Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option?

 and
and
Abstract
:1. Introduction
2. Bacterial Antiviral Defence
3. Classification of CRISPR/Cas Systems
4. Components and Mechanism of CRISPR/Cas9 System
5. DSB Repair Mechanisms
6. Applications of CRISPR/Cas9 System
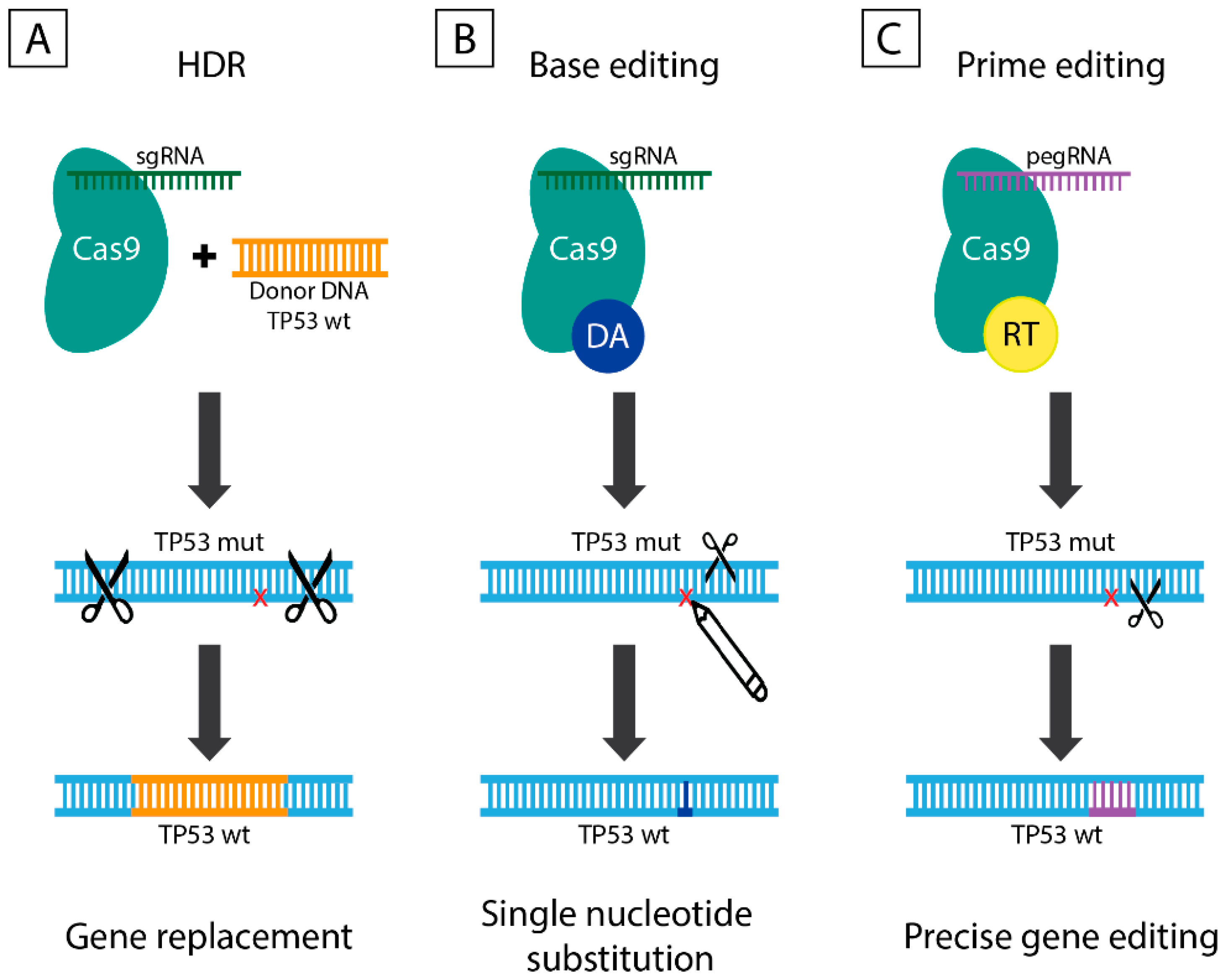
6.1. CRISPR/Cas9-Mediated DSB Repair Mechanisms
6.2. Base Editing
6.3. Prime Editing
7. CRISPR/Cas9 System for Gene Regulation
7.1. Transcriptional Activation and Repression
7.2. Epigenetic Regulation
8. Clinical Trials of CRISPR/Cas9 Therapeutic Genome Editing
9. CRISPR/Cas9 System and TP53 Pathway Genes
9.1. p53 Pathway Affects the Efficiency of CRISPR/Cas9 Gene Editing
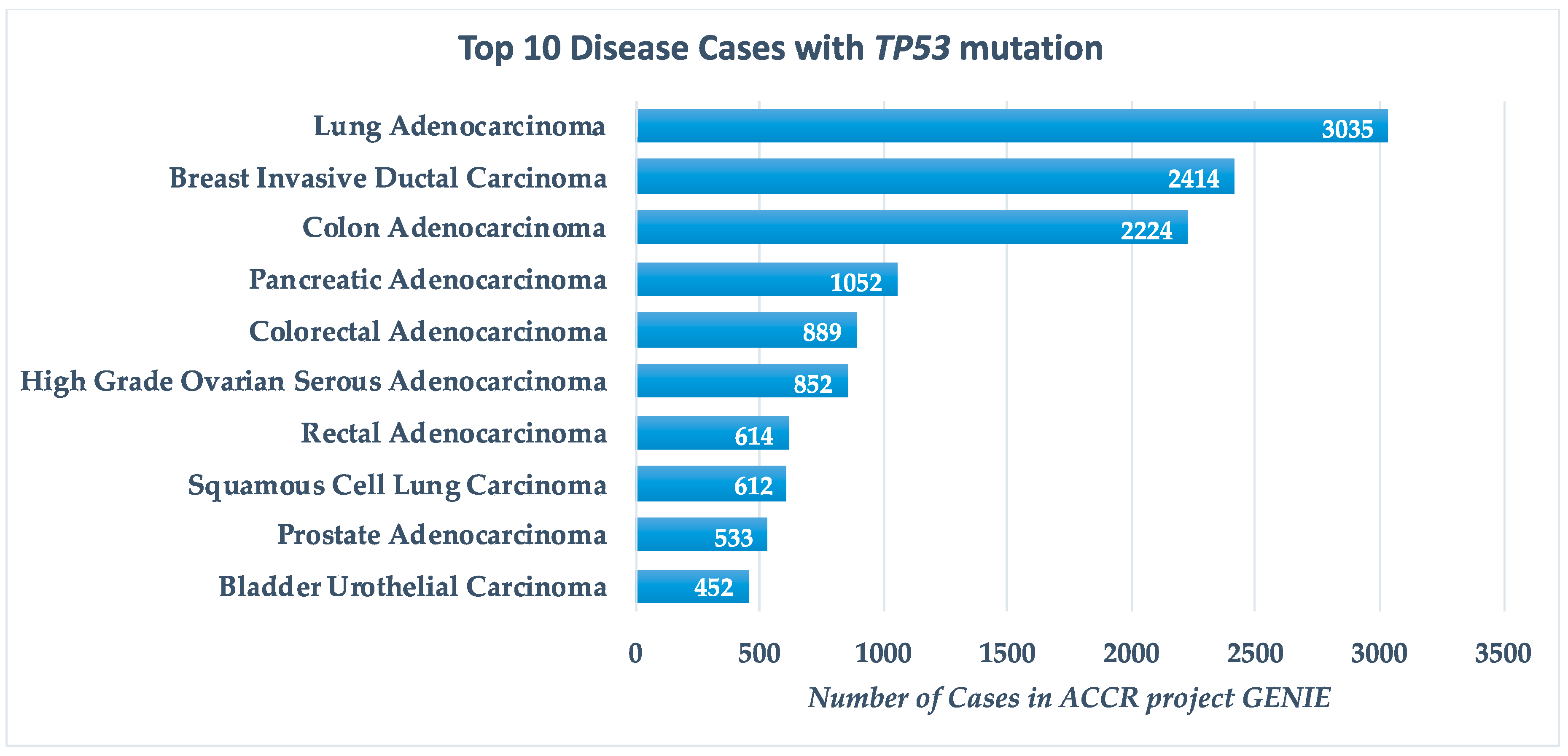
9.2. Perspectives of Applying CRISPR/Cas9 System for TP53 Editing and Regulation of p53-Pathway Genes
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- My Cancer Genome. Available online: https://www.mycancergenome.org/content/alteration/tp53-mutation/ (accessed on 9 June 2020).
- IARC TP53 Database. Available online: https://p53.iarc.fr/TP53SomaticMutations.aspx (accessed on 9 June 2020).
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-Awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulatov, E.; Sayarova, R.; Mingaleeva, R.; Miftakhova, R.; Gomzikova, M.; Ignatyev, Y.; Petukhov, A.; Davidovich, P.; Rizvanov, A.; Barlev, N.A. Isatin-schiff base-copper (II) complex induces cell death in p53-positive tumors. Cell Death Discov. 2018, 4, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulatov, E.; Valiullina, A.; Sayarova, R.; Rizvanov, A. Promising new therapeutic targets for regulation of inflammation and immunity: RING-type E3 ubiquitin ligases. Immunol. Lett. 2018, 202, 44–51. [Google Scholar] [CrossRef]
- Bulatov, E.; Zagidullin, A.; Valiullina, A.; Sayarova, R.; Rizvanov, A. Small molecule modulators of RING-type E3 ligases: MDM and cullin families as targets. Front. Pharmacol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bulatov, E.; Khaiboullina, S.; dos Reis, H.J.; Palotás, A.; Venkataraman, K.; Vijayalakshmi, M.; Rizvanov, A. Ubiquitin-Proteasome System: Promising Therapeutic Targets in Autoimmune and Neurodegenerative Diseases. Bionanoscience 2016, 6, 341–344. [Google Scholar] [CrossRef]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR—Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 22, 505–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; Van Der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef] [Green Version]
- Merienne, N.; Vachey, G.; de Longprez, L.; Meunier, C.; Zimmer, V.; Perriard, G.; Canales, M.; Mathias, A.; Herrgott, L.; Beltraminelli, T.; et al. The Self-Inactivating KamiCas9 System for the Editing of CNS Disease Genes. Cell Rep. 2017, 20, 2980–2991. [Google Scholar] [CrossRef] [Green Version]
- TP53 Gene. Available online: https://ghr.nlm.nih.gov/gene/TP53#sourcesforpage (accessed on 27 May 2020).
- Mojica, F.J.M.; Rodriguez-Valera, F. The discovery of CRISPR in archaea and bacteria. FEBS J. 2016, 283, 3162–3169. [Google Scholar] [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Dusko Ehrlich, S. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Makarova, K.S. Origins and evolution of CRISPR-Cas systems. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180087. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; Van Der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.E.; Dupuis, M.È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Redding, S.; Sternberg, S.H.; Marshall, M.; Gibb, B.; Bhat, P.; Guegler, C.K.; Wiedenheft, B.; Doudna, J.A.; Greene, E.C. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell 2015, 163, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Choi, J.; Bailey, S. Cut site selection by the two nuclease domains of the Cas9 RNA-guided endonuclease. J. Biol. Chem. 2014, 89, 13284–13294. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Tu, L.C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J. Cell Biol. 2016, 214, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.M.; Hur, J.W.; Kim, K. Evolution of CRISPR towards accurate and efficient mammal genome engineering. BMB Rep. 2019, 52, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The mechanism of DSB repair by the NHEJ. Annu. Rev. Biochem. 2011, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of double-strand breaks by end joining. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–21. [Google Scholar] [CrossRef]
- Saleh-Gohari, N.; Helleday, T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle un human cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef] [PubMed]
- Mcvey, M.; Lee, S.E.; Avenue, H.; Antonio, S. MMEJ repair of double-strand breaks: Deleted sequences and alternative endings. Trends Genet. 2017, 24, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.; Guschin, D.Y.; et al. Editing Using Zinc-Finger Nucleases. Nat. Biotechnol. 2012, 26, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Sheridan, C. Gene editing enters ’prime ’ time. Nat. Biotechnol. 2019, 40, BSR20200127. [Google Scholar] [CrossRef]
- Xu, X.; Qi, L.S. A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. XCRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Silverstein, N.J.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.C.; Gilbert, L.A. The Promise and Challenge of in Vivo Delivery for Genome Therapeutics. ACS Chem. Biol. 2018, 13, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Di Leonardo, A.; Linke, S.P.; Clarkin, K.; Wahl, G.M. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994, 8, 2540–2551. [Google Scholar] [CrossRef] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. P53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Guerra, K.B.; Cheng, K.; Leiserson, M.D.; Wilson, D.M.; Ryan, B.M.; Ze’ev, A.R.; Lee, J.S.; Deshpande, A.J.; Ruppin, E. Integrated computational and experimental identification of p53, KRAS and VHL mutant selection associated with CRISPR-Cas9 editing. bioRxiv 2019, 407767. [Google Scholar] [CrossRef]
- Bowden, A.R.; Juarez, D.A.M.; Sczaniecka-Clift, M.; Agudo, M.M.; Lukashchuk, N.; Thomas, J.C.; Jackson, S.P. Parallel CRISPR-Cas9 screens clarify impacts of p53 on screen performance. bioRxiv 2020, 9, e55325. [Google Scholar] [CrossRef] [PubMed]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, G.; Conti, A.; Ferrari, S.; Della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell 2019, 24, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do mutations turn p53 into an oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chira, S.; Gulei, D.; Hajitou, A.; Berindan-Neagoe, I. Restoring the p53 ’Guardian’ Phenotype in p53-Deficient Tumor Cells with CRISPR/Cas9. Trends Biotechnol. 2018, 36, 653–660. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, R.; Palange, N.J.; Satheka, A.C.; Togo, J.; An, Y.; Humphrey, M.; Ban, L.; Ji, Y.; Jin, H.; et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shao, Y.; Guan, Y.; Li, L.; Wu, L.; Chen, F.; Liu, M.; Chen, H.; Ma, Y.; Ma, X.; et al. Large genomic fragment deletion and functional gene cassette knock-in via Cas9 protein mediated genome editing in one-cell rodent embryos. Sci. Rep. 2015, 5, 17517. [Google Scholar] [CrossRef] [Green Version]
- Zhan, H.; Xie, H.; Zhou, Q.; Liu, Y.; Huang, W. Synthesizing a Genetic Sensor Based on CRISPR-Cas9 for Specifically Killing p53-Deficient Cancer Cells. ACS Synth. Biol. 2018, 7, 1798–1807. [Google Scholar] [CrossRef]
- Niu, X.; Deng, K.; Liu, L.; Yang, K.; Hu, X. A statistical framework for predicting critical regions of p53-dependent enhancers. Brief. Bioinform. 2020, bbaa053. [Google Scholar] [CrossRef]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanis-Lobato, G.; Zohren, J.; McCarthy, A.; Fogarty, N.M.; Kubikova, N.; Hardman, E.; Greco, M.; Wells, D.; Turner, J.M.; Niakan, K. Frequent loss-of-heterozygosity in CRISPR-Cas9-edited early human embryos. bioRxiv 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effector Module | Pre-crRNA Processing | Interference Stage (Target Cleavage) | ||
|---|---|---|---|---|
| Class 1 | Type I | Multi-subunit effector complex | Cas6 | Cas3 |
| Type III | Subunit of the processing complex | |||
| Type IV | Uncharacterized | |||
| Class 2 | Type II | Single multi-domain protein | RNase III | Nuclease domain of the effector protein |
| Type V | Nuclease domain of the effector protein | |||
| Type VI |
| Disease | Clinical Trial Number | Therapy | Study Start Date | Comment |
|---|---|---|---|---|
| Sickle cell disease | NCT03745287 | CTX001 (autologous CD34+ hHSPCs with CRISPR/Cas9-modified BCL11A gene) | November 2018 | Active |
| β-Thalassemia | NCT03655678 | CTX001 (autologous CD34+ hHSPCs with CRISPR/Cas9-modified BCL11A gene) | September 2018 | Active |
| β-Thalassemia | NCT03728322 | iHSCs with CRISPR/Cas9-modified HBB gene | January 2019 | Active |
| B cell leukaemia, B cell lymphoma | NCT03166878 | UCART019 (universal CRISPR/Cas9-edited anti-CD19 CAR-T cells) | June 2017 | Active |
| Prostate cancer | NCT02867345 | PD-1 knockout T cells | November 2016 | Withdrawn |
| Renal cell carcinoma | NCT02867332 | PD-1 knockout T cells | November 2016 | Withdrawn |
| Epstein–Barr virus (EBV) associated malignancies | NCT03044743 | PD-1 knockout T cells | April 2017 | Active |
| B cell malignancies | NCT04035434 | CTX110 (CRISPR/Cas9-edited T cells) | July 2019 | Active |
| HIV-1 | NCT03164135 | CRISPR/Cas9 CCR5 gene modified CD34+ hematopoietic stem/progenitor cells | May 2017 | Active |
| Multiple myeloma, melanoma, synovial sarcoma, myxoid/round cell liposarcoma | NCT03399448 | NY-ESO-1 redirected autologous T cells with CRISPR/Cas9-edited endogenous TCR and PD-1 | September 2018 | Terminated |
| Leber congenital amaurosis 10 | NCT03872479 | AGN-151587 (correction of c.2991+1655A>G mutation in CEP290 gene) | September 2019 | Not recruiting, active |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060704
Mirgayazova R, Khadiullina R, Chasov V, Mingaleeva R, Miftakhova R, Rizvanov A, Bulatov E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes. 2020; 11(6):704. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060704
Chicago/Turabian StyleMirgayazova, Regina, Raniya Khadiullina, Vitaly Chasov, Rimma Mingaleeva, Regina Miftakhova, Albert Rizvanov, and Emil Bulatov. 2020. "Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option?" Genes 11, no. 6: 704. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11060704