Identification of Novel Interspersed DNA Repetitive Elements in the Trypanosoma cruzi Genome Associated with the 3′UTRs of Surface Multigenic Families

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
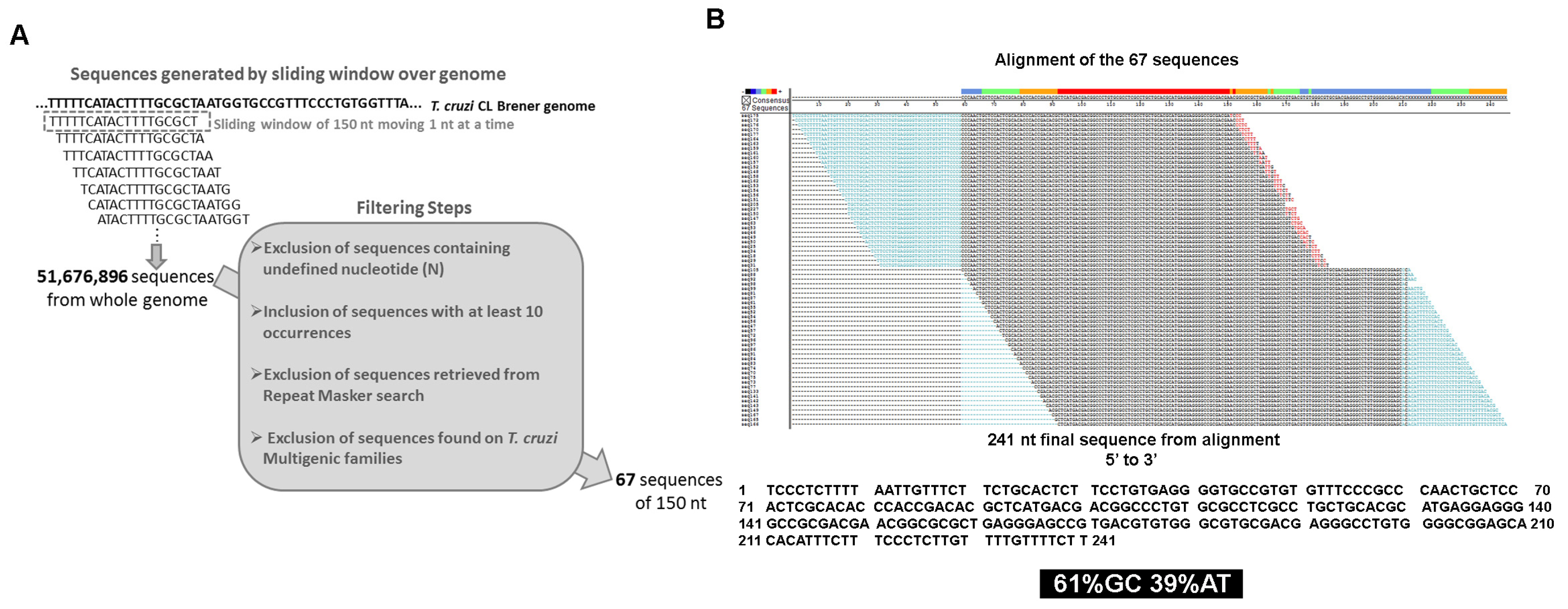
2.1. Filtering Steps for 150 bp Fragments
2.2. Search Terms in the TriTryp Database
2.3. Statistical Analysis
2.4. Genomes Analyzed
2.5. Monte Carlo Test of a 241 nt Repeat
2.6. RNA-seq Analysis
2.7. TPM Statistical Test
3. Results
3.1. Identification and Distribution of a Novel DNA Repeat on the T. cruzi Genome
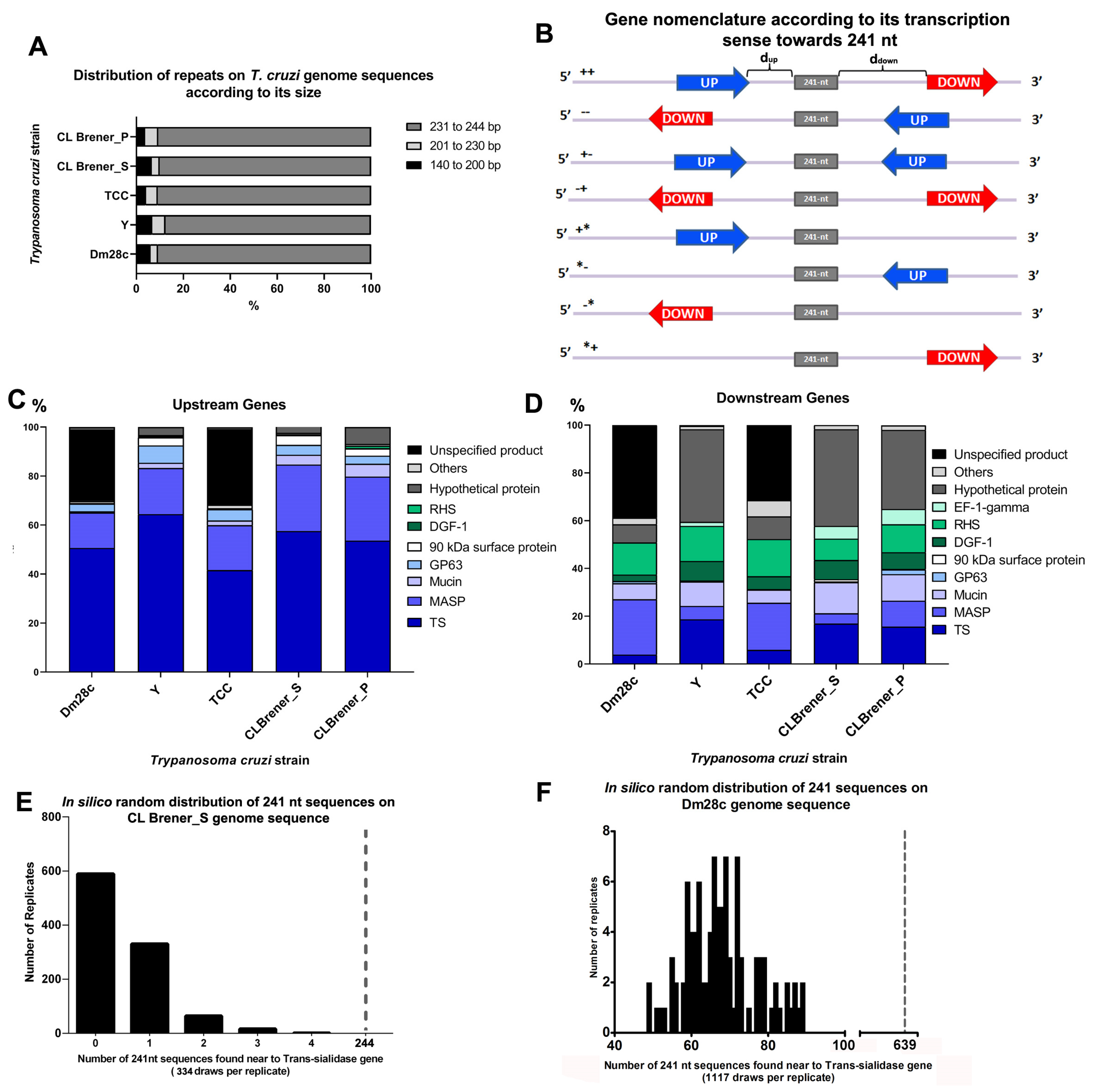
3.2. The 241 nt Repeat Is Enriched at Intergenic Regions of T. cruzi Genome Sequences
3.3. Regions Upstream and Downstream of the 241 nt Repeats Are Enriched in Surface Protein Genes
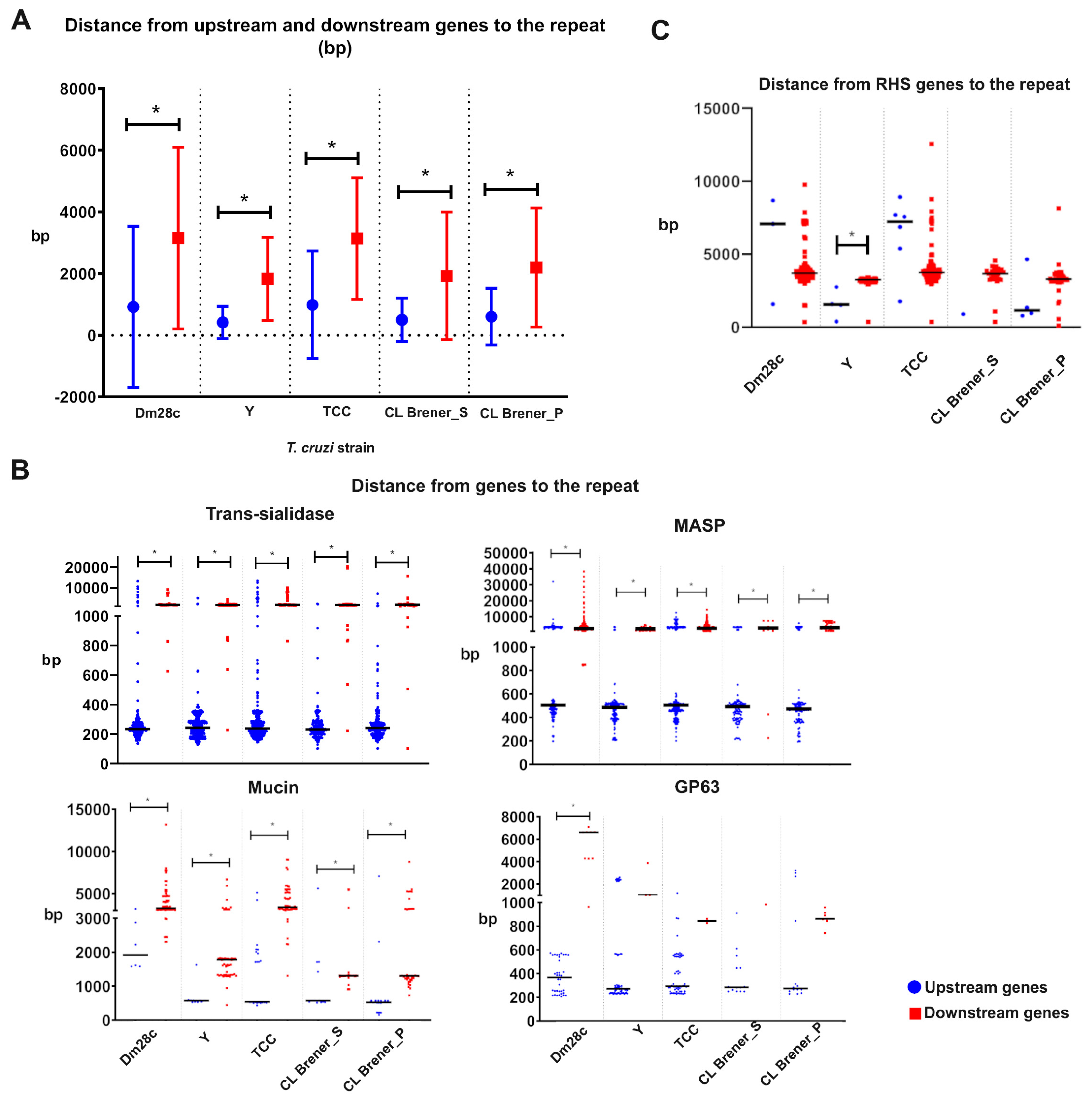
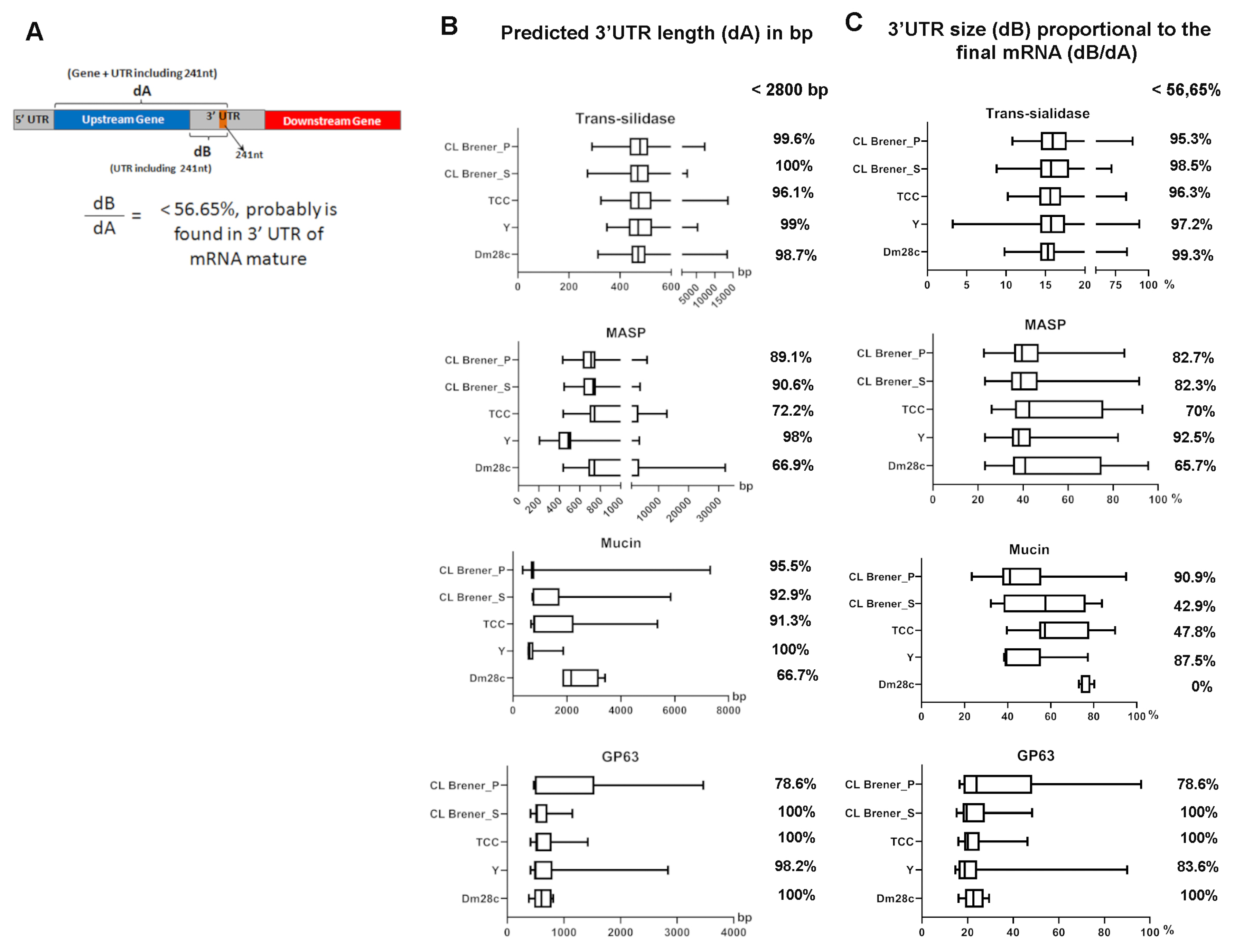
3.4. The 241 nt Repeat Is Found Closer to Upstream Genes and May be Part of the 3′UTR of Trans-Sialidase Gene mRNA
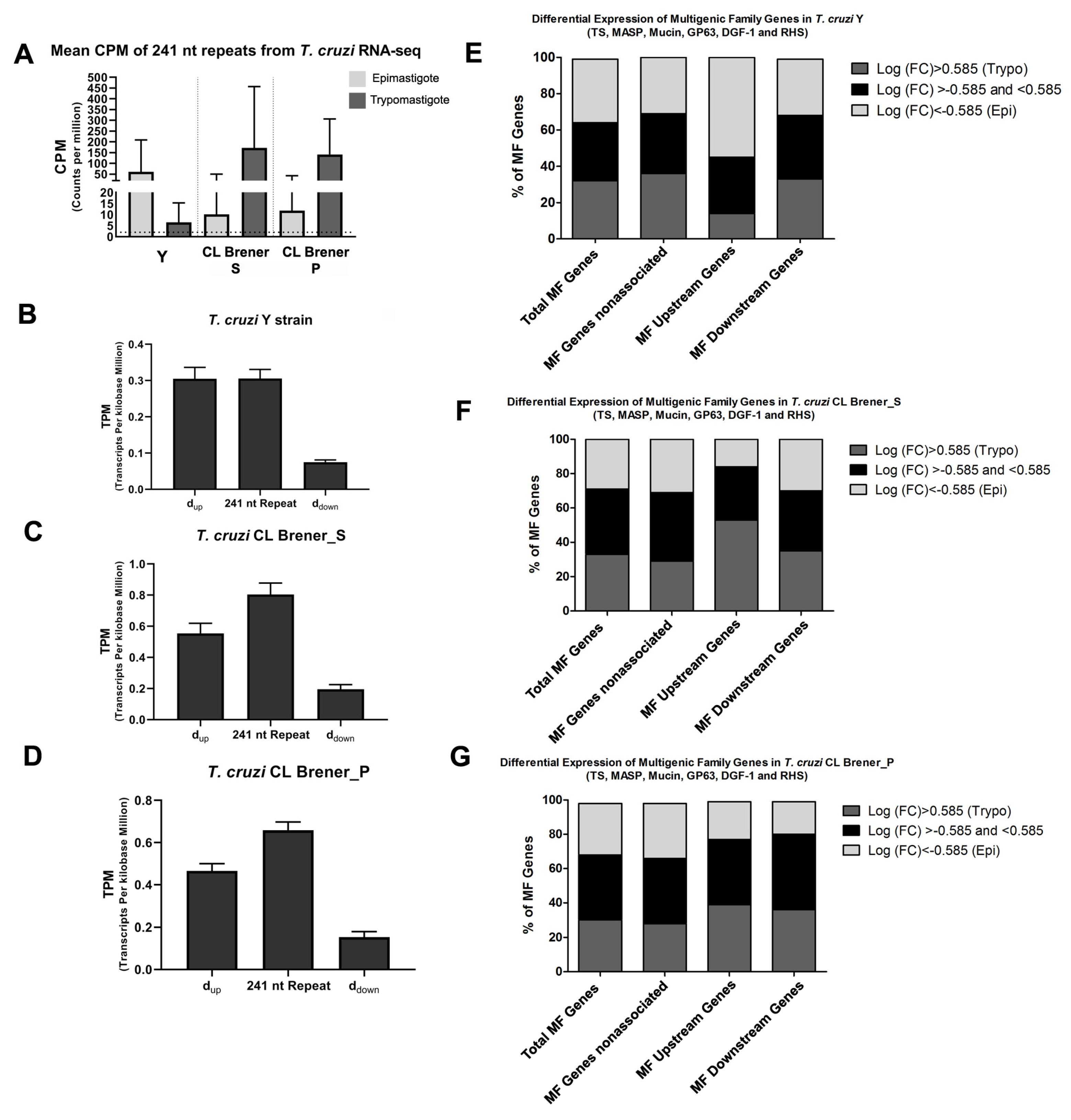
3.5. The 241 nt Repeats Are Found Significantly Expressed in Transcriptomes and Highly Correlated to the mRNA 3′UTR Sequence
- (i)
- The TPM parameter interprets the transcriptional abundances of determined regions, allowing comparison of the proportion of reads among mapped regions because the TPM normalizes the depth and length of the sequencing data.
- (ii)
- The dup encloses the 3′ UTR, while the ddown corresponds to the entire region between the nucleotide just after the 241 nt repeat to the last nucleotide before downstream gene. Therefore, the ddown encloses the segment transcribed from the genome but lost after trans-splicing as well as the 5′ UTR of the downstream gene. Thus, dup will have higher amounts of transcripts (higher TPM), and ddown will have lower amounts of transcripts (lower TPM).
3.6. Distinct Expression Profile Between the Epimastigote and Trypomastigote of Genes Is Associated with the 241 nt Repeat
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stanaway, J.D.; Roth, G. The burden of Chagas disease: Estimates and challenges. Glob. Heart 2015, 10, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, R.M.; Charneau, S.; Bastos, I.M.; Santana, J.M.; Sousa, M.V.; Roepstorff, P.; Ricart, C.A. Cell surface proteome analysis of human-hosted Trypanosoma cruzi life stages. J. Proteome Res. 2014, 13, 3530–3541. [Google Scholar] [CrossRef] [PubMed]
- Atwood, J.; Weatherly, D.B.; Minning, T.A.; Bundy, B.; Cavola, C.; Opperdoes, F.R.; Orlando, R.; Tarleton, R.L. The Trypanosoma cruzi proteome. Science 2005, 309, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Minning, T.A.; Weatherly, D.B.; Atwood, J.; Orlando, R.; Tarleton, R.L. The steady-state transcriptome of the four major life-cycle stages of Trypanosoma cruzi. BMC Genom. 2009, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Calvillo, S.; Vizuet-de-Rueda, J.C.; Florencio-Martinez, L.E.; Manning-Cela, R.G.; Figueroa-Angulo, E.E. Gene expression in trypanosomatid parasites. J. Biomed. Biotechnol. 2010, 2010, 525241. [Google Scholar] [CrossRef] [Green Version]
- Jager, A.V.; De Gaudenzi, J.G.; Cassola, A.; D’Orso, I.; Frasch, A.C. mRNA maturation by two-step trans-splicing/polyadenylation processing in trypanosomes. Proc. Natl. Acad. Sci. USA 2007, 104, 2035–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardelli, S.C.; Ávila, A.R.; Freund, A.; Motta, M.C.; Manhães, L.; De Jesus, T.C.L.; Schenkman, S.; Fragoso, S.P.; Krieger, M.A.; Goldenberg, S.; et al. Small-subunit rRNA processome proteins are translationally regulated during differentiation of Trypanosoma cruzi. Eukaryot. Cell 2007, 6, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Holetz, F.B.; Correa, A.; Avila, A.R.; Nakamura, C.V.; Krieger, M.A.; Goldenberg, S. Evidence of P-body-like structures in Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2007, 356, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Cassola, A.; De Gaudenzi, J.G.; Frasch, A.C. Recruitment of mRNAs to cytoplasmic ribonucleoprotein granules in trypanosomes. Mol. Microbiol. 2007, 65, 655–670. [Google Scholar] [CrossRef]
- McNicoll, F.; Müller, M.; Cloutier, S.; Boilard, N.; Rochette, A.; Dubé, M.; Papadopoulou, B. Distinct 3′-untranslated region elements regulate stage-specific mRNA accumulation and translation in Leishmania. J. Biol. Chem. 2005, 280, 35238–35246. [Google Scholar] [CrossRef] [Green Version]
- Jeacock, L.; Faria, J.; Horn, D. Codon usage bias controls mRNA and protein abundance in trypanosomatids. Elife 2018, 7, e32496. [Google Scholar] [CrossRef]
- Horn, D. Codon usage suggests that translational selection has a major impact on protein expression in trypanosomatids. BMC Genom. 2008, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, S. Trans-splicing in trypanosomes: Machinery and its impact on the parasite transcriptome. Future Microbiol. 2011, 6, 459–474. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.M.; Myler, P.J.; Bartholomeu, D.C.; Nilsson, D.; Aggarwal, G.; Tran, A.N.; Ghedin, E.; Worthey, E.A.; Delcher, A.L.; Blandin, G.; et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 2005, 309, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pita, S.; Diaz-Viraque, F.; Iraola, G.; Robello, C. The Tritryps comparative repeatome: Insights on repetitive element evolution in Trypanosomatid pathogens. Genome Biol. Evol. 2019, 11, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Callejas-Hernández, F.; Rastrojo, A.; Poveda, C.; Gironès, N.; Fresno, M. Genomic assemblies of newly sequenced Trypanosoma cruzi strains reveal new genomic expansion and greater complexity. Sci. Rep. 2018, 8, 14631. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, J.D.; Guhl, F.; Umezawa, E.S.; Morillo, C.A.; Rosas, F.; Marin-Neto, J.A.; Restrepo, S. Evaluation of adult chronic Chagas’ heart disease diagnosis by molecular and serological methods. J. Clin. Microbiol. 2009, 47, 3945–3951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pech-Canul, A.C.; Monteon, V.; Solis-Oviedo, R.L. A brief view of the surface membrane proteins from Trypanosoma cruzi. J. Parasitol. Res. 2017, 2017, 3751403. [Google Scholar] [CrossRef] [Green Version]
- Di Noia, J.M.; D’Orso, I.; Sanchez, D.O.; Frasch, A.C. AU-rich elements in the 3′-untranslated region of a new mucin-type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. J. Biol. Chem. 2000, 275, 10218–10227. [Google Scholar] [CrossRef] [Green Version]
- D’Orso, I.; Frasch, A.C. Functionally different AU-and G-rich cis-elements confer developmentally regulated mRNA stability in Trypanosoma cruzi by interaction with specific RNA-binding proteins. J. Biol. Chem. 2001, 276, 15783–15793. [Google Scholar] [CrossRef] [Green Version]
- Clayton, C.; Shapira, M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.H.R.; Green, P. Repeat Masker Open-4.0 2013–2015. Available online: www.repeatmasker.org (accessed on 22 June 2016).
- Aslett, M.; Aurrecoechea, C.; Berriman, M.; Brestelli, J.; Brunk, B.P.; Carrington, M.; Depledge, D.P.; Fischer, S.; Gajria, B.; Gao, X.; et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010, 38, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroese, D.P.; Brereton, T.; Taimre, T.; Botev, Z.I. Why the Monte Carlo method is so important today. WIREs Comput. Stat. 2014, 6, 386–392. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. A Language and Environment for Statistical Computing. R Found. Stat. Comput. 2020. Available online: https://www.R-project.org/ (accessed on 11 May 2020).
- Martin, C.; Baptista, C.S.; Ienne, S.; Cerqueira, G.C.; Bartholomeu, D.C.; Zingales, B. Genomic organization and transcription analysis of the 195-bp satellite DNA in Trypanosoma cruzi. Mol. Biochem. Parasitol. 2008, 160, 60–64. [Google Scholar] [CrossRef]
- Franzén, O.; Ochaya, S.; Sherwood, E.; Lewis, M.D.; Llewellyn, M.S.; Miles, M.A.; Andersson, B. Shotgun sequencing analysis of Trypanosoma cruzi I Sylvio X10/1 and comparison with T. cruzi VI CL Brener. PLoS Negl. Trop. Dis. 2011, 5, e984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandao, A.; Jiang, T. The composition of untranslated regions in Trypanosoma cruzi genes. Parasitol. Int. 2009, 58, 215–219. [Google Scholar] [CrossRef]
- Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [CrossRef] [Green Version]
- Li, Y.; Shah-Simpson, S.; Okrah, K.; Belew, A.T.; Choi, J.; Caradonna, K.L.; Padmanabhan, P.; Ndegwa, D.M.; Temanni, M.R.; Corrada Bravo, H.; et al. Transcriptome Remodeling in Trypanosoma cruzi and Human Cells during Intracellular Infection. PLoS Pathog. 2016, 12, e1005511. [Google Scholar] [CrossRef] [Green Version]
- Houston-Ludlam, G.A.; Belew, A.T.; El-Sayed, N.M. Comparative transcriptome profiling of human foreskin fibroblasts infected with the Sylvio and y strains of Trypanosoma cruzi. PLoS ONE 2016, 11, e0159197. [Google Scholar] [CrossRef] [Green Version]
- Reis-Cunha, J.L.; Rodrigues-Luiz, G.F.; Valdivia, H.O.; Baptista, R.P.; Mendes, T.A.; De Morais, G.L.; Guedes, R.; Macedo, A.M.; Bern, C.; Gilman, R.H.; et al. Chromosomal copy number variation reveals differential levels of genomic plasticity in distinct Trypanosoma cruzi strains. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Ghedin, E.; Bringaud, F.; Peterson, J.; Myler, P.; Berriman, M.; Ivens, A.; Andersson, B.; Bontempi, E.; Eisen, J.; Angiuoli, S.; et al. Gene synteny and evolution of genome architecture in trypanosomatids. Mol. Biochem. Parasitol. 2004, 134, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Berná, L.; Rodríguez, M.; Chiribao, M.L.; Parodi-Talice, A.; Pita, S.; Rijo, G.; Alvarez-Valin, F.; Robello, C. Expanding an expanded genome: Long-read sequencing of Trypanosoma cruzi. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, B.A.A.; Holetz, F.B.; Alves, L.R.; Goldenberg, S. RNA binding proteins and gene expression regulation in Trypanosoma cruzi. Front. Cell Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.A. Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006, 22, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Bakheet, T.; Frevel, M.; Williams, B.R.; Greer, W.; Khabar, K.S. ARED: Human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001, 29, 246–254. [Google Scholar] [CrossRef]
- D’Orso, I.; Frasch, A.C. TcUBP-1, a developmentally regulated U-rich RNA-binding protein involved in selective mRNA destabilization in trypanosomes. J. Biol. Chem. 2001, 276, 34801–34809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Orso, I.; De Gaudenzi, J.G.; Frasch, A.C. RNA-binding proteins and mRNA turnover in trypanosomes. Trends Parasitol. 2003, 19, 151–155. [Google Scholar] [CrossRef]
- De Gaudenzi, J.G.; Carmona, S.J.; Aguero, F.; Frasch, A.C. Genome-wide analysis of 3′-untranslated regions supports the existence of post-transcriptional regulons controlling gene expression in trypanosomes. PeerJ 2013, 1, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.H.; De Gaudenzi, J.G.; Alvarez, V.E.; Mendiondo, N.; Wang, H.; Kissinger, J.C.; Frasch, A.C.; Docampo, R. A 43-nucleotide U-rich element in 3′-untranslated region of large number of Trypanosoma cruzi transcripts is important for mRNA abundance in intracellular amastigotes. J. Biol. Chem. 2012, 287, 19058–19069. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DTU | B7 | TcI | TcII | TcV | TcVI | ||||
|---|---|---|---|---|---|---|---|---|---|
| T. cruzi strain | Marinkellei | Dm28c * | Brazil A4 | Sylvio X10/1 | Y C6 * | Bug2148 | TCC * | CL Brener S * | CL Brener P * |
| Chr count | 0 | 0 | 43 | 47 | 40 | 0 | 0 | 41 | 41 |
| Contig count | 16783 | 636 | 359 | 0 | 226 | 929 | 1236 | 0 | 0 |
| Genome size (Mbp) | 38.65 | 53.27 | 45.56 | 41.38 | 47.22 | 55.16 | 87.06 | 32.53 | 32.53 |
| Total gene count | 10282 | 19112 | 18779 | 20684 | 17713 | NA | 29302 | 10596 | 11106 |
| Y C6 | CL Brener | ||||
|---|---|---|---|---|---|
| S and P | S | P | |||
| Chr | Chr Size (kb) | N° of Repeats | Chr Size (kb) | N° of Repeats | N° of Repeats |
| 1 | 2,950,016 | 146 | 77,958 | 0 | 0 |
| 2 | 1,943,341 | 3 | 151.74 | 2 | 1 |
| 3 | 1608.8 | 73 | 196,644 | 1 | 0 |
| 4 | 1,578,048 | 2 | 200,401 | 3 | 1 |
| 5 | 1,465,819 | 45 | 227,319 | 0 | 1 |
| 6 | 1,365,397 | 24 | 389,024 | 3 | 2 |
| 7 | 1238.82 | 2 | 391,095 | 9 | 3 |
| 8 | 1,238,493 | 2 | 393,493 | 1 | 2 |
| 9 | 1,233,391 | 8 | 509,634 | 0 | 1 |
| 10 | 1,196,034 | 6 | 518,846 | 0 | 0 |
| 11 | 1,179,968 | 2 | 526.14 | 4 | 1 |
| 12 | 1,154,569 | 3 | 533,093 | 2 | 4 |
| 13 | 1,073,329 | 10 | 558,364 | 1 | 0 |
| 14 | 1041.73 | 6 | 598,625 | 5 | 7 |
| 15 | 973,991 | 10 | 612,853 | 4 | 17 |
| 16 | 931,817 | 0 | 646,207 | 11 | 16 |
| 17 | 919,065 | 3 | 648,584 | 8 | 4 |
| 18 | 889,019 | 10 | 655,081 | 28 | 23 |
| 19 | 879,731 | 15 | 671,453 | 7 | 1 |
| 20 | 835,455 | 23 | 656,799 | 15 | 5 |
| 21 | 802.19 | 3 | 704,149 | 4 | 9 |
| 22 | 794,882 | 13 | 710,778 | 3 | 2 |
| 23 | 771,598 | 18 | 655,477 | 5 | 7 |
| 24 | 748,092 | 8 | 779,922 | 6 | 10 |
| 25 | 747,041 | 8 | 822,374 | 9 | 24 |
| 26 | 713.53 | 17 | 801,422 | 9 | 6 |
| 27 | 704,292 | 0 | 850,241 | 0 | 0 |
| 28 | 683,656 | 4 | 853,233 | 12 | 27 |
| 29 | 683,261 | 6 | 870,934 | 12 | 18 |
| 30 | 618,893 | 9 | 863,882 | 2 | 2 |
| 31 | 613,739 | 9 | 947,473 | 4 | 6 |
| 32 | 587,789 | 6 | 968,069 | 0 | 1 |
| 33 | 572,88 | 19 | 1,041,172 | 3 | 13 |
| 34 | 565,606 | 1 | 1,065,764 | 4 | 1 |
| 35 | 563,146 | 6 | 1,186,946 | 1 | 4 |
| 36 | 542,602 | 21 | 1,180,744 | 2 | 1 |
| 37 | 354,446 | 3 | 1,355,803 | 3 | 2 |
| 38 | 332,206 | 0 | 1,444,805 | 24 | 67 |
| 39 | 241,231 | 2 | 1,854,104 | 3 | 3 |
| 40 | 239,696 | 5 | 2,036,759 | 34 | 8 |
| 41 | 2,371,736 | 90 | 108 | ||
| unplaced contig | 201 | \ | |||
| Total | 752 | 334 | 408 | ||
| TcI | TcII | TcVI | |||
|---|---|---|---|---|---|
| Dm28c | YC6 | TCC | CLBrener_S | CLBrener_P | |
| ++ | 472 | 349 | 472 | 150 | 190 |
| −− | 512 | 351 | 516 | 148 | 163 |
| +− | 94 | 38 | 103 | 22 | 45 |
| −+ | 2 | 1 | 11 | 2 | 3 |
| +* | 11 | 4 | 24 | 4 | 1 |
| *− | 18 | 6 | 30 | 8 | 6 |
| −* | 5 | 1 | 5 | 0 | 0 |
| *+ | 3 | 2 | 10 | 0 | 0 |
| Total | 1117 | 752 | 1171 | 334 | 408 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calderano, S.G.; Nishiyama Junior, M.Y.; Marini, M.; Nunes, N.d.O.; Reis, M.d.S.; Patané, J.S.L.; da Silveira, J.F.; da Cunha, J.P.C.; Elias, M.C. Identification of Novel Interspersed DNA Repetitive Elements in the Trypanosoma cruzi Genome Associated with the 3′UTRs of Surface Multigenic Families. Genes 2020, 11, 1235. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101235
Calderano SG, Nishiyama Junior MY, Marini M, Nunes NdO, Reis MdS, Patané JSL, da Silveira JF, da Cunha JPC, Elias MC. Identification of Novel Interspersed DNA Repetitive Elements in the Trypanosoma cruzi Genome Associated with the 3′UTRs of Surface Multigenic Families. Genes. 2020; 11(10):1235. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101235
Chicago/Turabian StyleCalderano, Simone Guedes, Milton Yutaka Nishiyama Junior, Marjorie Marini, Nathan de Oliveira Nunes, Marcelo da Silva Reis, José Salvatore Leister Patané, José Franco da Silveira, Julia Pinheiro Chagas da Cunha, and Maria Carolina Elias. 2020. "Identification of Novel Interspersed DNA Repetitive Elements in the Trypanosoma cruzi Genome Associated with the 3′UTRs of Surface Multigenic Families" Genes 11, no. 10: 1235. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11101235