Of Drugs and Trypanosomatids: New Tools and Knowledge to Reduce Bottlenecks in Drug Discovery

 , and
, and 
Abstract
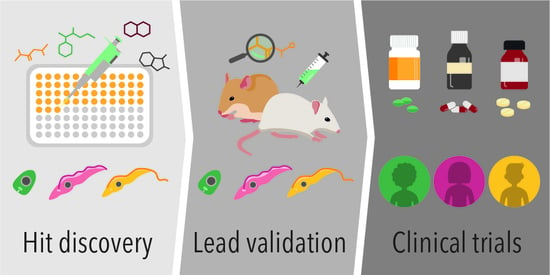
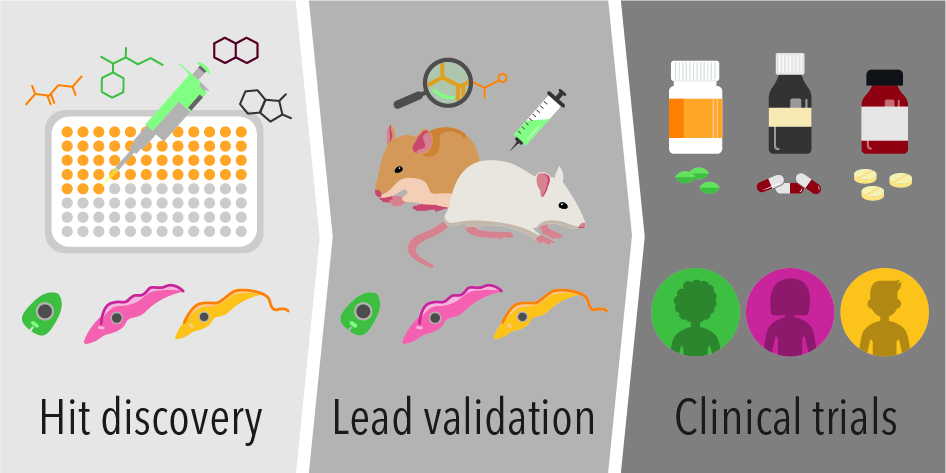
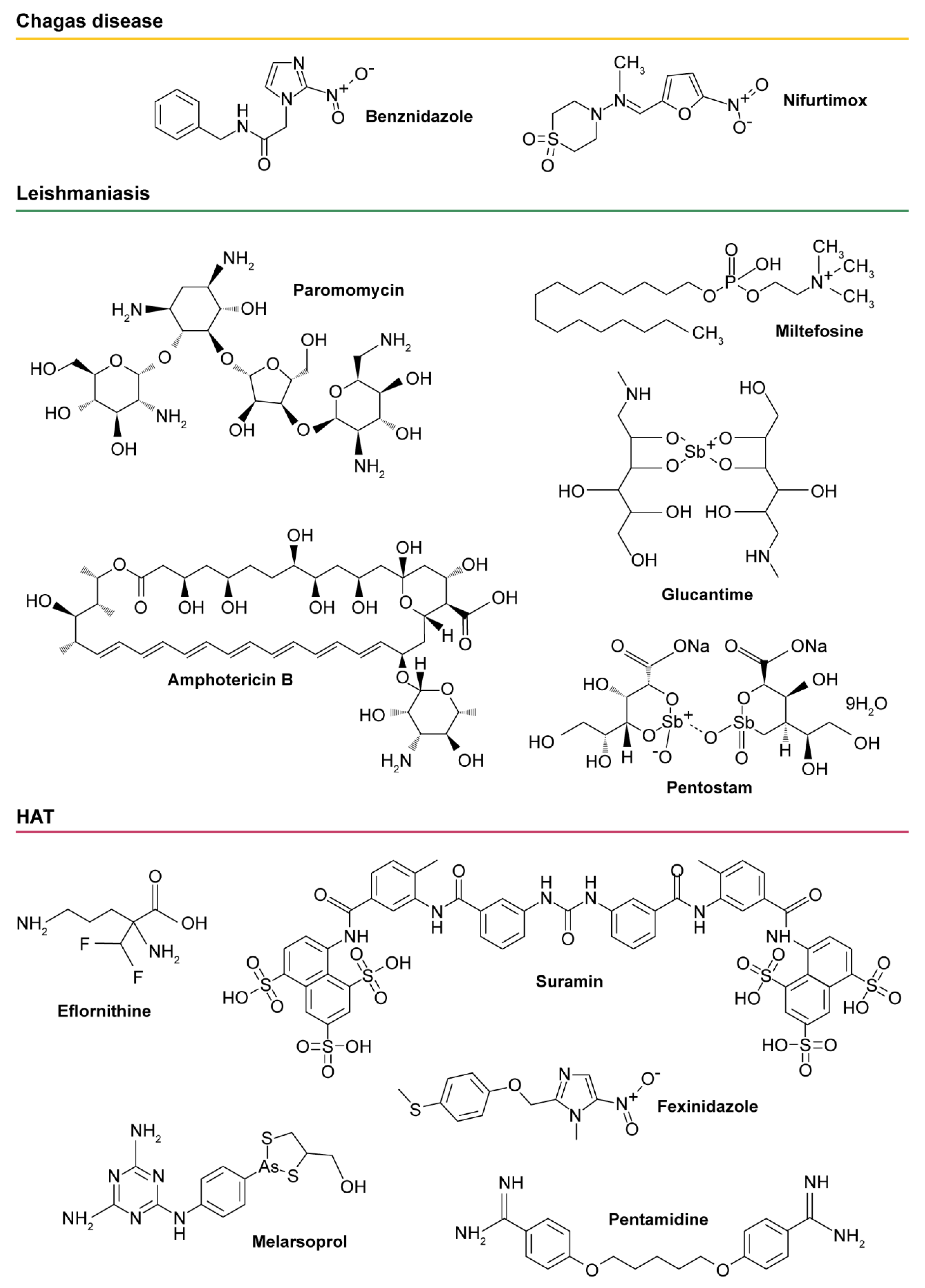
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Status and Impact of Trypanosomatid-Borne Infections
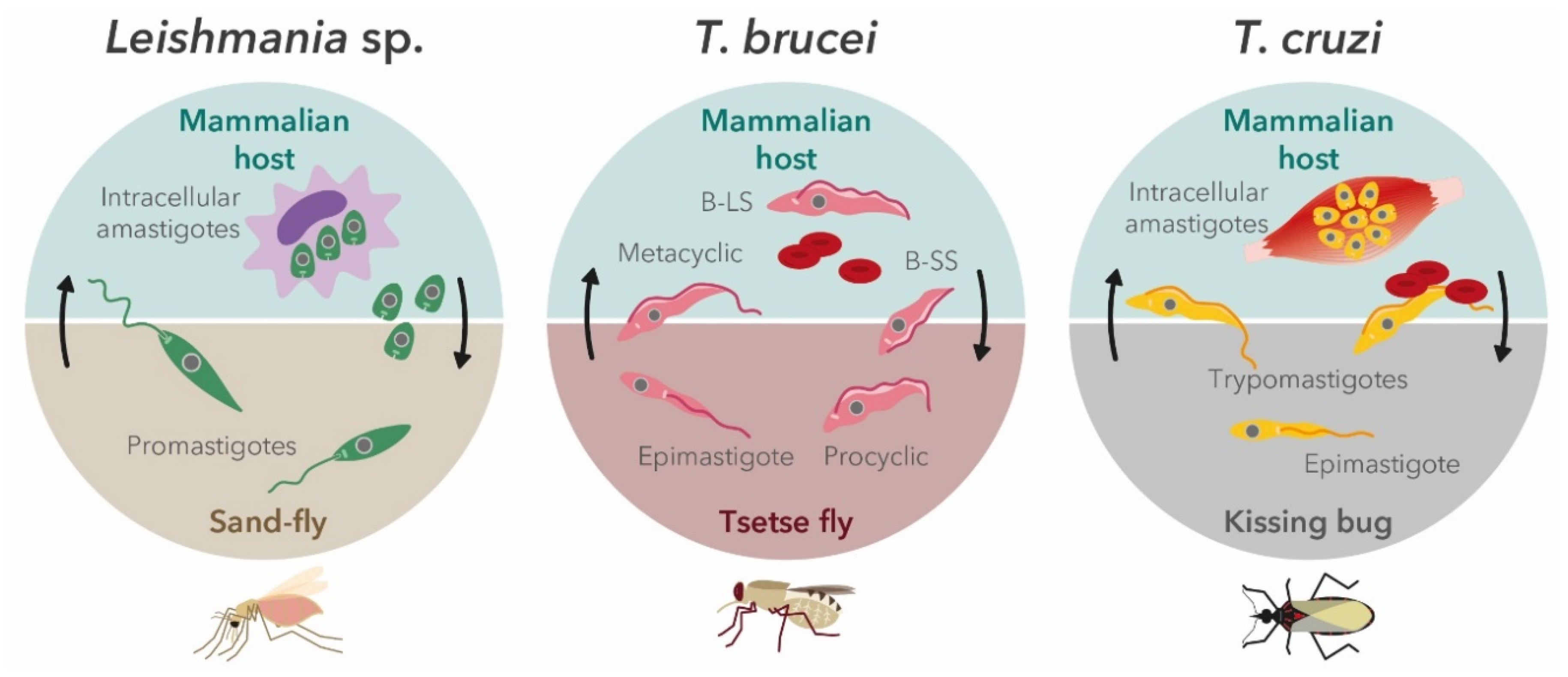
2. Trypanosomatids′ Life Cycle in the Context of In Vitro Screening Assays
2.1. Leishmania Parasites
2.2. Trypanosoma brucei
2.3. Trypanosoma cruzi
3. Animal Models in Drug Discovery and Development against Trypanosomatids
4. Cheminformatics in Drug Discovery
5. Quiescence, a Double-Edged Sword in the Quest of New Trypanocidal Drugs
6. Cytology-Driven MoA Profiling
7. Genome-Wide Approaches in Target and Resistance (Resistomics)
8. Metabolomics in Drug Screening
9. Theranostic Approaches
10. Case Study: Proteasomal Inhibitors against Leishmania
11. Perspectives and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fernandez-Prada, C.; Minguez-Menendez, A.; Pena, J.; Tunes, L.G.; Pires, D.E.V.; Monte-Neto, R. Repurposed molecules: A new hope in tackling neglected infectious diseases. In Silico Drug Design 1st Edition: Repurposing Techniques and Methodologies; Roy, K., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 119–160. [Google Scholar]
- Arce, A.; Estirado, A.; Ordobas, M.; Sevilla, S.; Garcia, N.; Moratilla, L.; de la Fuente, S.; Martinez, A.M.; Perez, A.M.; Aranguez, E.; et al. Re-emergence of leishmaniasis in Spain: Community outbreak in Madrid, Spain, 2009 to 2012. Eurosurveillance 2013, 18, 20546. [Google Scholar] [CrossRef] [Green Version]
- Uranw, S.; Hasker, E.; Roy, L.; Meheus, F.; Das, M.L.; Bhattarai, N.R.; Rijal, S.; Boelaert, M. An outbreak investigation of visceral leishmaniasis among residents of Dharan town, eastern Nepal, evidence for urban transmission of Leishmania donovani. BMC Infect. Dis. 2013, 13, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubakar, A.; Ruiz-Postigo, J.A.; Pita, J.; Lado, M.; Ben-Ismail, R.; Argaw, D.; Alvar, J. Visceral leishmaniasis outbreak in South Sudan 2009-2012: Epidemiological assessment and impact of a multisectoral response. PLoS Negl. Trop Dis. 2014, 8, e2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babuadze, G.; Alvar, J.; Argaw, D.; de Koning, H.P.; Iosava, M.; Kekelidze, M.; Tsertsvadze, N.; Tsereteli, D.; Chakhunashvili, G.; Mamatsashvili, T.; et al. Epidemiology of visceral leishmaniasis in Georgia. PLoS Negl. Trop Dis. 2014, 8, e2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas disease: From discovery to a worldwide health problem. Front Pub. Health 2019, 7, 166. [Google Scholar] [CrossRef]
- Lee, B.Y.; Bacon, K.M.; Bottazzi, M.E.; Hotez, P.J. Global economic burden of Chagas disease: A computational simulation model. Lancet Infect. Dis. 2013, 13, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, V.; Dias, N.; Paiva, T.; Hagstrom-Bex, L.; Nitz, N.; Pratesi, R.; Hecht, M. Current trends in the pharmacological management of Chagas disease. Int. J. Parasitol. Drugs Drug Resist. 2019, 12, 7–17. [Google Scholar] [CrossRef]
- Bern, C.; Montgomery, S.P.; Herwaldt, B.L.; Rassi, A.; Marin-Neto, J.A.; Dantas, R.O.; Maguire, J.H.; Acquatella, H.; Morillo, C.; Kirchhoff, L.V.; et al. Evaluation and treatment of Chagas disease in the united states a systematic review. JAMA 2007, 298, 2171–2181. [Google Scholar] [CrossRef] [Green Version]
- Meymandi, S.; Hernandez, S.; Park, S.; Sanchez, D.R.; Forsyth, C. Treatment of Chagas disease in the United States. Curr. Treat Options Infect. Dis. 2018, 10, 373–388. [Google Scholar] [CrossRef] [Green Version]
- Viotti, R.; Alarcon de Noya, B.; Araujo-Jorge, T.; Grijalva, M.J.; Guhl, F.; Lopez, M.C.; Ramsey, J.M.; Ribeiro, I.; Schijman, A.G.; Sosa-Estani, S.; et al. Latin American network for Chagas disease, NHEPACHA. Towards a paradigm shift in the treatment of chronic Chagas disease. Antimicrob. Agents Chemother. 2014, 58, 635–639. [Google Scholar] [CrossRef] [Green Version]
- Alsford, S.; Kelly, J.M.; Baker, N.; Horn, D. Genetic dissection of drug resistance in trypanosomes. Parasitology 2013, 140, 1478–1491. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.R.; Kelly, J.M. Trypanocidal drugs: Mechanisms, resistance and new targets. Expert Rev. Mol. Med. 2009, 11, e31. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Hotez, P.J. Global urbanization and the neglected tropical diseases. PLoS Negl. Trop Dis. 2017, 11, e0005308. [Google Scholar] [CrossRef] [PubMed]
- Booth, M. Climate change and the neglected tropical diseases. Adv. Parasitol. 2018, 100, 39–126. [Google Scholar]
- Robertson, S.A.; Renslo, A.R. Drug discovery for neglected tropical diseases at the Sandler Center. Future Med. Chem. 2011, 3, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Berenstein, A.J.; Magarinos, M.P.; Chernomoretz, A.; Aguero, F. A multilayer network approach for guiding drug repositioning in neglected diseases. PLoS Negl. Trop Dis. 2016, 10, e0004300. [Google Scholar] [CrossRef] [Green Version]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.R.; Sullivan, D.J.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharm. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br. J. Pharm. 2018, 175, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, A.; Ellis, J.; Stark, D.; Barratt, J. The evolution of trypanosomatid taxonomy. Parasit. Vectors 2017, 10, 287. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukes, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votypka, J.; Yurchenko, V. Trypanosomatids are much more than just trypanosomes: Clues from the expanded family tree. Trends Parasitol. 2018, 34, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Goyal, N.; Rastogi, A.K. In vitro cultivation and characterization of axenic amastigotes of Leishmania. Trends Parasitol. 2001, 17, 150–153. [Google Scholar] [CrossRef]
- Moreira, E.S.; Soares, R.M.; Petrillo-Peixoto Mde, L. Glucantime susceptibility of Leishmania promastigotes under variable growth conditions. Parasitol. Res. 1995, 81, 291–295. [Google Scholar]
- De Rycker, M.; Hallyburton, I.; Thomas, J.; Campbell, L.; Wyllie, S.; Joshi, D.; Cameron, S.; Gilbert, I.H.; Wyatt, P.G.; Frearson, J.A.; et al. Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrob. Agents Chemother. 2013, 57, 2913–2922. [Google Scholar] [CrossRef] [Green Version]
- Vermeersch, M.; da Luz, R.I.; Tote, K.; Timmermans, J.P.; Cos, P.; Maes, L. In vitro susceptibilities of Leishmania donovani promastigote and amastigote stages to antileishmanial reference drugs: Practical relevance of stage-specific differences. Antimicrob. Agents Chemother. 2009, 53, 3855–3859. [Google Scholar] [CrossRef] [Green Version]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- Brochu, C.; Wang, J.; Roy, G.; Messier, N.; Wang, X.Y.; Saravia, N.G.; Ouellette, M. Antimony uptake systems in the protozoan parasite Leishmania and accumulation differences in antimony-resistant parasites. Antimicrob. Agents Chemother. 2003, 47, 3073–3079. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Prada, C.; Vincent, I.M.; Brotherton, M.C.; Roberts, M.; Roy, G.; Rivas, L.; Leprohon, P.; Smith, T.K.; Ouellette, M. Different mutations in a P-type ATPase transporter in Leishmania parasites are associated with cross-resistance to two leading drugs by distinct mechanisms. PLoS Negl. Trop Dis. 2016, 10, e0005171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zhao, Y.; Ni, B.; Yao, C.; Zhou, Y.; Xu, W.; Wang, Z.; Qiao, Z. Comparison of the expression profiles of promastigotes and axenic amastigotes in Leishmania donovani using serial analysis of gene expression. Parasitol. Res. 2008, 103, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Shadab, M.; Das, S.; Banerjee, A.; Sinha, R.; Asad, M.; Kamran, M.; Maji, M.; Jha, B.; Deepthi, M.; Kumar, M.; et al. RNA-Seq Revealed expression of many novel genes associated with Leishmania donovani persistence and clearance in the host macrophage. Front Cell Infect. Microbiol. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Alvarez, E.; Alvarez-Velilla, R.; Fernandez-Prada, C.; Balana-Fouce, R.; Reguera, R.M. Trypanosomatids see the light: Recent advances in bioimaging research. Drug Discov. Today 2015, 20, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tegazzini, D.; Diaz, R.; Aguilar, F.; Pena, I.; Presa, J.L.; Yardley, V.; Martin, J.J.; Coteron, J.M.; Croft, S.L.; Cantizani, J. A replicative in vitro assay for drug discovery against Leishmania donovani. Antimicrob. Agents Chemother. 2016, 60, 3524–3532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada, C.F.; Alvarez-Velilla, R.; Balana-Fouce, R.; Prieto, C.; Calvo-Alvarez, E.; Escudero-Martinez, J.M.; Requena, J.M.; Ordonez, C.; Desideri, A.; Perez-Pertejo, Y.; et al. Gimatecan and other camptothecin derivatives poison Leishmania DNA-topoisomerase IB leading to a strong leishmanicidal effect. Biochem. Pharm. 2013, 85, 1433–1440. [Google Scholar] [CrossRef] [Green Version]
- Balana-Fouce, R.; Prada, C.F.; Requena, J.M.; Cushman, M.; Pommier, Y.; Alvarez-Velilla, R.; Escudero-Martinez, J.M.; Calvo-Alvarez, E.; Perez-Pertejo, Y.; Reguera, R.M. Indotecan (LMP400) and AM13-55: Two novel indenoisoquinolines show potential for treating visceral leishmaniasis. Antimicrob. Agents Chemother. 2012, 56, 5264–5270. [Google Scholar] [CrossRef] [Green Version]
- Seifert, K.; Escobar, P.; Croft, S.L. In vitro activity of anti-leishmanial drugs against Leishmania donovani is host cell dependent. J. Antimicrob. Chemother. 2010, 65, 508–511. [Google Scholar] [CrossRef] [Green Version]
- Ginouves, M.; Simon, S.; Nacher, M.; Demar, M.; Carme, B.; Couppie, P.; Prevot, G. In vitro sensitivity of cutaneous Leishmania promastigote isolates circulating in French Guiana to a set of drugs. Am. J. Trop. Med. Hyg. 2017, 96, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deep, D.K.; Singh, R.; Bhandari, V.; Verma, A.; Sharma, V.; Wajid, S.; Sundar, S.; Ramesh, V.; Dujardin, J.C.; Salotra, P. Increased miltefosine tolerance in clinical isolates of Leishmania donovani is associated with reduced drug accumulation, increased infectivity and resistance to oxidative stress. PLoS Negl. Trop Dis. 2017, 11, e0005641. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, C.E.; Braillard, S.; Speed, W.; Glossop, P.A.; Whitlock, G.A.; Gibson, K.R.; Mills, J.E.; Brown, A.D.; Gardner, J.M.; Cao, Y.; et al. Novel amino-pyrazole ureas with potent in vitro and in vivo antileishmanial activity. J. Med. Chem. 2015, 58, 9615–9624. [Google Scholar] [CrossRef] [PubMed]
- Van den Kerkhof, M.; Mabille, D.; Chatelain, E.; Mowbray, C.E.; Braillard, S.; Hendrickx, S.; Maes, L.; Caljon, G. In vitro and in vivo pharmacodynamics of three novel antileishmanial lead series. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 81–86. [Google Scholar] [CrossRef]
- Tunes, L.G.; Morato, R.E.; Garcia, A.; Schmitz, V.; Steindel, M.; Correa-Junior, J.D.; Dos Santos, H.F.; Frezard, F.; de Almeida, M.V.; Silva, H.; et al. Preclinical gold complexes as oral drug candidates to treat leishmaniasis are potent trypanothione reductase inhibitors. ACS Infect. Dis. 2020, 6, 1121–1139. [Google Scholar] [CrossRef] [PubMed]
- Osorio, Y.; Travi, B.L.; Renslo, A.R.; Peniche, A.G.; Melby, P.C. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl. Trop Dis. 2011, 5, e962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osorio, Y.E.; Travi, B.L.; Melby, P.C. An ex vivo splenic explant model system for the identification of small molecule therapeutics for visceral leishmaniasis. FASEB J. 2008, 22, 1122–1136. [Google Scholar]
- Hefnawy, A.; Cantizani, J.; Pena, I.; Manzano, P.; Rijal, S.; Dujardin, J.C.; De Muylder, G.; Martin, J. Importance of secondary screening with clinical isolates for anti-leishmania drug discovery. Sci. Rep. 2018, 8, 11765. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.K.; Bringaud, F.; Nolan, D.P.; Figueiredo, L.M. Metabolic reprogramming during the Trypanosoma brucei life cycle. F1000Research 2017, 6, 683. [Google Scholar] [CrossRef]
- MacGregor, P.; Szoor, B.; Savill, N.J.; Matthews, K.R. Trypanosomal immune evasion, chronicity and transmission: An elegant balancing act. Nat. Rev. Microbiol. 2012, 10, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Mackey, Z.B.; Baca, A.M.; Mallari, J.P.; Apsel, B.; Shelat, A.; Hansell, E.J.; Chiang, P.K.; Wolff, B.; Guy, K.R.; Williams, J.; et al. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem. Biol. Drug Des. 2006, 67, 355–363. [Google Scholar] [CrossRef]
- Sykes, M.L.; Avery, V.M. A luciferase based viability assay for ATP detection in 384-well format for high throughput whole cell screening of Trypanosoma brucei brucei bloodstream form strain 427. Parasit. Vectors 2009, 2, 54. [Google Scholar] [CrossRef] [Green Version]
- Sykes, M.L.; Baell, J.B.; Kaiser, M.; Chatelain, E.; Moawad, S.R.; Ganame, D.; Ioset, J.R.; Avery, V.M. Identification of compounds with anti-proliferative activity against Trypanosoma brucei brucei strain 427 by a whole cell viability based HTS campaign. PLoS Negl. Trop Dis. 2012, 6, e1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, J.; Moraes, C.B.; Song, R.; Pascoalino, B.S.; Lee, N.; Siqueira-Neto, J.L.; Cruz, D.J.; Parkinson, T.; Ioset, J.R.; Cordeiro-da-Silva, A.; et al. Drug discovery for human African trypanosomiasis: Identification of novel scaffolds by the newly developed HTS SYBR Green assay for Trypanosoma brucei. J. Biomol. Screen 2015, 20, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berninger, M.; Erk, C.; Fuss, A.; Skaf, J.; Al-Momani, E.; Israel, I.; Raschig, M.; Guntzel, P.; Samnick, S.; Holzgrabe, U. Fluorine walk: The impact of fluorine in quinolone amides on their activity against African sleeping sickness. Eur. J. Med. Chem. 2018, 152, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazue, G.; Bray, M.A.; Pecoul, B. Fexinidazole: A new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop Dis. 2010, 4, e923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [Green Version]
- Villalta, F.; Rachakonda, G. Advances in preclinical approaches to Chagas disease drug discovery. Expert Opin. Drug Discov. 2019, 14, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- de Souza, W.; de Carvalho, T.M.; Barrias, E.S. Review on Trypanosoma cruzi: Host cell interaction. Int. J. Cell Biol. 2010, 2010, 295394. [Google Scholar] [CrossRef] [Green Version]
- Reyes, P.A.; Vallejo, M. Trypanocidal drugs for late stage, symptomatic Chagas disease (Trypanosoma cruzi infection). Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Chatelain, E.; Ioset, J.R. Phenotypic screening approaches for Chagas disease drug discovery. Expert Opin. Drug Discov. 2018, 13, 141–153. [Google Scholar] [CrossRef]
- Bot, C.; Hall, B.S.; Bashir, N.; Taylor, M.C.; Helsby, N.A.; Wilkinson, S.R. Trypanocidal activity of aziridinyl nitrobenzamide prodrugs. Antimicrob. Agents Chemother. 2010, 54, 4246–4252. [Google Scholar] [CrossRef] [Green Version]
- Canavaci, A.M.; Bustamante, J.M.; Padilla, A.M.; Perez Brandan, C.M.; Simpson, L.J.; Xu, D.; Boehlke, C.L.; Tarleton, R.L. In vitro and in vivo high-throughput assays for the testing of anti-Trypanosoma cruzi compounds. PLoS Negl. Trop Dis. 2010, 4, e740. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.C.; Ang, K.K.; Chen, S.; Arkin, M.R.; McKerrow, J.H.; Doyle, P.S. Image-based high-throughput drug screening targeting the intracellular stage of Trypanosoma cruzi, the agent of Chagas′ disease. Antimicrob. Agents Chemother. 2010, 54, 3326–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, I.; Pilar Manzano, M.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I.; et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015, 5, 8771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Padilla, J.; Cotillo, I.; Presa, J.L.; Cantizani, J.; Pena, I.; Bardera, A.I.; Martin, J.J.; Rodriguez, A. Automated high-content assay for compounds selectively toxic to Trypanosoma cruzi in a myoblastic cell line. PLoS Negl. Trop Dis. 2015, 9, e0003493. [Google Scholar] [CrossRef]
- De Rycker, M.; Thomas, J.; Riley, J.; Brough, S.J.; Miles, T.J.; Gray, D.W. Identification of trypanocidal activity for known clinical compounds using a new Trypanosoma cruzi hit-discovery screening cascade. PLoS Negl. Trop Dis. 2016, 10, e0004584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, L.M.; Thomas, J.; Lewis, M.D.; Cotillo, I.; Gray, D.W.; De Rycker, M. Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl. Trop Dis. 2018, 12, e0006612. [Google Scholar] [CrossRef]
- Bernatchez, J.A.; Chen, E.; Hull, M.V.; McNamara, C.W.; McKerrow, J.H.; Siqueira-Neto, J.L. High-throughput screening of the ReFRAME library identifies potential drug repurposing candidates for Trypanosoma cruzi. Microorganisms 2020, 8, 472. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.N. Visceral leishmaniasis: Experimental models for drug discovery. Indian J. Med. Res. 2011, 133, 27–39. [Google Scholar]
- Loria-Cervera, E.N.; Andrade-Narvaez, F.J. Animal models for the study of leishmaniasis immunology. Rev. Inst. Med. Trop. Sao Paulo 2014, 56, 1–11. [Google Scholar] [CrossRef]
- Sacks, D.L.; Melby, P.C. Animal models for the analysis of immune responses to leishmaniasis. Curr. Protoc. Immunol. 2015, 108, 19.2.1–19.2.24. [Google Scholar] [CrossRef] [Green Version]
- Loeuillet, C.; Banuls, A.L.; Hide, M. Study of Leishmania pathogenesis in mice: Experimental considerations. Parasit. Vectors 2016, 9, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mears, E.R.; Modabber, F.; Don, R.; Johnson, G.E. A review: The current in vivo models for the discovery and utility of new anti-leishmanial drugs targeting cutaneous leishmaniasis. PLoS Negl. Trop Dis. 2015, 9, e0003889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, C.I.; Brodskyn, C.I. The immunobiology of Leishmania braziliensis infection. Front Immunol. 2012, 3, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwell, J.M. Genetic susceptibility to leishmanial infections: Studies in mice and man. Parasitology 1996, 112, S67–S74. [Google Scholar] [CrossRef]
- Leon, C.M.; Montilla, M.; Vanegas, R.; Castillo, M.; Parra, E.; Ramirez, J.D. Murine models susceptibility to distinct Trypanosoma cruzi I genotypes infection. Parasitology 2017, 144, 512–519. [Google Scholar] [CrossRef]
- Torres-Vargas, J.; Jimenez-Coello, M.; Guzman-Marin, E.; Acosta-Viana, K.Y.; Yadon, Z.E.; Gutierrez-Blanco, E.; Guillermo-Cordero, J.L.; Garg, N.J.; Ortega-Pacheco, A. Quantitative and histological assessment of maternal-fetal transmission of Trypanosoma cruzi in guinea pigs: An experimental model of congenital Chagas disease. PLoS Negl. Trop Dis. 2018, 12, e0006222. [Google Scholar] [CrossRef] [Green Version]
- Becvar, T.; Siriyasatien, P.; Bates, P.; Volf, P.; Sadlova, J. Development of Leishmania (Mundinia) in guinea pigs. Parasit. Vectors 2020, 13, 181. [Google Scholar] [CrossRef] [Green Version]
- Paranaiba, L.F.; Pinheiro, L.J.; Macedo, D.H.; Menezes-Neto, A.; Torrecilhas, A.C.; Tafuri, W.L.; Soares, R.P. An overview on Leishmania (Mundinia) enriettii: Biology, immunopathology, LRV and extracellular vesicles during the host-parasite interaction. Parasitology 2018, 145, 1265–1273. [Google Scholar] [CrossRef]
- McCarroll, C.S.; Rossor, C.L.; Morrison, L.R.; Morrison, L.J.; Loughrey, C.M. A Pre-clinical animal model of Trypanosoma brucei infection demonstrating cardiac dysfunction. PLoS Negl. Trop Dis. 2015, 9, e0003811. [Google Scholar] [CrossRef]
- Fulton, J.D.; Joyner, L.P.; Chandler, R.J. Studies on protozoa. Part II: The golden hamster (Cricetus auratus) and cotton rat (Sigmodon hispidus) as experimental hosts for Leishmania donovani. Trans R. Soc. Trop. Med. Hyg. 1950, 44, 105–112. [Google Scholar] [CrossRef]
- Gomes-Silva, A.; Valverde, J.G.; Ribeiro-Romao, R.P.; Placido-Pereira, R.M.; Da-Cruz, A.M. Golden hamster (Mesocricetus auratus) as an experimental model for Leishmania (Viannia) braziliensis infection. Parasitology 2013, 140, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Chan, J.V.; Aguilar-Cetina Adel, C.; Villanueva-Lizama, L.E.; Martinez-Vega, P.P.; Ramirez-Sierra, M.J.; Rosado-Vallado, M.E.; Guillermo-Cordero, J.L.; Dumonteil, E. A canine model of experimental infection with Leishmania (L.) mexicana. Parasit. Vectors 2014, 7, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probst, R.J.; Wellde, B.T.; Lawyer, P.G.; Stiteler, J.S.; Rowton, E.D. Rhesus monkey model for Leishmania major transmitted by Phlebotomus papatasi sandfly bites. Med. Vet. Entomol. 2001, 15, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Freidag, B.L.; Mendez, S.; Cheever, A.W.; Kenney, R.T.; Flynn, B.; Sacks, D.L.; Seder, R.A. Immunological and pathological evaluation of rhesus macaques infected with Leishmania major. Exp. Parasitol. 2003, 103, 160–168. [Google Scholar] [CrossRef]
- Souza-Lemos, C.; de-Campos, S.N.; Teva, A.; Porrozzi, R.; Grimaldi, G.J. In situ characterization of the granulomatous immune response with time in nonhealing lesional skin of Leishmania braziliensis-infected rhesus macaques (Macaca mulatta). Vet. Immunol. Immunopathol. 2011, 142, 147–155. [Google Scholar] [CrossRef]
- Sathler-Avelar, R.; Vitelli-Avelar, D.M.; Mattoso-Barbosa, A.M.; Perdigao-de-Oliveira, M.; Costa, R.P.; Eloi-Santos, S.M.; Gomes Mde, S.; Amaral, L.R.; Teixeira-Carvalho, A.; Martins-Filho, O.A.; et al. Phenotypic features of circulating leukocytes from non-human primates naturally infected with Trypanosoma cruzi resemble the major immunological findings observed in human Chagas disease. PLoS Negl. Trop Dis. 2016, 10, e0004302. [Google Scholar] [CrossRef]
- Folmer, R.H. Integrating biophysics with HTS-driven drug discovery projects. Drug Discov. Today 2016, 21, 491–498. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Dos Santos, R.N.; Ferreira, L.G.; Andricopulo, A.D. Practices in molecular docking and structure-based virtual screening. Methods Mol. Biol. 2018, 1762, 31–50. [Google Scholar]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Saeed, Y.; Bahram, H. Chemometrics tools in QSAR/QSPR studies: A historical perspective. Chemom. Intell. Lab. Syst. 2015, 149, 177–204. [Google Scholar]
- Gourley, D.G.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Crystallization of recombinant Leishmania major pteridine reductase 1 (PTR1). Acta Cryst. D Biol. Cryst. 1999, 55, 1608–1610. [Google Scholar] [CrossRef] [PubMed]
- Rashid, U.; Sultana, R.; Shaheen, N.; Hassan, S.F.; Yaqoob, F.; Ahmad, M.J.; Iftikhar, F.; Sultana, N.; Asghar, S.; Yasinzai, M.; et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 2016, 115, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, S.; Perdih, A.; Sencanski, M.; Glisic, S.; Duarte, M.; Tomas, A.M.; Sena, F.V.; Sousa, F.M.; Pereira, M.M.; Solmajer, T. In silico discovery of a substituted 6-methoxy-quinalidine with leishmanicidal activity in Leishmania infantum. Molecules 2018, 23, 772. [Google Scholar] [CrossRef] [Green Version]
- De Luca, L.; Ferro, S.; Buemi, M.R.; Monforte, A.M.; Gitto, R.; Schirmeister, T.; Maes, L.; Rescifina, A.; Micale, N. Discovery of benzimidazole-based Leishmania mexicana cysteine protease CPB2.8DeltaCTE inhibitors as potential therapeutics for leishmaniasis. Chem. Biol. Drug Des. 2018, 92, 1585–1596. [Google Scholar] [CrossRef]
- Romero-Estudillo, I.; Viveros-Ceballos, J.L.; Cazares-Carreno, O.; Gonzalez-Morales, A.; de Jesus, B.F.; Lopez-Castillo, M.; Razo-Hernandez, R.S.; Castaneda-Corral, G.; Ordonez, M. Synthesis of new alpha-aminophosphonates: Evaluation as anti-inflammatory agents and QSAR studies. Bioorg. Med. Chem. 2019, 27, 2376–23861. [Google Scholar] [CrossRef]
- Purohit, P.; Pandey, A.K.; Singh, D.; Chouhan, P.S.; Ramalingam, K.; Shukla, M.; Goyal, N.; Lal, J.; Chauhan, P.M.S. An insight into tetrahydro-beta-carboline-tetrazole hybrids: Synthesis and bioevaluation as potent antileishmanial agents. Medchemcomm 2017, 8, 1824–1834. [Google Scholar] [CrossRef]
- Wyllie, S.; Thomas, M.; Patterson, S.; Crouch, S.; De Rycker, M.; Lowe, R.; Gresham, S.; Urbaniak, M.D.; Otto, T.D.; Stojanovski, L.; et al. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 2018, 560, 192–197. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Mandal, S.; Njikan, S.; Kumar, A.; Early, J.V.; Parish, T. The relevance of persisters in tuberculosis drug discovery. Microbiology 2019, 165, 492–499. [Google Scholar] [CrossRef]
- Barrett, M.P.; Kyle, D.E.; Sibley, L.D.; Radke, J.B.; Tarleton, R.L. Protozoan persister-like cells and drug treatment failure. Nat. Rev. Microbiol. 2019, 17, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Markus, M.B. Malaria Eradication and the Hidden Parasite Reservoir. Trends Parasitol. 2017, 33, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Le Rutte, E.A.; Zijlstra, E.E.; de Vlas, S.J. Post-Kala-Azar dermal leishmaniasis as a reservoir for visceral leishmaniasis transmission. Trends Parasitol. 2019, 35, 590–592. [Google Scholar] [CrossRef] [Green Version]
- Kloehn, J.; Saunders, E.C.; O’Callaghan, S.; Dagley, M.J.; McConville, M.J. Characterization of metabolically quiescent Leishmania parasites in murine lesions using heavy water labeling. PLoS Pathog. 2015, 11, e1004683. [Google Scholar] [CrossRef] [Green Version]
- Mandell, M.A.; Beverley, S.M. Continual renewal and replication of persistent Leishmania major parasites in concomitantly immune hosts. Proc. Natl. Acad. Sci. USA 2017, 114, E801–E810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jara, M.; Berg, M.; Caljon, G.; de Muylder, G.; Cuypers, B.; Castillo, D.; Maes, I.; Orozco, M.D.C.; Vanaerschot, M.; Dujardin, J.C.; et al. Macromolecular biosynthetic parameters and metabolic profile in different life stages of Leishmania braziliensis: Amastigotes as a functionally less active stage. PLoS ONE 2017, 12, e0180532. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Valdez, F.J.; Padilla, A.; Wang, W.; Orr, D.; Tarleton, R.L. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. Elife 2018, 7, e34039. [Google Scholar] [CrossRef]
- Jara, M.; Maes, I.; Imamura, H.; Domagalska, M.A.; Dujardin, J.C.; Arevalo, J. Tracking of quiescence in Leishmania by quantifying the expression of GFP in the ribosomal DNA locus. Sci. Rep. 2019, 9, 18951. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, M.S.; Rudenko, G. TDP1 is an HMG chromatin protein facilitating RNA polymerase I transcription in African trypanosomes. Nucleic. Acids. Res. 2013, 41, 2981–2992. [Google Scholar] [CrossRef] [Green Version]
- Ferrick, D.A.; Neilson, A.; Beeson, C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov. Today 2008, 13, 268–274. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring mitochondrial function in permeabilized cells using the Seahorse XF Analyzer or a Clark-Type oxygen electrode. Curr. Protoc. Toxicol. 2014, 60, 25.2.1–25.2.16. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ortiz, L.M.; Sanchez-Villamil, J.P.; Celis-Rodriguez, M.A.; Lineros, G.; Sanabria-Barrera, S.; Serrano, N.C.; Rincon, M.Y.; Bautista-Nino, P.K. Measuring mitochondrial respiration in adherent cells infected with Trypanosoma cruzi Chagas, 1909 using Seahorse extracellular flux analyser. Folia Parasitol. 2019, 66, 16. [Google Scholar] [CrossRef]
- Furtado, C.; Kunrath-Lima, M.; Rajao, M.A.; Mendes, I.C.; de Moura, M.B.; Campos, P.C.; Macedo, A.M.; Franco, G.R.; Pena, S.D.; Teixeira, S.M.; et al. Functional characterization of 8-oxoguanine DNA glycosylase of Trypanosoma cruzi. PLoS ONE 2012, 7, e42484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Shah-Simpson, S.; Okrah, K.; Belew, A.T.; Choi, J.; Caradonna, K.L.; Padmanabhan, P.; Ndegwa, D.M.; Temanni, M.R.; Corrada Bravo, H.; et al. Transcriptome remodeling in Trypanosoma cruzi and human cells during intracellular infection. PLoS Pathog. 2016, 12, e1005511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah-Simpson, S.; Pereira, C.F.; Dumoulin, P.C.; Caradonna, K.L.; Burleigh, B.A. Bioenergetic profiling of Trypanosoma cruzi life stages using Seahorse extracellular flux technology. Mol. Biochem. Parasitol. 2016, 208, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Rufener, R.; Dick, L.; D’Ascoli, L.; Ritler, D.; Hizem, A.; Wells, T.N.C.; Hemphill, A.; Lundstrom-Stadelmann, B. Repurposing of an old drug: In vitro and in vivo efficacies of buparvaquone against Echinococcus multilocularis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 440–450. [Google Scholar] [CrossRef]
- Field, M.C.; Allen, C.L.; Dhir, V.; Goulding, D.; Hall, B.S.; Morgan, G.W.; Veazey, P.; Engstler, M. New approaches to the microscopic imaging of Trypanosoma brucei. Microsc. Microanal. 2004, 10, 621–636. [Google Scholar] [CrossRef] [Green Version]
- Aulner, N.; Danckaert, A.; Rouault-Hardoin, E.; Desrivot, J.; Helynck, O.; Commere, P.H.; Munier-Lehmann, H.; Spath, G.F.; Shorte, S.L.; Milon, G.; et al. High content analysis of primary macrophages hosting proliferating Leishmania amastigotes: Application to anti-leishmanial drug discovery. PLoS Negl. Trop Dis. 2013, 7, e2154. [Google Scholar] [CrossRef] [Green Version]
- Proto, W.R.; Coombs, G.H.; Mottram, J.C. Cell death in parasitic protozoa: Regulated or incidental? Nat. Rev. Microbiol. 2013, 11, 58–66. [Google Scholar] [CrossRef]
- Thomas, J.A.; Baker, N.; Hutchinson, S.; Dominicus, C.; Trenaman, A.; Glover, L.; Alsford, S.; Horn, D. Insights into antitrypanosomal drug mode-of-action from cytology-based profiling. PLoS Negl. Trop Dis. 2018, 12, e0006980. [Google Scholar] [CrossRef]
- Mishra, A.; Khan, M.I.; Jha, P.K.; Kumar, A.; Das, S.; Das, P.; Das, P.; Sinha, K.K. Oxidative stress-mediated overexpression of uracil DNA glycosylase in Leishmania donovani confers tolerance against antileishmanial drugs. Oxid. Med. Cell Longev. 2018, 2018, 4074357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glover, L.; Horn, D. Trypanosomal histone gammaH2A and the DNA damage response. Mol. Biochem. Parasitol. 2012, 183, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Bellofatto, V.; Palenchar, J.B. RNA interference as a genetic tool in trypanosomes. Methods Mol. Biol. 2008, 442, 83–94. [Google Scholar] [PubMed]
- Alsford, S.; Turner, D.J.; Obado, S.O.; Sanchez-Flores, A.; Glover, L.; Berriman, M.; Hertz-Fowler, C.; Horn, D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011, 21, 915–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.; Glover, L.; Munday, J.C.; Aguinaga Andres, D.; Barrett, M.P.; de Koning, H.P.; Horn, D. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 10996–11001. [Google Scholar] [CrossRef] [Green Version]
- Glover, L.; Alsford, S.; Baker, N.; Turner, D.J.; Sanchez-Flores, A.; Hutchinson, S.; Hertz-Fowler, C.; Berriman, M.; Horn, D. Genome-scale RNAi screens for high-throughput phenotyping in bloodstream-form African trypanosomes. Nat. Protoc. 2015, 10, 106–133. [Google Scholar] [CrossRef]
- Marquis, N.; Gourbal, B.; Rosen, B.P.; Mukhopadhyay, R.; Ouellette, M. Modulation in aquaglyceroporin AQP1 gene transcript levels in drug-resistant Leishmania. Mol. Microbiol. 2005, 57, 1690–1699. [Google Scholar] [CrossRef]
- Gazanion, E.; Fernandez-Prada, C.; Papadopoulou, B.; Leprohon, P.; Ouellette, M. Cos-Seq for high-throughput identification of drug target and resistance mechanisms in the protozoan parasite Leishmania. Proc. Natl. Acad. Sci. USA 2016, 113, E3012–E3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Prada, C.; Sharma, M.; Plourde, M.; Bresson, E.; Roy, G.; Leprohon, P.; Ouellette, M. High-throughput Cos-Seq screen with intracellular Leishmania infantum for the discovery of novel drug-resistance mechanisms. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 165–173. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Leprohon, P.; Bigot, S.; Padmanabhan, P.K.; Mukherjee, A.; Roy, G.; Gingras, H.; Mestdagh, A.; Papadopoulou, B.; Ouellette, M. Coupling chemical mutagenesis to next generation sequencing for the identification of drug resistance mutations in Leishmania. Nat. Commun. 2019, 10, 5627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beneke, T.; Madden, R.; Makin, L.; Valli, J.; Sunter, J.; Gluenz, E. A CRISPR Cas9 high-throughput genome editing toolkit for kinetoplastids. R. Soc. Open Sci. 2017, 4, 170095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martel, D.; Beneke, T.; Gluenz, E.; Spath, G.F.; Rachidi, N. Characterisation of casein kinase 1.1 in Leishmania donovani using the CRISPR Cas9 toolkit. Biomed. Res. Int. 2017, 2017, 4635605. [Google Scholar] [CrossRef] [Green Version]
- Beneke, T.; Demay, F.; Hookway, E.; Ashman, N.; Jeffery, H.; Smith, J.; Valli, J.; Becvar, T.; Myskova, J.; Lestinova, T.; et al. Genetic dissection of a Leishmania flagellar proteome demonstrates requirement for directional motility in sand fly infections. PLoS Pathog. 2019, 15, e1007828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negreira, G.H.; Monsieurs, P.; Imamura, H.; Maes, I.; Kuk, N.; Yagoubat, A.; den Broeck, F.V.; Sterkers, Y.; Dujardin, J.-C.; Domagalska, M.A. Exploring the evolution and adaptive role of mosaic aneuploidy in a clonal Leishmania donovani population using high throughput single cell genome sequencing. bioRxiv 2020, 2020, 976233v1. Available online: www.biorxiv.org/content/10.1101/2020.03.05.976233v1 (accessed on 15 May 2020).
- Ubeda, J.M.; Raymond, F.; Mukherjee, A.; Plourde, M.; Gingras, H.; Roy, G.; Lapointe, A.; Leprohon, P.; Papadopoulou, B.; Corbeil, J.; et al. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 2014, 12, e1001868. [Google Scholar] [CrossRef] [Green Version]
- Dumetz, F.; Cuypers, B.; Imamura, H.; Zander, D.; D’Haenens, E.; Maes, I.; Domagalska, M.A.; Clos, J.; Dujardin, J.C.; De Muylder, G. Molecular preadaptation to antimony resistance in Leishmania donovani on the Indian Subcontinent. MSphere 2018, 3, e00548-17. [Google Scholar] [CrossRef] [Green Version]
- Poinar, G.J.; Poinar, R. Fossil evidence of insect pathogens. J. Invertebr. Pathol. 2005, 89, 243–250. [Google Scholar] [CrossRef]
- Vargas, D.A.; Prieto, M.D.; Martinez-Valencia, A.J.; Cossio, A.; Burgess, K.E.V.; Burchmore, R.J.S.; Gomez, M.A. Pharmacometabolomics of meglumine antimoniate in patients with cutaneous leishmaniasis. Front Pharm. 2019, 10, 657. [Google Scholar] [CrossRef] [PubMed]
- Hennig, K.; Abi-Ghanem, J.; Bunescu, A.; Meniche, X.; Biliaut, E.; Ouattara, A.D.; Lewis, M.D.; Kelly, J.M.; Braillard, S.; Courtemanche, G.; et al. Metabolomics, lipidomics and proteomics profiling of myoblasts infected with Trypanosoma cruzi after treatment with different drugs against Chagas disease. Metabolomics 2019, 15, 117. [Google Scholar] [CrossRef]
- Ty, M.C.; Loke, P.; Alberola, J.; Rodriguez, A.; Rodriguez-Cortes, A. Immuno-metabolic profile of human macrophages after Leishmania and Trypanosoma cruzi infection. PLoS ONE 2019, 14, e0225588. [Google Scholar] [CrossRef]
- McCall, L.I.; Tripathi, A.; Vargas, F.; Knight, R.; Dorrestein, P.C.; Siqueira-Neto, J.L. Experimental Chagas disease-induced perturbations of the fecal microbiome and metabolome. PLoS Negl. Trop Dis. 2018, 12, e0006344. [Google Scholar] [CrossRef] [PubMed]
- McCall, L.I.; Morton, J.T.; Bernatchez, J.A.; de Siqueira-Neto, J.L.; Knight, R.; Dorrestein, P.C.; McKerrow, J.H. Mass spectrometry-based chemical cartography of a cardiac parasitic infection. Anal. Chem. 2017, 89, 10414–10421. [Google Scholar] [CrossRef] [PubMed]
- Gazos-Lopes, F.; Martin, J.L.; Dumoulin, P.C.; Burleigh, B.A. Host triacylglycerols shape the lipidome of intracellular trypanosomes and modulate their growth. PLoS Pathog. 2017, 13, e1006800. [Google Scholar] [CrossRef]
- Creek, D.J.; Nijagal, B.; Kim, D.H.; Rojas, F.; Matthews, K.R.; Barrett, M.P. Metabolomics guides rational development of a simplified cell culture medium for drug screening against Trypanosoma brucei. Antimicrob. Agents Chemother. 2013, 57, 2768–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trochine, A.; Creek, D.J.; Faral-Tello, P.; Barrett, M.P.; Robello, C. Benznidazole biotransformation and multiple targets in Trypanosoma cruzi revealed by metabolomics. PLoS Negl. Trop Dis. 2014, 8, e2844. [Google Scholar] [CrossRef]
- Rojo, D.; Canuto, G.A.; Castilho-Martins, E.A.; Tavares, M.F.; Barbas, C.; Lopez-Gonzalvez, A.; Rivas, L. A multiplatform metabolomic approach to the basis of antimonial action and resistance in Leishmania infantum. PLoS ONE 2015, 10, e0130675. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, M.C.; Bourassa, S.; Leprohon, P.; Legare, D.; Poirier, G.G.; Droit, A.; Ouellette, M. Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. PLoS ONE 2013, 8, e81899. [Google Scholar] [CrossRef] [Green Version]
- Vincent, I.M.; Barrett, M.P. Metabolomic-based strategies for anti-parasite drug discovery. J. Biomol. Screen 2015, 20, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Alves-Ferreira, M.; Guimaraes, A.C.; Capriles, P.V.; Dardenne, L.E.; Degrave, W.M. A new approach for potential drug target discovery through in silico metabolic pathway analysis using Trypanosoma cruzi genome information. Mem. Inst. Oswaldo Cruz. 2009, 104, 1100–1110. [Google Scholar] [CrossRef] [Green Version]
- Coelho, E.A.; Chavez-Fumagalli, M.A.; Costa, L.E.; Tavares, C.A.; Soto, M.; Goulart, L.R. Theranostic applications of phage display to control leishmaniasis: Selection of biomarkers for serodiagnostics, vaccination, and immunotherapy. Rev. Soc. Bras. Med. Trop. 2015, 48, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, L.E.; Alves, P.T.; Carneiro, A.P.; Dias, A.C.S.; Fujimura, P.T.; Araujo, G.R.; Tavares, G.S.V.; Ramos, F.F.; Duarte, M.C.; Menezes-Souza, D.; et al. Leishmania infantum beta-tubulin identified by reverse engineering technology through phage display applied as theranostic marker for human visceral leishmaniasis. Int. J. Mol. Sci. 2019, 20, 1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, O.P.; Gedda, M.R.; Mudavath, S.L.; Srivastava, O.N.; Sundar, S. Envisioning the innovations in nanomedicine to combat visceral leishmaniasis: For future theranostic application. Nanomedicine 2019, 14, 1911–1927. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.; Myburgh, E.; Gao, M.Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, S.; Brand, S.; Thomas, M.; De Rycker, M.; Chung, C.W.; Pena, I.; Bingham, R.P.; Bueren-Calabuig, J.A.; Cantizani, J.; Cebrian, D.; et al. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. USA 2019, 116, 9318–9323. [Google Scholar] [CrossRef] [Green Version]
- Ardal, C.; Balasegaram, M.; Laxminarayan, R.; McAdams, D.; Outterson, K.; Rex, J.H.; Sumpradit, N. Antibiotic development–economic, regulatory and societal challenges. Nat. Rev. Microbiol. 2020, 18, 267–274. [Google Scholar] [CrossRef]
- Pecoul, B. New drugs for neglected diseases: From pipeline to patients. PLoS Med. 2004, 1, e6. [Google Scholar] [CrossRef] [Green Version]
- Croft, S.L. Neglected tropical diseases in the genomics era: Re-evaluating the impact of new drugs and mass drug administration. Genome Biol. 2016, 17, 46. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, A.; Corbeil, A.; do Monte-Neto, R.L.; Fernandez-Prada, C. Of Drugs and Trypanosomatids: New Tools and Knowledge to Reduce Bottlenecks in Drug Discovery. Genes 2020, 11, 722. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070722
Bhattacharya A, Corbeil A, do Monte-Neto RL, Fernandez-Prada C. Of Drugs and Trypanosomatids: New Tools and Knowledge to Reduce Bottlenecks in Drug Discovery. Genes. 2020; 11(7):722. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070722
Chicago/Turabian StyleBhattacharya, Arijit, Audrey Corbeil, Rubens L. do Monte-Neto, and Christopher Fernandez-Prada. 2020. "Of Drugs and Trypanosomatids: New Tools and Knowledge to Reduce Bottlenecks in Drug Discovery" Genes 11, no. 7: 722. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11070722