Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Breeds and Samples
2.2. DNA Extraction
2.3. Microsatellite Genotyping and Detection of Consensus Genotypes
2.4. Statistical Data Analysis
3. Results
3.1. Estimation of Consensus Genotypes
3.2. Genetic Variability
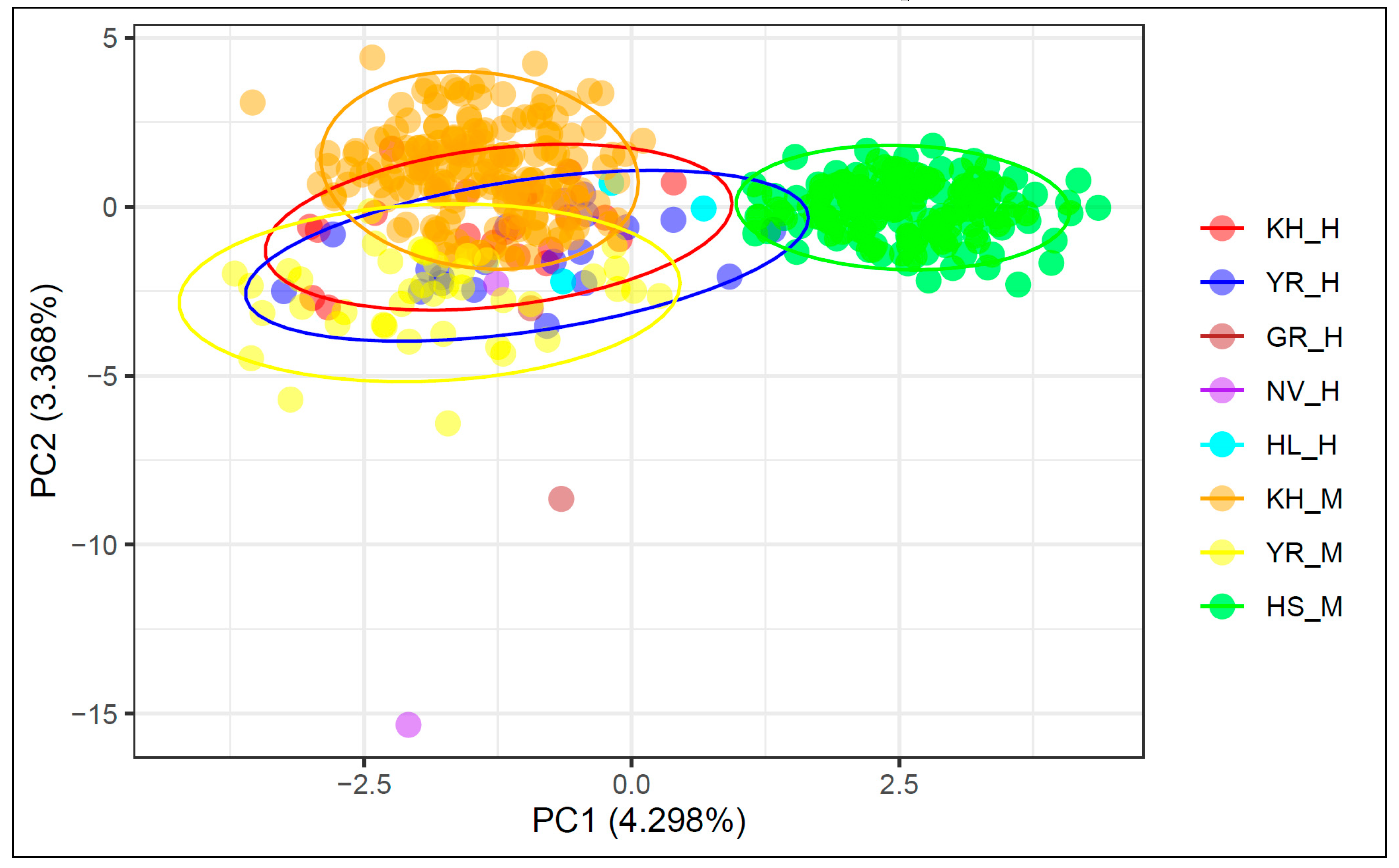
3.3. Principal Component Analysis
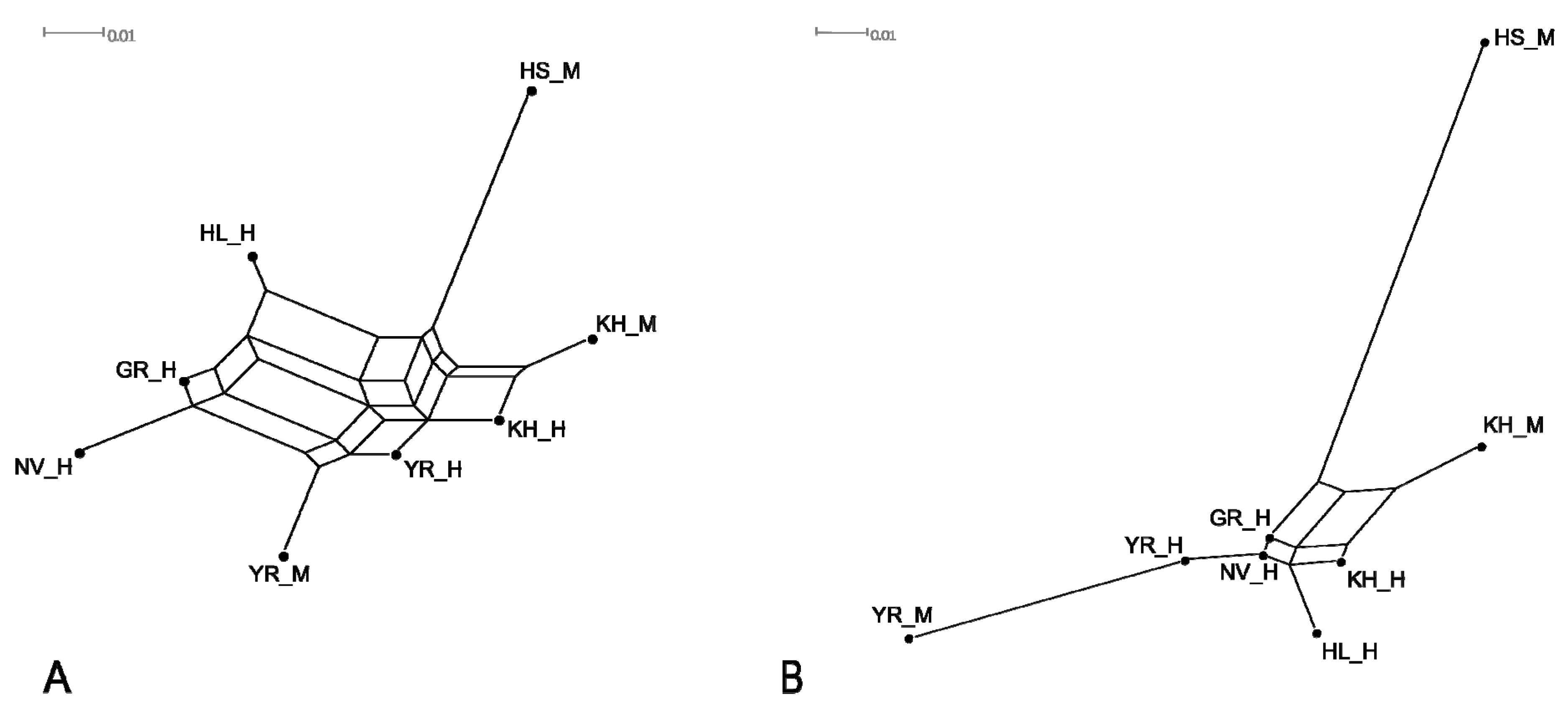
3.4. Genetic Relationship between Populations
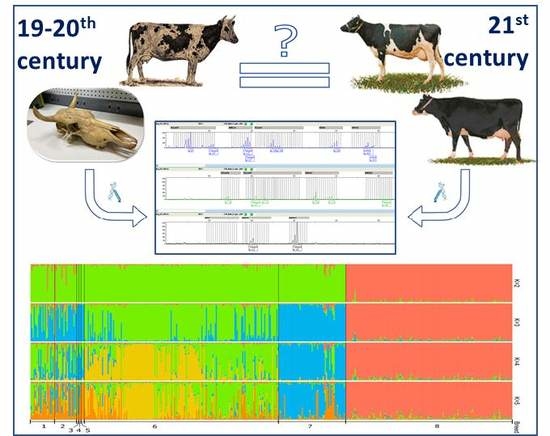
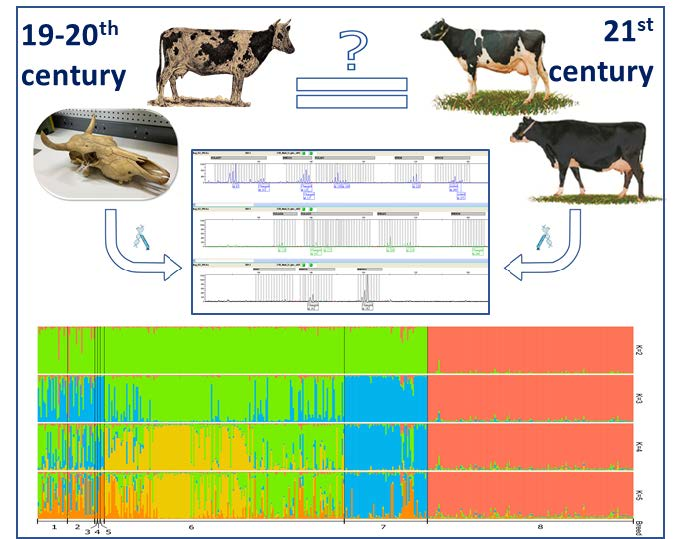
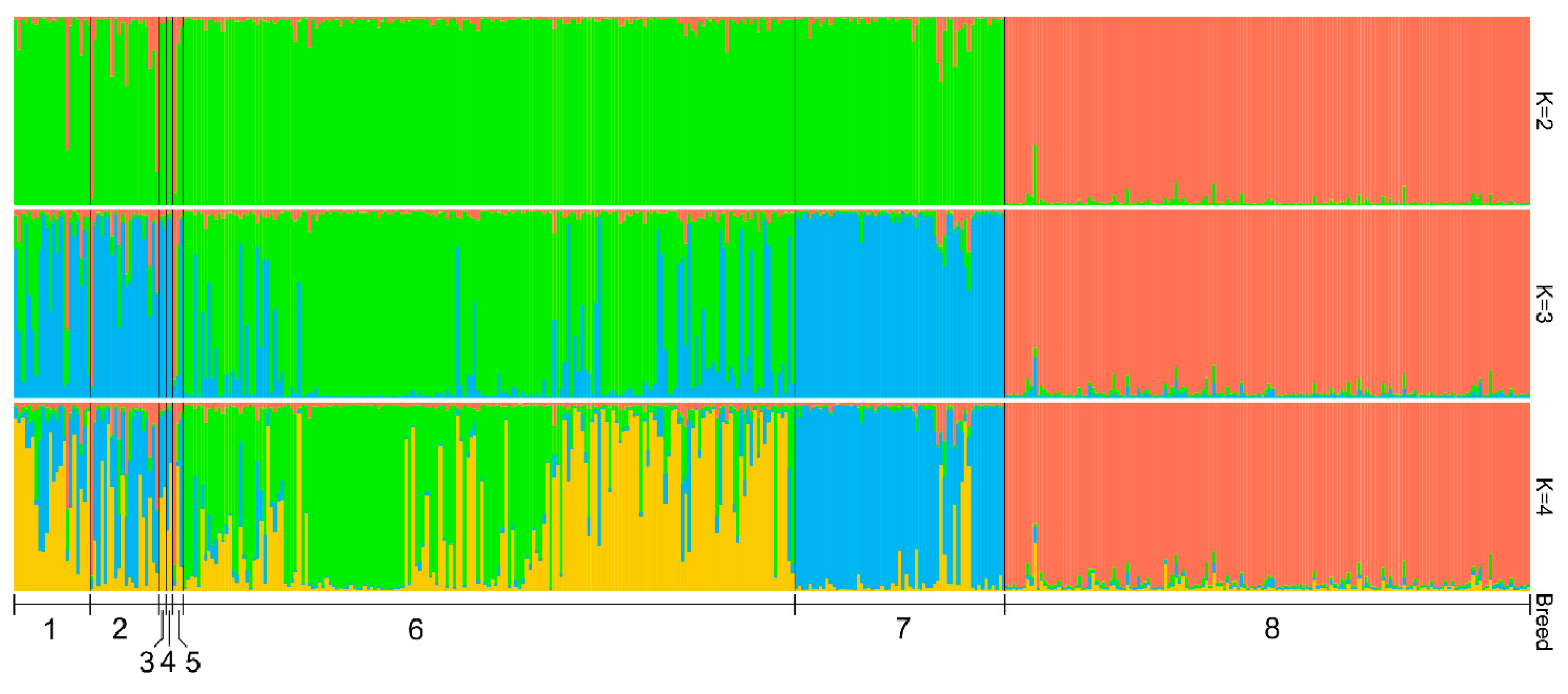
3.5. Genetic Structure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Larkin, D.M.; Daetwyler, H.D.; Hernandez, A.G.; Wright, C.L.; Hetrick, L.A.; Boucek, L.; Bachman, S.L.; Band, M.R.; Akraiko, T.V.; Cohen-Zinder, M.; et al. Whole-genome resequencing of two elite sires for the detection of haplotypes under selection in dairy cattle. Proc. Natl. Acad. Sci. USA 2012, 109, 7693–7698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO. The Second Report on the State of the World’s Animal Genetic Resources for Food and Agriculture; Scherf, B.D., Pilling, D., Eds.; FAO Commission on Genetic Resources for Food and Agriculture Assessments: Rome, Italy, 2015; Available online: http://www.fao.org/3/a-i4787e/index.html (accessed on 14 April 2020).
- DAD-IS. Domestic Animal Diversity Information System (DAD-IS). Available online: http://www.fao.org/dad-is/ (accessed on 14 April 2020).
- Ajmone-Marsan, P.; The GLOBALDIV Consortium. A global view of livestock biodiversity and conservation-GLOBALDIV. Anim. Genet. 2010, 41, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Felius, M.; Theunissen, B.; Lenstra, J. Conservation of cattle genetic resources: The role of breeds. J. Agric. Sci. 2015, 153, 152–162. [Google Scholar] [CrossRef]
- Liskun, E.F. Otechestvennye Porody Krupnogo Rogatogo Skota (Native Breeds of Cattle); GISL: Moskva, Russia, 1949. (In Russian) [Google Scholar]
- Liskun, E.F. What is Good about Russian Northern Cattle; Publishing House of the People’s Commissariat of Agriculture: Petrograd, Russia, 1919; p. 23. Available online: https://www.booksite.ru/fulltext/liskun1/text.pdf (accessed on 22 April 2020).
- Li, M.-H.; Kantanen, J. Genetic structure of Eurasian cattle (Bos taurus) based on microsatellites: Clarification for their breed classification. Anim. Genet. 2010, 41, 150–158. [Google Scholar] [CrossRef]
- Dmitriev, N.G.; Ernst, L.K. Animal Genetic Resources of the USSR; FAO and UNEP: Rome, Italy, 1989; Available online: http://www.fao.org/3/ah759e/AH759E07.htm (accessed on 22 April 2020).
- Zinovieva, N.A.; Dotsev, A.V.; Sermyagin, A.A.; Wimmers, K.; Reyer, H.; Sölkner, J.; Deniskova, T.E.; Brem, G. Study of genetic diversity and population structure of five Russian cattle breeds using whole-genome SNP analysis. Agric. Biol. 2016, 51, 788–800. [Google Scholar] [CrossRef] [Green Version]
- Dotsev, A.V.; Sermyagin, A.A.; Shakhin, A.V.; Paronyan, I.A.; Plemyashov, K.V.; Reyer, H.; Wimmers, K.; Brem, G.; Zinovieva, N.A. Evaluation of current gene pool of Kholmogor and Black-and-white cattle breeds based on whole genome SNP analysis. Vestn. VOGiS 2018, 22, 742–747. [Google Scholar] [CrossRef]
- Sermyagin, A.A.; Dotsev, A.V.; Gladyr, E.A.; Traspov, A.A.; Deniskova, T.E.; Kostyunina, O.V.; Reyer, H.; Wimmers, K.; Barbato, M.; Paronyan, I.A.; et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet. Sel. Evol. 2018, 50, 37. [Google Scholar] [CrossRef] [Green Version]
- Yurchenko, A.; Yudin, N.; Aitnazarov, R.; Plyusnina, A.; Brukhin, V.; Soloshenko, V.; Lhasaranov, B.; Popov, R.; Paronyan, I.A.; Plemyashov, K.V.; et al. Genome-wide genotyping uncovers genetic profiles and history of the Russian cattle breeds. Heredity 2018, 120, 125–137. [Google Scholar] [CrossRef]
- Zinovieva, N.A.; Sermyagin, A.A.; Dotsev, A.V.; Boronetslaya, O.I.; Petrikeeva, L.V.; Abdelmanova, A.S.; Brem, G. Animal genetic resources: Developing the research of allele pool of Russian cattle breeds—Minireview. Agric. Biol. 2019, 54, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Groeneveld, L.F.; Lenstra, J.A.; Eding, H.; Toro, M.A.; Scherf, B.; Pilling, D.; Negrini, R.; Finlay, E.K.; Jianlin, H.; Groeneveld, E.; et al. Genetic diversity in farm animals—A review. Anim. Genet. 2010, 41 (Suppl. 1), 6–31. [Google Scholar] [CrossRef] [Green Version]
- McTavish, E.J.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F.; Hillis, D.M. New World cattle show ancestry from multiple independent domestication events. Proc. Natl. Acad. Sci. USA 2013, 110, E1398–E1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, J.E.; McKay, S.D.; Rolf, M.M.; Kim, J.; Molina Alcalá, A.; Sonstegard, T.S.; Hanotte, O.; Götherström, A.; Seabury, C.M.; Praharani, L.; et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014, 10, e1004254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugo, G.P.; Dover, M.J.; MacHugh, D.E. Unlocking the origins and biology of domestic animals using ancient DNA and paleogenomics. BMC Biol. 2019, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eusebi, P.G.; Martinez, A.; Cortes, O. Genomic tools for effective conservation of livestock breed diversity. Diversity 2019, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Putman, A.I.; Carbone, I. Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol. Evol. 2014, 4, 4399–4428. [Google Scholar] [CrossRef]
- Laoun, A.; Harkat, S.; Lafri, M.; Gaouar, S.B.S.; Belabdi, I.; Ciani, E.; De Groot, M.; Blanquet, V.; Leroy, G.; Rognon, X.; et al. Inference of breed structure in farm animals: Empirical comparison between SNP and microsatellite performance. Genes 2020, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Brenig, B.; Schütz, E. Recent development of allele frequencies and exclusion probabilities of microsatellites used for parentage control in the German Holstein Friesian cattle population. BMC Genet. 2016, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- Edwards, C.J.; Connellan, J.; Wallace, P.F.; Park, S.D.E.; McCormick, F.M.; Olsaker, I.; Eythórsdóttir, E.; MacHugh, D.E.; Bailey, J.F.; Bradley, D.G. Feasibility and utility of microsatellite markers in archaeological cattle remains from a Viking Age settlement in Dublin. Anim. Genet. 2003, 34, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Gargani, M.; Pariset, L.; Lenstra, J.A.; De Minicis, E.; European Cattle Genetic Diversity Consortium; Valentini, A. Microsatellite genotyping of medieval cattle from central Italy suggests an old origin of Chianina and Romagnola cattle. Front. Genet. 2015, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Rowe, K.C.; Singhal, S.; Macmanes, M.D.; Ayroles, J.F.; Morelli, T.L.; Rubidge, E.M.; Bi, K.; Moritz, C.C. Museum genomics: Low-cost and high-accuracy genetic data from historical specimens. Mol. Ecol. Resour. 2011, 11, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Billerman, S.M.; Walsh, J. Historical DNA as a tool to address key questions in avian biology and evolution: A review of methods, challenges, applications, and future directions. Mol. Ecol. Resour. 2019, 19, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Höss, M.; Pääbo, S. DNA extraction from Pleistocene bones by a silica-based purification method. Nucl. Acids Res. 1993, 21, 3913–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, A.J.; Mitchell, D.L.; Wiuf, C.; Paniker, L.; Brand, T.B.; Binladen, J.; Gilichinsky, D.A.; Rønn, R.; Willerslev, E. Crosslinks rather than strand breaks determine access to ancient DNA sequences from frozen sediments. Genetics 2006, 173, 1175–1179. [Google Scholar] [CrossRef] [Green Version]
- Abdelmanova, A.S.; Mishina, A.I.; Volkova, V.V.; Chinarov, R.Y.; Sermyagin, A.A.; Dotsev, A.V.; Boronetskaya, O.I.; Petrikeeva, L.V.; Kostyunina, O.V.; Brem, G.; et al. Comparative study of different methods of DNA extraction from cattle bones specimens maintained in a craniological collection. Agric. Biol. 2019, 54, 1110–1121. [Google Scholar] [CrossRef]
- Hansen, H.B.; Damgaard, P.B.; Margaryan, A.; Stenderup, J.; Lynnerup, N.; Willerslev, E.; Allentoft, M.E. Comparing ancient DNA preservation in petrous bone and tooth cementum. PLoS ONE 2017, 12, e0170940. [Google Scholar] [CrossRef] [Green Version]
- Rohland, N.; Glocke, I.; Aximu-Petri, A.; Meyer, M. Extraction of highly degraded DNA from ancient bones, teeth and sediments for high-throughput sequencing. Nat. Protoc. 2018, 13, 2447–2461. [Google Scholar] [CrossRef]
- Scheu, A.; Powell, A.; Bollongino, R.; Vigne, J.-D.; Tresset, A.; Çakırlar, C.; Benecke, N.; Burger, J. The genetic prehistory of domesticated cattle from their origin to the spread across Europe. BMC Genet. 2015, 16, 54. [Google Scholar] [CrossRef] [Green Version]
- Robin, E.D.; Wong, R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 1988, 136, 507–513. [Google Scholar] [CrossRef]
- Ballard, J.W.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [Green Version]
- Cañón, J.; Alexandrino, P.; Bessa, I.; Carleos, C.; Carretero, Y.; Dunner, S.; Ferran, N.; Garcia, D.; Jordana, J.; Laloë, D.; et al. Genetic diversity measures of local European beef cattle breeds for conservation purposes. Genet. Sel. Evol. 2001, 33, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taberlet, P.; Griffin, S.; Goossens, B.; Questiau, S.; Manceau, V.; Escaravage, N.; Waits, L.P.; Bouvet, J. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996, 24, 3189–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taberlet, P.; Waits, L.P.; Luikart, G. Noninvasive genetic sampling: Look before you leap. Trends Ecol. Evol. 1999, 14, 323–327. [Google Scholar] [CrossRef]
- Schlötterer, C.; Tautz, D. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 1992, 20, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Navidi, W.; Arnheim, N.; Waterman, M.S. A multiple-tube approach for accurate genotyping of very small DNA samples by using PCR: Statistical considerations. Am. J. Hum. Genet. 1992, 50, 347–359. [Google Scholar]
- FAO. Molecular Genetic Characterization of Animal Genetic Resources; FAO Animal Production and Health Guidelines: Rome, Italy, 2011; pp. 68–69. Available online: http://www.fao.org/3/i2413e/i2413e00.pdf (accessed on 15 July 2020).
- Bishop, M.D.; Kappes, S.M.; Keele, J.W.; Stone, R.T.; Sunden, S.L.; Hawkins, G.A.; Toldo, S.S.; Fries, R.; Grosz, M.D.; Yoo, J.; et al. A genetic linkage map for cattle. Genetics 1994, 136, 619–639. [Google Scholar]
- Toldo, S.S.; Fries, R.; Steffen, P.; Neiberg, H.L.; Barendse, W.; Womack, J.E.; Hetzel, D.J.S.; Stranzinger, G. Physically mapped, cosmid-derived microsatellite markers as anchor loci on bovine chromosomes. Mamm. Genome 1993, 4, 720–727. [Google Scholar] [CrossRef]
- Steffen, P.; Eggen, A.; Stranzinger, G.; Fries, R.; Dietz, A.B.; Womack, J.E. Isolation and mapping of polymorphic microsatellites in cattle. Anim. Genet. 2009, 24, 121–124. [Google Scholar] [CrossRef]
- Vaiman, D.; Mercier, D.; Moazami-Goudarzi, K.; Eggen, A.; Ciampolini, R.; Lepingle, A.; Velmala, R.; Kaukinen, J.; Varvio, S.L.; Martin, P.; et al. A set of 99 cattle microsatellites: Characterization, synteny mapping, and polymorphism. Mamm. Genome 1994, 5, 288–297. [Google Scholar] [CrossRef]
- Moore, S.S.; Byrne, K.; Berger, K.T.; Barendse, W.; McCarthy, F.; Womack, J.E.; Hetzel, D.J.S. Characterization of 65 bovine microsatellites. Mamm. Genome 1994, 5, 84–90. [Google Scholar] [CrossRef]
- Barendse, W.; Armitage, S.M.; Kossarek, L.M.; Shalom, A.; Kirkpatrick, B.W.; Ryan, A.M.; Clayton, D.; Li, L.; Neibergs, H.L.; Zhang, N.; et al. A genetic linkage map of the bovine genome. Nat. Genet. 1994, 6, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Mondol, S.; Karanth, K.U.; Kumar, N.S.; Gopalaswamy, A.M.; Andheria, A.; Ramakrishnan, U. Evaluation of non-invasive genetic sampling methods for estimating tiger population size. Biol. Conserv. 2009, 142, 2350–2360. [Google Scholar] [CrossRef]
- Modi, S.; Habib, B.; Ghaskadbi, P.; Nigam, P.; Mondol, S. Standardization and validation of a panel of cross-species microsatellites to individually identify the Asiatic wild dog (Cuon alpinus). PeerJ 2019, 7, e7453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, C.; Bellemain, E.; Poillot, C.; Bessière, J.; Durand, A.; Taberlet, P. Quality indexes to assess the reliability of genotypes in studies using noninvasive sampling and multiple-tube approach. Mol. Ecol. Notes 2006, 6, 985–988. [Google Scholar] [CrossRef]
- Broquet, T.; Petit, E. Quantifying genotyping errors in noninvasive population genetics. Mol. Ecol. 2004, 13, 3601–3608. [Google Scholar] [CrossRef]
- He, G.; Huang, K.; Guo, S.T.; Ji, W.H.; Qi, X.G.; Ren, Y.; Jin, X.L.; Li, B.G. Evaluating the reliability of microsatellite genotyping from low-quality DNA templates with a polynomial distribution model. Chin. Sci. Bull. 2011, 56, 2523–2530. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Chakraborty, R.; Kimmel, M.; Stivers, D.N.; Davison, L.J.; Deka, R. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc. Natl. Acad. Sci. USA 1997, 94, 1041–1046. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Ritland, K.; Dunn, D.W.; Qi, X.; Guo, S.; Li, B. Estimating relatedness in the presence of null alleles. Genetics 2016, 202, 247–260. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis (Use R!); Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 12 May 2020).
- Jost, L. GST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. Available online: http://clumpak.tau.ac.il (accessed on 8 July 2020). [CrossRef] [PubMed] [Green Version]
- MacHugh, D.E.; Troy, C.S.; McCormick, F.; Olsaker, I.; Eythórsdóttir, E.; Bradle, D.G. Early medieval cattle remains from a Scandinavian settlement in Dublin: Genetic analysis and comparison with extant breeds. Philos. Trans. R. Soc. B 1999, 354, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Bollongino, R.; Edwards, C.J.; Alt, K.W.; Burger, J.; Bradley, D.G. Early history of European domestic cattle as revealed by ancient DNA. Biol. Lett. 2006, 2, 155–159. [Google Scholar] [CrossRef]
- Beja-Pereira, A.; Caramelli, D.; Lalueza-Fox, C.; Vernesi, C.; Ferrand, N.; Casoli, A.; Goyache, F.; Royo, L.J.; Conti, S.; Lari, M.; et al. The origin of European cattle: Evidence from modern and ancient DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 8113–8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Zhang, N.; Shao, X.; Sun, W.; Zhu, S.; Yang, D.Y. New ancient DNA data on the origins and spread of sheep and cattle in northern China around 4000 BP. Asian Archaeol. 2018, 2, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Siripan, S.; Wonnapinij, P.; Auetrakulvit, P.; Wangthongchaicharoen, N.; Surat, W. Origin of prehistoric cattle excavated from four archaeological sites in central and northeastern Thailand. Mitochondrial DNA Part A 2019, 30, 609–617. [Google Scholar] [CrossRef]
- Larson, G.; Burger, J. A population genetics view of animal domestication. Trends Genet. 2013, 29, 197–205. [Google Scholar] [CrossRef]
- Liskun, E.F. Russkie Otrod’ya Krupno-Rogatogo Skota; Novyj agronom: Moskva, Russia, 1928. (In Russian) [Google Scholar]
- Arandjelovic, M.; Guschanski, K.; Schubert, G.; Harris, T.R.; Thalmann, O.; Siedel, H.; Vigilant, L. Two-step multiplex polymerase chain reaction improves the speed and accuracy of genotyping using DNA from noninvasive and museum samples. Mol. Ecol. Resour. 2009, 9, 28–36. [Google Scholar] [CrossRef]
- Butler, J.M.; Shen, Y.; McCord, B.R. The development of reduced size STR amplicons as tools for analysis of degraded DNA. J. Forensic Sci. 2003, 48, 1054–1064. Available online: www.astm.org (accessed on 8 August 2020). [CrossRef]
- Sefc, K.M.; Payne, R.B.; Sorenson, M.D. Microsatellite amplification from museum feather samples: Effects of fragment size and template concentration on genotyping errors. Auk 2003, 120, 982–989. [Google Scholar] [CrossRef]
- Polanc, P.; Sindičić, M.; Jelenčič, M.; Gomerčič, T.; Kos, I.; Huber, Đ. Genotyping success of historical Eurasian lynx (Lynx lynx L.) samples. Mol. Ecol. Resour. 2012, 2, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kantanen, J.; Edwards, C.J.; Bradley, D.G.; Viinalass, H.; Thessler, S.; Ivanova, Z.; Kiselyova, T.; Ćinkulov, M.; Popov, R.; Stojanović, S.; et al. Maternal and paternal genealogy of Eurasian taurine cattle (Bos taurus). Heredity 2009, 103, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamborg, C.; Sandøe, P. Sustainable farm animal breeding: A review. Livest. Prod. Sci. 2005, 92, 221–231. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Locus | Observed Allele Ranges, bp a | Number of Alleles | Amplification Failure, % | ADO Rate, % | FA Rate, % | ER, % |
|---|---|---|---|---|---|---|---|
| 1 | BM2113 | 121–141 | 10 | 0.00 | 1.29 | 0.37 | 1.47 |
| 2 | BM1824 | 178–188 | 5 | 0.74 | 3.24 | 0.74 | 2.96 |
| 3 | ETH10 | 211–225 | 8 | 2.94 | 3.23 | 0.38 | 2.65 |
| 4 | ETH225 | 140–160 | 8 | 1.84 | 1.03 | 0.37 | 1.12 |
| 5 | INRA023 | 198–216 | 9 | 5.88 | 2.29 | 1.17 | 3.13 |
| 6 | SPS115 | 248–260 | 6 | 5.51 | 4.07 | 1.17 | 3.89 |
| 7 | TGLA122 | 139–181 | 16 | 0.74 | 1.99 | 1.11 | 2.59 |
| 8 | TGLA126 | 115–127 | 7 | 2.57 | 3.69 | 0.75 | 3.77 |
| 9 | TGLA227 | 77–103 | 12 | 0.74 | 0.90 | 1.11 | 1.85 |
| Mean value | 9.00 ± 3.35 | 2.33 ± 0.31 | 2.35 ± 0.36 | 0.79 ± 0.18 | 2.59 ± 0.33 | ||
| Population | n | HO (M ± SE) | UHE (M ± SE) | AR (M ± SE) | UFIS (CI) |
|---|---|---|---|---|---|
| KH_H | 22 | 0.783 ± 0.030 | 0.726 ± 0.026 | 2.716 ± 0.092 | −0.080 (−0.131; −0.029) |
| YR_H | 20 | 0.789 ± 0.036 | 0.774 ± 0.023 | 2.893 ± 0.088 | −0.020 (−0.099; 0.059) |
| GR_H | 2 | 0.722 ± 0.121 | 0.852 ± 0.043 | 3.111 ± 0.261 | 0.167 (−0.118; 0.452) |
| NV_H | 2 | 0.611 ± 0.111 | 0.722 ± 0.104 | 2.778 ± 0.324 | 0.137 (−0.088; 0.362) |
| HL_H | 3 | 0.556 ± 0.111 | 0.644 ± 0.100 | 2.533 ± 0.268 | 0.147 (−0.023; 0.317) |
| KH_M | 177 | 0.714 ± 0.027 | 0.708 ± 0.031 | 2.661 ± 0.106 | −0.011 (−0.030; 0.008) |
| YR_M | 61 | 0.674 ± 0.045 | 0.739 ± 0.032 | 2.758 ± 0.106 | 0.080 (−0.039; 0.199) |
| HS_M | 152 | 0.705 ± 0.035 | 0.697 ± 0.031 | 2.616 ± 0.104 | −0.011 (−0.045; 0.023) |
| Population | KH_H | YR_H | GR_H | NV_H | HL_H | KH_M | YR_M | HS_M |
|---|---|---|---|---|---|---|---|---|
| KH_H | - | 0.017 | 0.072 | 0.068 | 0.060 | 0.018 | 0.047 | 0.076 |
| YR_H | 0.024 | - | 0.033 | 0.060 | 0.061 | 0.041 | 0.027 | 0.062 |
| GR_H | 0.025 | 0.003 | - | −0.061 | 0.006 | 0.089 | 0.034 | 0.079 |
| NV_H | 0.008 | 0.034 | −0.018 | - | 0.067 | 0.107 | 0.063 | 0.133 |
| HL_H | 0.021 | 0.031 | −0.008 | −0.001 | - | 0.064 | 0.082 | 0.085 |
| KH_M | 0.016 | 0.081 | 0.020 | 0.048 | 0.067 | - | 0.071 | 0.075 |
| YR_M | 0.070 | 0.056 | 0.001 | 0.045 | 0.123 | 0.174 | - | 0.105 |
| HS_M | 0.146 | 0.125 | 0.016 | 0.082 | 0.156 | 0.127 | 0.256 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Mishina, A.I.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Petrikeeva, L.V.; Chinarov, R.Y.; Brem, G.; et al. Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes 2020, 11, 940. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080940
Abdelmanova AS, Kharzinova VR, Volkova VV, Mishina AI, Dotsev AV, Sermyagin AA, Boronetskaya OI, Petrikeeva LV, Chinarov RY, Brem G, et al. Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis. Genes. 2020; 11(8):940. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080940
Chicago/Turabian StyleAbdelmanova, Alexandra S., Veronika R. Kharzinova, Valeria V. Volkova, Arina I. Mishina, Arsen V. Dotsev, Alexander A. Sermyagin, Oxana I. Boronetskaya, Lidia V. Petrikeeva, Roman Yu Chinarov, Gottfried Brem, and et al. 2020. "Genetic Diversity of Historical and Modern Populations of Russian Cattle Breeds Revealed by Microsatellite Analysis" Genes 11, no. 8: 940. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11080940