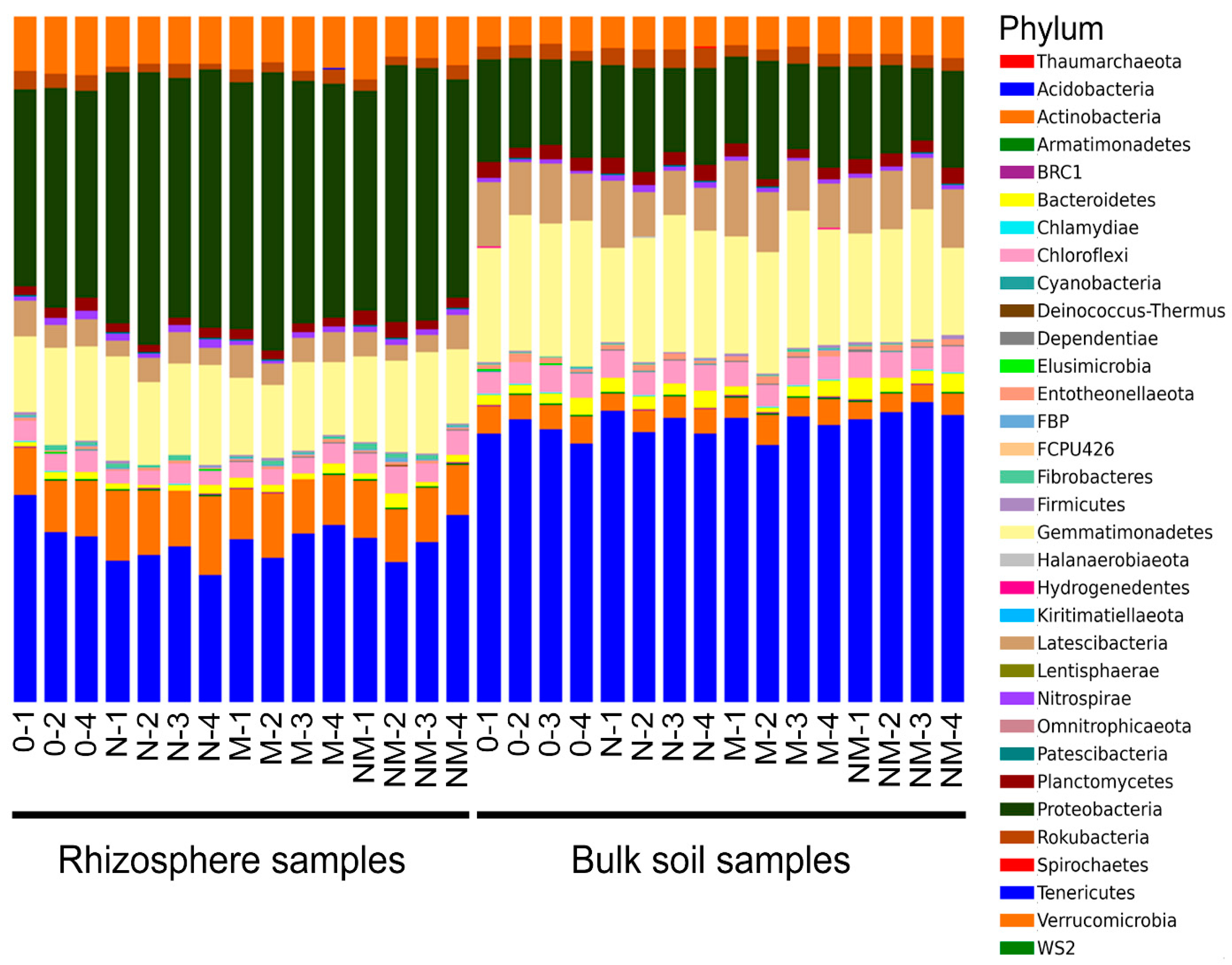
3.1. IonTorrent Sequencing Analysis
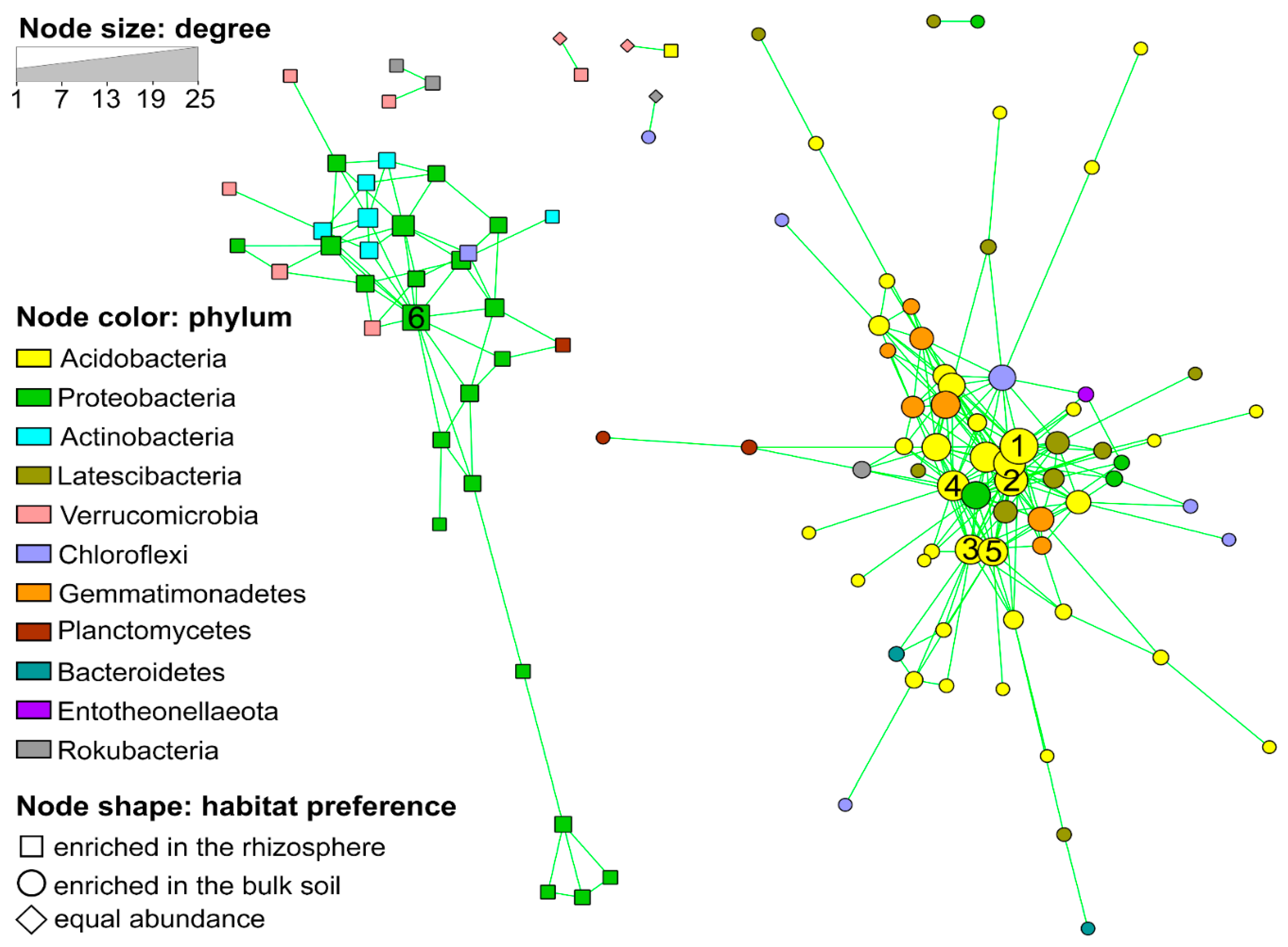
3.2. Co-Occurrence Network Analysis
3.3. Soil Chemical Analyses
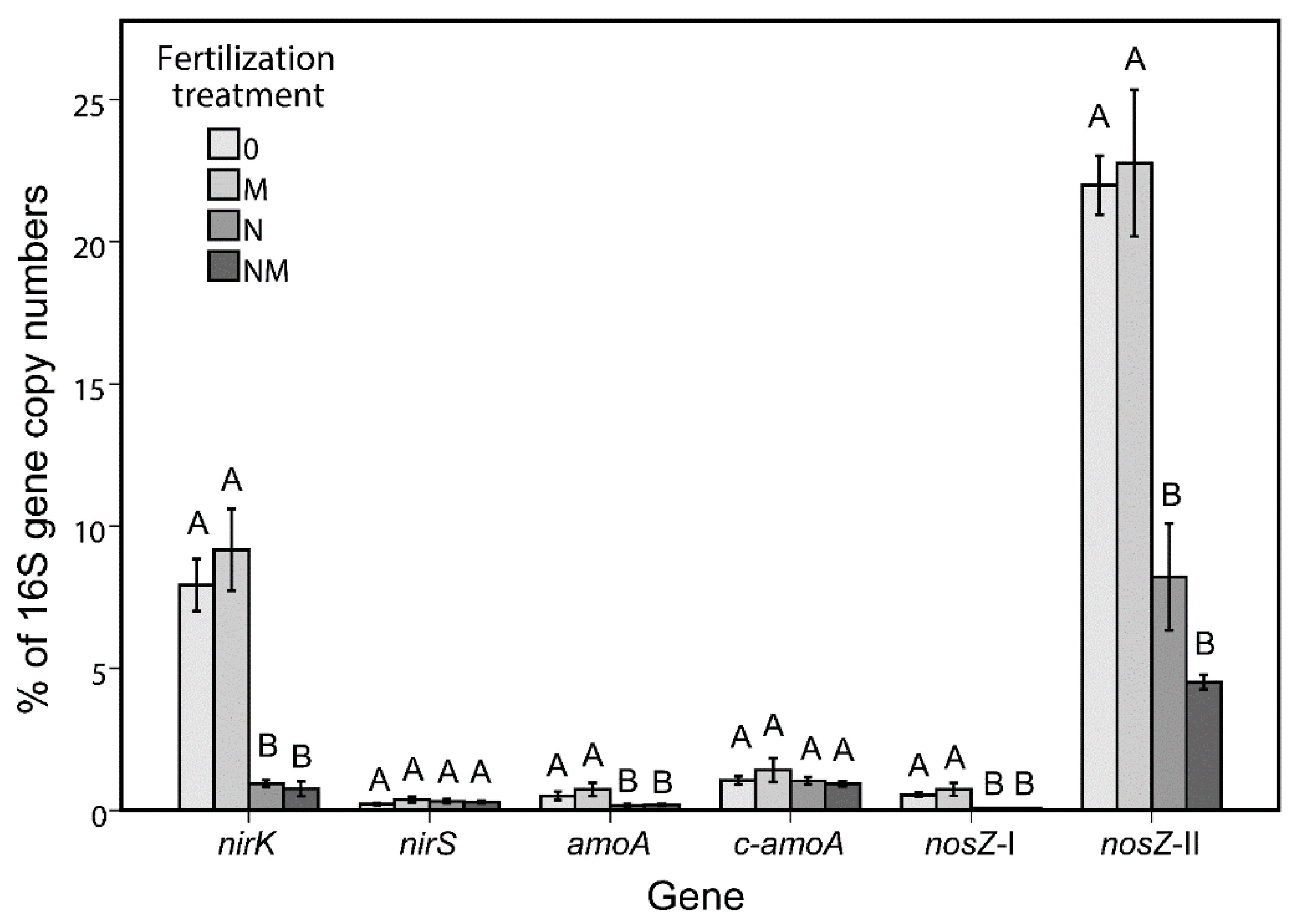
3.4. Quantitative PCR
4. Discussion
5. Conclusions
- In the long-term fertilization experiment in Giessen (Germany), the different nitrogen fertilization regimes affected the soil microbiota differentially depending on the soil habitat. Thus, it should be considered that it is not possible to extrapolate general conclusions when only one soil habitat is analyzed.
- Bulk soil was the soil habitat most affected by different nitrogen fertilizations. For future studies, we suggest using this soil habitat for investigating the effects of long-term fertilization regimes on the structure and function of the soil microbiota.
- The soil microbiota appeared organized in strongly interconnected clusters, which were also taxonomically coherent and soil habitat-specific (habitat complementarity). The cluster enriched in the bulk soil was characterized by Acidobacteria and appeared more strongly interconnected.
- Acidobacteria were the hub taxa in the bulk soil. These hub taxa deserve future attention, as they appear to be functional key regulators of the soil microbiome.
- The soil microbiome acts as a single functional unit. Therefore, a better understanding of the factors driving the interactions between microbial species, and how these correlate with the physico-chemical parameters of the soil is needed to optimize soil management practices for a more sustainable agriculture. Here, we showed that, despite the strong influence of the soil habitat on the general microbiome structure and function, the N-fertilization regime (in particular, the mineral + organic) significantly influenced two of the five most important OTUs of the soil microbial network of the bulk soil.
- Mineral-N fertilization has a strong negative impact on the functional genes related to the nitrogen cycle. However, it seems this is an indirect effect, which can be attributed to the stimulation of opportunistic microbes, which use the provided nitrogen.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Körschens, M. Die wichtigsten dauerfeldversuche der welt übersicht, bedeutung, ergebnisse. Arch. Agron. Soil Sci. 1997, 42, 157–168. [Google Scholar] [CrossRef]
- Richter, D.D.; Hofmockel, M.; Callaham, M.A.; Powlson, D.S.; Smith, P. Long-term soil experiments: Keys to managing Earth’s rapidly changing ecosystems. Soil Sci. Soc. Am. J. 2007, 71, 266–279. [Google Scholar] [CrossRef]
- Berzsenyi, Z.; Győrffy, B.; Lap, D. Effect of crop rotation and fertilisation on maize and wheat yields and yield stability in a long-term experiment. Eur. J. Agron. 2000, 13, 225–244. [Google Scholar] [CrossRef]
- Černý, J.; Balík, J.; Kulhánek, M.; Čásová, K.; Nedvěd, V. Mineral and organic fertilization efficiency in long-term stationary experiments. Plant Soil Environ. 2010, 56, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.Q.; Li, X.Y.; Li, X.P.; Shi, X.J.; Huang, S.M.; Wang, B.R.; Poulton, P. Long-term fertilizer experiment network in China: Crop yields and soil nutrient trends. Agron. J. 2010, 102, 216–230. [Google Scholar] [CrossRef]
- Hartmann, M.; Frey, B.; Mayer, J.; Mäder, P.; Widmer, F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 2015, 9, 1177–1194. [Google Scholar] [CrossRef] [Green Version]
- Allison, S.D.; Weintraub, M.N.; Gartner, T.B.; Waldrop, M.P. Evolutionary-economic principles as regulators of soil enzyme production and ecosystem function. In Soil Enzymology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 229–243. [Google Scholar]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef]
- Gruber, N.; Galloway, J.N. An Earth-system perspective of the global nitrogen cycle. Nature 2008, 451, 293–296. [Google Scholar] [CrossRef]
- Vitousek, P.M.; Porder, S.; Houlton, B.Z.; Chadwick, O.A. Terrestrial phosphorus limitation: Mechanisms, implications, and nitrogen–phosphorus interactions. Ecol. Appl. 2010, 20, 5–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauke, S.L.; von Sperber, C.; Tamburini, F.; Gocke, M.I.; Honermeier, B.; Schweitzer, K.; Amelung, W. Subsoil phosphorus is affected by fertilization regime in long-term agricultural experimental trials. Eur. J. Soil Sci. 2018, 69, 103–112. [Google Scholar] [CrossRef]
- Van Der Heijden, M.G.; Bardgett, R.D.; Van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Berg, G. Plant–microbe interactions promoting plant growth and health: Perspectives for controlled use of microorganisms in agriculture. Appl. Microbiol. Biotechnol. 2009, 84, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chisholm, S.T.; Coaker, G.; Day, B.; Staskawicz, B.J. Host-microbe interactions: Shaping the evolution of the plant immune response. Cell 2006, 124, 803–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solano, B.R.; Maicas, J.B.; Mañero, F.G. Physiological and molecular mechanisms of plant growth promoting rhizobacteria (PGPR). In Plant-Bacteria Interactions: Strategies and Techniques to Promote Plant Growth; Wiley: Weinheim, Germany, 2008; pp. 41–54. [Google Scholar]
- Fageria, V.D. Nutrient interactions in crop plants. J. Plant Nutr. 2001, 24, 1269–1290. [Google Scholar] [CrossRef]
- Galloway, J.N.; Dentener, F.J.; Capone, D.G.; Boyer, E.W.; Howarth, R.W.; Seitzinger, S.P.; Karl, D.M. Nitrogen cycles: Past, present, and future. Biogeochemistry 2004, 70, 153–226. [Google Scholar] [CrossRef]
- Bernhard, A. The nitrogen cycle: Processes, players, and human impact. Nat. Educ. Knowl. 2010, 3, 25–33. [Google Scholar]
- Vitousek, P.M.; Howarth, R.W. Nitrogen limitation on land and in the sea: How can it occur? Biogeochemistry 1991, 13, 87–115. [Google Scholar] [CrossRef]
- Xia, J.; Wan, S. Global response patterns of terrestrial plant species to nitrogen addition. New Phytol. 2008, 179, 428–439. [Google Scholar] [CrossRef]
- Böhme, L.; Langer, U.; Böhme, F. Microbial biomass, enzyme activities and microbial community structure in two European long-term field experiments. Agric. Ecosyst. Environ. 2005, 109, 141–152. [Google Scholar] [CrossRef]
- Chu, H.; Lin, X.; Fujii, T.; Morimoto, S.; Yagi, K.; Hu, J.; Zhang, J. Soil microbial biomass, dehydrogenase activity, bacterial community structure in response to long-term fertilizer management. Soil Biol. Biochem. 2007, 39, 2971–2976. [Google Scholar] [CrossRef]
- Gu, Y.; Zhang, X.; Tu, S.; Lindström, K. Soil microbial biomass, crop yields, and bacterial community structure as affected by long-term fertilizer treatments under wheat-rice cropping. Eur. J. Soil Biol. 2009, 45, 239–246. [Google Scholar] [CrossRef]
- Chu, H.; Fujii, T.; Morimoto, S.; Lin, X.; Yagi, K. Population size and specific nitrification potential of soil ammonia-oxidizing bacteria under long-term fertilizer management. Soil Biol. Biochem. 2008, 40, 1960–1963. [Google Scholar] [CrossRef]
- Enwall, K.; Nyberg, K.; Bertilsson, S.; Cederlund, H.; Stenström, J.; Hallin, S. Long-term impact of fertilization on activity and composition of bacterial communities and metabolic guilds in agricultural soil. Soil Biol. Biochem. 2007, 39, 106–115. [Google Scholar] [CrossRef]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkilä, J.; Marchesi, J.R.; Smidt, H.; O’Toole, P.W. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE 2009, 4, e6669. [Google Scholar] [CrossRef] [Green Version]
- Engelbrektson, A.; Kunin, V.; Wrighton, K.C.; Zvenigorodsky, N.; Chen, F.; Ochman, H.; Hugenholtz, P. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010, 4, 642–647. [Google Scholar] [CrossRef]
- Ambika Manirajan, B.; Ratering, S.; Rusch, V.; Schwiertz, A.; Geissler-Plaum, R.; Cardinale, M.; Schnell, S. Bacterial microbiota associated with flower pollen is influenced by pollination type, and shows a high degree of diversity and species-specificity. Environ. Microbiol. 2016, 18, 5161–5174. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArdle, B.H.; Anderson, M.J. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Volterra, V. Variazioni e fluttuazioni del numero d’individui in specie animali conviventi. Mem. R. Accad. Naz. dei Lincei. 1926, 6, 31–113. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Konopka, A.; Lindemann, S.; Fredrickson, J. Dynamics in microbial communities: Unraveling mechanisms to identify principles. ISME J. 2015, 9, 1488–1495. [Google Scholar] [CrossRef]
- Bak, F.; Scheff, G.; Jansen, K.H. A rapid and sensitive ion chromatographic technique for the determination of sulfate and sulfate reduction rates in freshwater lake sediments. FEMS Microbiol. Lett. 1991, 85, 23–30. [Google Scholar] [CrossRef]
- Kandeler, E.; Gerber, H. Short-term assay of soil urease activity using colorimetric determination of ammonium. Biol. Fertil. Soils 1988, 6, 68–72. [Google Scholar] [CrossRef]
- Kampmann, K.; Ratering, S.; Kramer, I.; Schmidt, M.; Zerr, W.; Schnell, S. Unexpected stability of Bacteroidetes and Firmicutes communities in laboratory biogas reactors fed with different defined substrates. Appl. Environ. Microbiol. 2012, 78, 2106–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.; Diouf, D.; Kane, A.; Moulin, L. Influence of plant genotype and soil on the wheat rhizosphere microbiome: Identification of a core microbiome across eight African and European soils. bioRxiv 2019, 777383. [Google Scholar] [CrossRef] [PubMed]
- Neumann, D.; Heuer, A.; Hemkemeyer, M.; Martens, R.; Tebbe, C.C. Response of microbial communities to long-term fertilization depends on their microhabitat. FEMS Microbiol. Ecol. 2013, 86, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Baudoin, E.; Benizri, E.; Guckert, A. Impact of artificial root exudates on the bacterial community structure in bulk soil and maize rhizosphere. Soil Biol. Biochem. 2003, 35, 1183–1192. [Google Scholar] [CrossRef]
- Marschner, P.; Yang, C.H.; Lieberei, R.; Crowley, D.E. Soil and plant specific effects on bacterial community composition in the rhizosphere. Soil Biol. Biochem. 2001, 33, 1437–1445. [Google Scholar] [CrossRef]
- Ward, N.L.; Challacombe, J.F.; Janssen, P.H.; Henrissat, B.; Coutinho, P.M.; Wu, M.; Barabote, R.D. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl. Environ. Microbiol. 2009, 75, 2046–2056. [Google Scholar] [CrossRef] [Green Version]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The ecology of Acidobacteria: Moving beyond genes and genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Kim, H.; Yoon, S. Nitrous oxide reduction by an obligate aerobic bacterium, Gemmatimonas aurantiaca strain T-27. Appl. Environ. Microbiol. 2017, 83, e00502-17. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, H.; Ratering, S.; Felix-Henningsen, P.; Schnell, S. Stability of in situ immobilization of trace metals with different amendments revealed by microbial 13C-labelled wheat root decomposition and efflux-mediated metal resistance of soil bacteria. Sci. Total Environ. 2019, 659, 1082–1089. [Google Scholar] [CrossRef]
- Semenov, A.V.; e Silva, M.C.P.; Szturc-Koestsier, A.E.; Schmitt, H.; Salles, J.F.; van Elsas, J.D. Impact of incorporated fresh 13C potato tissues on the bacterial and fungal community composition of soil. Soil Biol. Biochem. 2012, 49, 88–95. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.J.; Howe, A.; Hofmockel, K.S. Demonstrating microbial co-occurrence pattern analyses within and between ecosystems. Front. Microbiol. 2014, 5, 358. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, M.; Grube, M.; Erlacher, A.; Quehenberger, J.; Berg, G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ. Microbiol. 2015, 17, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.A.; Schröder, P.; Armbruster, M.; Schloter, M. Organic amendments in a long-term field trial—Consequences for the bulk soil bacterial community as revealed by network analysis. Microb. Ecol. 2018, 76, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.X.; Haudenshield, J.S.; Bowen, C.R.; Hartman, G.L. Metagenome-wide association study and machine learning prediction of bulk soil microbiome and crop productivity. Front. Microbiol. 2017, 8, 519. [Google Scholar] [CrossRef]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef] [Green Version]
- Leininger, S.; Urich, T.; Schloter, M.; Schwark, L.; Qi, J.; Nicol, G.W.; Schleper, C. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 2006, 442, 806–809. [Google Scholar] [CrossRef]
- Jones, C.M.; Graf, D.R.; Bru, D.; Philippot, L.; Hallin, S. The unaccounted yet abundant nitrous oxide-reducing microbial community: A potential nitrous oxide sink. ISME J. 2013, 7, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, L.H.; Rodriguez-R, L.M.; Higgins, S.; Chee-Sanford, J.C.; Sanford, R.A.; Ritalahti, K.M.; Konstantinidis, K.T. Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: Method development and implications for the nitrogen cycle. MBio 2014, 5, e01193-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Soil habitat | Fertilization Treatment | Shannon Index (Diversity) | N. of OTUs (Richness) |
|---|---|---|---|
| Bulk soil | N | 7.92 ± 0.07 BC | 767.0 ± 21.7 BCD |
| M | 7.80 ± 0.06 C | 730.3 ± 17.1 D | |
| NM | 7.93 ± 0.07 BC | 768.5 ± 15.5 BCD | |
| 0 | 7.87 ± 0.07 C | 746.8 ± 19.3 CD | |
| Rhizosphere | N | 8.23 ± 0.02 A | 824.3 ± 3.83 ABC |
| M | 8.33 ± 0.06 AB | 861.5 ± 16.6 A | |
| NM | 8.34 ± 0.06 A | 850.8 ± 14.7 AB | |
| 0 | 8.42 ± 0.06 A | 869.7 ± 21.8 A |
| Soil Habitat | Alpha-Diversity (ANOVA) | Beta-Diversity (Adonis) | N. of OTUs Affected by Treatment (% of Total Reads) 1 | |||
|---|---|---|---|---|---|---|
| Shannon | Phil. Div. | Richness | Bray–Curtis | Weighted Unifrac | ||
| Rhizosphere | 0.038 | 0.366 | 0.213 | 0.025 | 0.035 | 1 (0.55 %) |
| Bulk | 0.536 | 0.200 | 0.377 | 0.030 | 0.020 | 66 (33.1 %) |
| Network Parameter | Rhizosphere Cluster | Bulk Soil Cluster |
|---|---|---|
| N. of nodes | 33 | 65 |
| Avg. N. of neighbors | 4.12 | 6.46 |
| Clustering coefficient | 0.396 | 0.389 |
| Network centralization | 0.329 | 0.299 |
| Network density | 0.129 | 0.101 |
| Soil Habitat | Fertilization Treatment | Nitrate µmol NO3 * g soildw−1 | Ammonium µg NH4 –N * g soildw−1 |
|---|---|---|---|
| Bulk soil | N | 71.2 ± 24.9 AB | 8.38 ± 2.69 A |
| M | 30.6 ± 4.1 B | 0.90 ± 0.06 B | |
| NM | 66.4 ± 22.4 AB | 2.82 ± 0.60 AB | |
| 0 | 41.6 ± 8.8 B | 0.95 ± 0.08 B | |
| Rhizosphere | N | 107.6 ± 22.2 AB | 2.31 ± 0.58 AB |
| M | 66.5 ± 7.4 AB | 0.94 ± 0.05 B | |
| NM | 136.7 ± 15.1 A | 1.95 ± 0.44 AB | |
| 0 | 29.7 ± 5.7 B | 0.67 ± 0.05 B |
| Gene | N. of Positively Correlated OTUs | N. of Negatively Correlated OTUs |
|---|---|---|
| amoA | 0 | 1 |
| nirK | 0 | 11 |
| nosZ-I | 0 | 4 |
| nosZ-II | 0 | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardinale, M.; Ratering, S.; Sadeghi, A.; Pokhrel, S.; Honermeier, B.; Schnell, S. The Response of the Soil Microbiota to Long-Term Mineral and Organic Nitrogen Fertilization is Stronger in the Bulk Soil than in the Rhizosphere. Genes 2020, 11, 456. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040456
Cardinale M, Ratering S, Sadeghi A, Pokhrel S, Honermeier B, Schnell S. The Response of the Soil Microbiota to Long-Term Mineral and Organic Nitrogen Fertilization is Stronger in the Bulk Soil than in the Rhizosphere. Genes. 2020; 11(4):456. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040456
Chicago/Turabian StyleCardinale, Massimiliano, Stefan Ratering, Aitak Sadeghi, Sushil Pokhrel, Bernd Honermeier, and Sylvia Schnell. 2020. "The Response of the Soil Microbiota to Long-Term Mineral and Organic Nitrogen Fertilization is Stronger in the Bulk Soil than in the Rhizosphere" Genes 11, no. 4: 456. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11040456