A Review of the Important Role of CYP2D6 in Pharmacogenomics
by
 , , and
, , and
Christopher Taylor
1,2,* ,
,
Ian Crosby
2,
Vincent Yip
1,
Peter Maguire
2,
Munir Pirmohamed
1 and
Richard M. Turner
1 1
Wolfson Centre for Personalised Medicine, University of Liverpool, Liverpool L69 3BX, UK
2
MC Diagnostics, St Asaph Business Park, Saint Asaph LL17 0LJ, UK
*
Author to whom correspondence should be addressed.
Genes 2020, 11(11), 1295; https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111295
Submission received: 26 September 2020
/
Revised: 25 October 2020
/
Accepted: 28 October 2020
/
Published: 30 October 2020
(This article belongs to the Special Issue Pharmacogenomic Determinants of Interindividual Drug Response Variability: From Discovery to Implementation)
Abstract
:Cytochrome P450 2D6 (CYP2D6) is a critical pharmacogene involved in the metabolism of ~20% of commonly used drugs across a broad spectrum of medical disciplines including psychiatry, pain management, oncology and cardiology. Nevertheless, CYP2D6 is highly polymorphic with single-nucleotide polymorphisms, small insertions/deletions and larger structural variants including multiplications, deletions, tandem arrangements, and hybridisations with non-functional CYP2D7 pseudogenes. The frequency of these variants differs across populations, and they significantly influence the drug-metabolising enzymatic function of CYP2D6. Importantly, altered CYP2D6 function has been associated with both adverse drug reactions and reduced drug efficacy, and there is growing recognition of the clinical and economic burdens associated with suboptimal drug utilisation. To date, pharmacogenomic clinical guidelines for at least 48 CYP2D6-substrate drugs have been developed by prominent pharmacogenomics societies, which contain therapeutic recommendations based on CYP2D6-predicted categories of metaboliser phenotype. Novel algorithms to interpret CYP2D6 function from sequencing data that consider structural variants, and machine learning approaches to characterise the functional impact of novel variants, are being developed. However, CYP2D6 genotyping is yet to be implemented broadly into clinical practice, and so further effort and initiatives are required to overcome the implementation challenges and deliver the potential benefits to the bedside.
1. Introduction
1.1. Background to Cytochrome P450 Enzymes
The cytochrome P450 (CYP) superfamily is an ancient family of enzymes identified in hundreds of eukaryote and prokaryote species [1,2], and are named thus because they strongly absorb 450 nm wavelength light when reduced and bound by carbon monoxide. CYP standard nomenclature is the prefix, “CYP”, followed by a number for the family (proteins sharing more than 40% amino acid sequence identity), a letter for the subfamily (at least 55% identity) and a number for the specific gene; for example, CYP2D6 [3]. The human genome encodes 57 putatively functional CYP genes along with 58 pseudogenes [4]. Of these 57 functional human CYPs, 12 are implicated in the metabolism of 70–80% of commonly used drugs; specifically, these are: CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2J2, CYP3A4 and CYP3A5 [4].
CYPs are hemoproteins and there is extensive diversity of CYP gene sequences [5,6]. However, the three-dimensional (3D) structure of the common CYP fold is highly conserved, being predominantly α-helical (usually 12–13 α-helices) with a small number of β-sheets [5,6,7]. The structural domains that surround the heme complex form the substrate recognition and access channel [3]. CYP enzymes are typically the terminal oxidase enzymes in electron transfer chains. The major electron donor for microsomal CYPs is P450 reductase (POR), which donates the first and often both of the electrons required by a CYP for catalysis [8,9]. The canonical reaction catalysed by CYPs is monooxygenation, although they can also catalyse other reactions including hydroxylation and dealkylation. Human and other mammalian CYP enzymes are membrane-anchored proteins embedded in the phospholipid bilayers of the endoplasmic reticulum or mitochondria [5,10].
1.2. Background to Pharmacogenomics
Interindividual variability in drug response is a growing concern for healthcare systems across the world, since it can blunt drug efficacy and precipitate adverse drug reactions (ADRs) leading to patient harm, poorer health outcomes, and inefficient consumption of limited healthcare resources. It is estimated that for many drugs used across a range of diseases, only 50–75% of patients experience a beneficial response [11]. Studies in European populations have demonstrated that 3.5–6.5% of hospitalised patients are admitted because of ADRs, and one or more ADRs can occur in almost 15% of hospital inpatients [12,13,14]. Additionally, the risk of ADRs increases in patients administered multiple drugs [15], contributing to the clinical burden of (potentially inappropriate) polypharmacy. ADRs differ in severity with the majority of adverse events considered mild (e.g., headaches, fatigue, constipation and/or nausea), although the overall number of cases is believed to be underreported [16]. Nevertheless, non-severe and non-serious cases still present an avoidable burden to healthcare systems. Notably, a 2015 National Institute for Health and Care Excellence (NICE) statement concluded that 72% of ADRs are potentially avoidable, with the estimated cost of these ADRs in excess of £500 million in the UK alone [17]. Therefore, it is important to understand and mitigate drug response variation.
Variation in drug response is, in general, multifactorial with demographic, environmental, clinical, and genomic components. Pharmacogenomics is the study and clinical application of the genetic determinants of drug response. The aim of pharmacogenomics is to guide dose selection, drug selection, and/or prioritise patients for closer monitoring through stratification of patients based on genetic variants in order to improve drug efficacy and/or safety, and this is at the vanguard of the precision medicine initiative [18,19,20]. Pharmacogenomic studies have evolved from candidate gene investigations to genome-wide association studies (GWAS) and increasingly to sequencing based projects; sample sizes have similarly grown although, in general, remain notably smaller compared to genomic studies of polygenic diseases. Nevertheless, the magnitude of drug–gene associations is often notably larger than disease–gene associations [21], presumably reflecting less constrained evolutionary selection pressures. Currently, pharmacogenomic information is contained somewhere within the drug label of 298 drugs approved by the US Food & Drug Administration (FDA) [22]. Moreover, to date, the clinical application of gene associations with 123 drugs has been reviewed and, of these, clinical guidelines for just over 80 drugs have been developed that contain clinically actionable pharmacogenomic recommendations [23]. The majority of this evidence assessment and guideline writing has been conducted by the Royal Dutch Association for the Advancement of Pharmacy-Pharmacogenetics Working Group (DPWG) [24] and the Clinical Pharmacogenetics Implementation Consortium (CPIC) [25], although other guideline-writing committees also exist such as the Canadian Pharmacogenomics Network for Drug Safety (CPNDS) [26], the Réseau National de Pharmacogénétique (RNPGx) [27], and the NHS England Pharmacogenomics working group.
1.3. Background to CYP2D6
CYP2D6 is a particularly important and well-studied member of the CYP superfamily. In the late 1970s, the metabolism of debrisoquine, and separately sparteine, were both shown to be highly variable yet controlled by a single autosomal gene, which we now know to by CYP2D6 [28,29]. The human CYP2D6 gene is relatively short, spanning just ~4.3 Kbps on the long arm of chromosome 22 (22q13.2), is encoded by nine exons, is translated into CYP2D6 protein that localises to the endoplasmic reticulum, and is highly expressed in liver, brain, intestinal tissue and lymphoid cells [10,30]. CYP2D6 has numerous endogenous substrates including tyramine in the brain and lymphocytes [31,32], and 5-hydroxyindoleacetic acid in brain tissue and possibly cerebrospinal fluid [33,34]. Importantly, although CYP2D6 constitutes just 2–4% of total hepatic CYP content [35], it is a cardinal drug-metabolising enzyme involved in the metabolism of approximately 20% of commonly used drugs [36,37]. Thus, CYP2D6 can metabolise a wide range of substrates including analgesics (e.g., codeine, tramadol), antidepressants (e.g., paroxetine, tricyclic antidepressants), antihypertensives (e.g., metoprolol, bisoprolol) and the anti-cancer agent, tamoxifen [38,39,40,41].
Overall, to date, 72 different drugs have CYP2D6 mentioned within their FDA-approved product label [42]. Furthermore, the clinical relevance of CYP2D6 to at least 48 drugs has been reviewed by pharmacogenomic guideline committees and actionable recommendations based on CYP2D6 have been developed for 26 drugs to date [23], as summarised in Table 1. It is clear that CYP2D6 is a promiscuous pharmacogene implicated in drug response across a large number of medical specialities (Figure 1).
The rate of CYP2D6-mediated microsomal metabolism varies at least 60-fold between individuals [43]. CYP2D6 is highly polymorphic and its genetic complexity is an important contributor to its functional variation. Therefore, the genetic complexity of CYP2D6 and its pivotal role in the metabolism of multiple drugs makes accurate and effective CYP2D6 genotype-based clinical prescribing a key milestone in any pharmacogenomics implementation effort. Thus, this review will focus on describing CYP2D6 genomic variation, the functional consequences of this variation, laboratory methods to detect and analyse CYP2D6 variants, the clinical impact of CYP2D6 pharmacogenomics, and highlight cutting edge developments in parsing CYP2D6 genotype-to-phenotype translation. This review has been written for an intended audience that includes both non-geneticists and healthcare prescribers as well as those proficient in pharmacogenomics.
2. An Overview of CYP2D6 Variation
Variation in CYP2D6 includes single-nucleotide variants (SNVs), indels, whole-gene deletions, multiplications, tandem arrangements and hybridisations [40,44]; it can also be affected by larger chromosomal deletions or other structural alterations as part of the rare 22q13 deletion syndrome, known as Phelan-McDermid syndrome. Currently, there are 133 CYP2D6 “star” (“*”) alleles listed on the PharmVar data repository [45], as detailed within a recent and in-depth review of 2D6 nomenclature and 2D6 allele designation by Nofziger et al. [46]. In essence, the star allele nomenclature system represents a useful haplotype-based system that is well established and recognised within the pharmacogenomic field [46]. Many of the listed variants contribute in part or entirely to altered rates of CYP2D6-associated drug metabolism. The majority of identified CYP2D6 variants are SNVs, which encompass both single-nucleotide polymorphisms (SNPs, defined as SNVs with a minor allele frequency, MAF ≥ 1%) and rarer (MAF < 1%) nucleotide substitutions. Like other genes, CYP2D6 SNVs can be in the non-coding regions (e.g., the promoter or introns) with variable effects on protein expression, or located in the coding regions, leading to synonymous or nonsynonymous (both missense and nonsense) alterations. Small base insertions/deletions (indels) may be of little consequence when occurring in non-coding (e.g., intronic) gene regions, although they could perturb transcription factor binding. However, when indels affect coding DNA and are not a multiple of three, they can change the reading frame and result in frameshift mutations. Importantly, beyond these short forms of variation, the genetic architecture of CYP2D6 differs from that of other CYP pharmacogenes by the extent and complexity of larger structural variation that can occur, as briefly highlighted below. For further information, the reader is directed to the Pharmacogene Variation Consortium (PharmVar) CYP2D6 Structural Variation document [47], which provides an excellent up-to-date and detailed overview of CYP2D6 structural variation.
2.1. Pseudogenes
One major complication of analysing CYP2D6 is resolving the issue of the highly similar non-functional pseudogenes, CYP2D7 and CYP2D8, which are located nine and 19 kbp upstream of CYP2D6 and have 94.2% and 89.1% sequence similarity to CYP2D6, respectively [36]. All three genes consist of nine exons, and CYP2D6 and CYP2D7 share a common downstream element that is 0.6 kbp in length [43,47]. Moreover, CYP2D6 and CYP2D7 have near-identical repetitive sequences termed REP6 and REP7, respectively, although REP7 is separated by an additional short length of sequence from the common element in CYP2D7, unlike REP6 in CYP2D6 [47] (see Figure 2A). This level of similarity, particularly between CYP2D7 and CYP2D6, requires any genotype detection method to be highly specific to CYP2D6 [36]. However, further complicating this problem is that many haplotype-defining CYP2D6 SNPs, such as rs35742686 (*3), have identical flanking sequences in CYP2D6 and CYP2D7, and thus present a challenge to distinguish between using probe-based and microarray testing techniques.
2.2. Copy Number Variation
Copy number variation (CNV) is a type of larger structural variation and, in a diploid genome, generally refers to the presence of more or less than two copies of a gene. CNV is common in CYP2D6 and individuals, with one or three CYP2D6 copies occurs at a frequency of 12–23% depending on population ethnicity [40]. The significance of this type of variation on the metaboliser phenotype of CYP2D6 depends on the functionality of the duplicated CYP2D6 allele. Indeed, it has been shown in a population of patients from Hong Kong that fewer than 20% of duplicated CYP2D6 alleles were duplications of functional alleles [44]. However, an increased copy number of functional CYP2D6 alleles increases the rate of metabolism of associated drugs [39], with some studies reporting as many as 13 copies of CYP2D6 in a single individual’s genome [48]. Thus, an individual with more than two functional copies of CYP2D6 is considered to have ultra-rapid metabolism for CYP2D6 substrate drugs. CYP2D6*1, *2 and *4 are the most common haplotypes in which duplications or multiplications are observed and are denoted as ‘CYP2D6*1xN’ ‘*2xN’ and ‘*4xN’, respectively [40]. Other rarer duplications have been identified, including CYP2D6*6xN, *10xN, *17xN, *36xN, *41xN, *43xN and *45xN [49].
CYP2D6*5 represents a whole allele deletion with a corresponding complete loss of function of the allele. Overall, this variant is present in 3–6% of African, European and East Asian populations (Table 2). However, it is more diverse within sub-populations and so, for example, gradually increases from 1 to 6%, moving from southern to northern European countries, respectively [50]. CYP2D6*5 deletions are derived from breakpoints in the highly repetitive REP6 and REP7 sequences [38], leaving CYP2D6*5 with a REPdel sequence consisting of fused REP6 and REP7 elements [51].
2.3. Hybridisations, Tandem Arrangements and Conversions
Structural hybridisation describes the rearrangement of a gene sequence to form an alternate gene sequence, which often leads to a protein with reduced or no function. Many different hybridisations of CYP2D6 have been observed and typically involve the joining of CYP2D6 and CYP2D7 sequences together to make a recombinant hybrid gene (Figure 2B) [51,52,53,54]. CYP2D6-2D7 and CYP2D7-2D6 are two distinct forms of hybridisation (see below). It is also possible that a sequence from CYP2D7 can be inserted into CYP2D6 at a specific point, which is termed a gene conversion [36,47].
CYP2D6-2D7 hybrids are formed when the 5′ section is from CYP2D6 and the 3′ section is from CYP2D7, forming a hybrid structure in place of wildtype CYP2D6. Examples of this include *36 and *61. Similarly, in CYP2D7-2D6 hybrid genes, the 5′ element of the hybrid is from CYP2D7 and the 3′ part is from CYP2D6; CYP2D7-2D6 arrangements lack CYP2D7 5′ to the recombinant gene and so could originate from the deletion of the intervening sequence [53]. Several CYP2D7-2D6 hybrids that conform to this pattern have been identified; although they differ in the exact 2D7-2D6 joining point between the two genes, they are collectively grouped together under the CYP2D6*13 designation [47].
Tandem arrangements contain two or more copies of a gene unit on the same chromosome featuring non-identical variation between the gene units. Gene duplications and multiplications are distinguished from tandems by the gene units of duplications/multiplications being the same [47]. In most tandems, at least one gene copy is a CYP2D7-2D6 or CYP2D6-2D7 hybrid arrangement. CYP2D6*36 + *10 is the most common tandem arrangement, and is particularly prominent in East Asian populations with an MAF of 34.1% [44]. In this tandem arrangement, the 5′ upstream gene unit is usually a 2D6-2D7 hybrid (*36), whilst the 3′ downstream gene unit contains the missense SNP that defines *10 (rs1065852, p.P34S). Evidence suggests that the *36 + *10 tandem is associated with decreased function, reflecting that *36 alone (as a single gene unit) has no function and *10 alone has decreased function [36,55,56].
2.4. Structural Impact of Star Alleles
X-ray crystallography has revealed that the 3D structural conformation of CYP2D6 differs between ligand-bound and ligand-free forms. The most distinct structural changes observed include the closing of the active site by reorganisation of helix F and Gly-218 [57,58]. The structural changes caused by the non-synonymous, frameshifts and splicing mutations, which define the CYP2D6 star allele, drive the change in function, thus driving the metabolism phenotype. However, several of the most common function effecting point mutations do not occur near amino acids associated near the active site, instead occurring towards the extremities of the molecule, distant form the central heme iron (Figure 3).
3. CYP2D6 Metaboliser Status
The current categorisation of CYP2D6 metaboliser status is based on activity scoring of known haplotypes [46]. Activity scores (AS) serve as a useful tool to translate information regarding the function of individual haplotypes into an overall predicted metaboliser status for a given diplotype, and thus an individual. At present, CYP2D6 categories remain relatively discrete and are broadly grouped into poor metaboliser (PM, AS = 0), intermediate metaboliser (IM, AS = 0.25–1), extensive (normal) metaboliser (EM, AS = 1.25–2.25) and ultra-rapid metaboliser (UM, AS > 2.25) strata [60]. This pragmatic approach is useful and is periodically updated as evidence regarding the metaboliser function of individual haplotypes accumulates. CYP2D6 pharmacogenomic guidelines use these metaboliser categories to provide clinicians with drug-specific recommendations with the aim of avoiding doses or drugs expected to be ineffective or harmful in patients that have a particular CYP2D6 metaboliser status.
4. CYP2D6 Star Allele Frequencies
Table 2 shows population frequencies for several CYP2D6 star alleles (that generally represent haplotypes) that are common (MAF ≥ 1%) in at least one major population group and (except *2) affect CYP2D6 function. However, importantly, these crude frequencies do not account for the complexities of CYP2D6 haplotype impact on metabolism. For example, rare variants are not considered. Furthermore, in-depth studies assessing overall frequencies of CYP2D6 metabolism status found that >20% of Ashkenazi Jewish and Europeans populations have non-functional haplotypes, 45% of East Asian Individuals had reduced function haplotypes and >10% Oceania individuals have increased function haplotypes [61]. This high haplotype frequency disparity can also be seen in other sub-populations and some haplotypes are only present at high frequency in specific populations. For example, CYP2D6*4 was found to be present in 22% of Jewish individuals [61] and CYP2D6*5 was found to be present in 16% of north Indian individuals; however, both CYP2D6*4 and CYP2D6*5 were significantly lower in other populations [62]. These population frequencies have also been stratified by AS, which varies depending on the combination of haplotypes and number of duplicates/deletions present. These data showed the high frequency of AS 1.5–2 across most major populations, and AS > 2 in over 10% of Oceanian, Middle Eastern and Ashkenazi Jewish populations [61].
5. Detection and Interpretation of CYP2D6 Genotype
Several existing CYP2D6 haplotype detection methods exist, including methods which can detect hybrid arrangements and quantify CNVs. Many of these methods include an initial sequencing step to specifically amplify CYP2D6 prior to genotyping, or as a comparison to other assays. It is important to note that, at present, there is no agreed consensus on which CYP2D6 variants should be always included when interrogating CYP2D6. Therefore, whilst different commercial solutions usually test some major CYP2D6 alleles (*2, *4, *5, *10, *17), different combinations of other CYP2D6 variants are provided by different products [65].
Long-range polymerase chain reaction (PCR) or extra-long-range PCR methods are designed to amplify the entire CYP2D6 gene and therefore can detect multiple copies or whole-gene deletions [66]. Some studies further developed this approach by directly sequencing the PCR product to ensure high sequence fidelity within exonic regions [67]. Pyrosequencing in particular has been established as an inexpensive high-throughput sequencing method in comparison to traditional Sanger sequencing [68] and can been used as a sequencing step for CYP2D6 variant detection [69]. This sequencing step is then followed by a CYP2D6-specific mutant probe detection assay [67]. Combined, these methods are able to accurately determine the presence of a small number of CNVs and has the potential to capture multitudes of other SNPs of interest. However, in its current form, this process is better suited for small-scale research projects, as it consists of multiple PCR steps, and is expensive and labour-intensive.
Taqman methods use bioluminescent tagged probes which can be designed to capture specific sequences with a high degree of precision. qPCR amplification using Taqman probes enables the relative quantification of target sequences, making it ideal for detection of CNVs [70]. However, this method is expensive and labour-intensive if testing a wide variety of haplotypes and has limited multiplexing potential.
Many companies offer custom microarrays which can capture SNPs; however, not all are able to determine CNVs and thus have limited use for clinically meaningful CYP2D6 genetic analysis. More specialised products are capable of detecting CYP2D6 CNVs [71]. The Amplichip CYP2D6 genechip platform is a microarray hybridisation assay that can detect CYP2D6*2, *3, *4, *5, *6, *9, *10, *35 and *41 and duplicates of these SNPs [72]. Similar to Ampligene, CYP2D6 Genochip is a microarray that can detect CYP2D6*3, *4, *6, *7, *8, *9, *10, *11, *17, *29, *41, gene deletion (*5) and gene multiplication (*xN). Comparisons between these platforms have shown a high degree of concordance; however, the Ampligene platform appears able to elucidate CYP2D6 diplotypes more precisely [73].
Several algorithms have been developed to infer CYP2D6 haplotype from whole-genome sequencing data [74,75,76]. Most recently, the Cyrius software algorithm has been shown to be the most accurate with 96.5% concordance between predicted and reported haplotype [76]. This approach could prove to be a valuable way to predict an individual’s metabolism using existing data as whole-genome sequencing becomes more affordable and more commonly undertaken.
Sequencing and genotyping of the somatic genome is becoming increasingly available in oncology. However, there are challenges to using (stored) tumour tissue to determine an individual’s (germline) pharmacogenomic profile. Of note, cancer mutagenesis can potentially alter the genotype of pharmacogenes within the somatic genome, leading to misclassification of the predicted germline genotype. Furthermore, in breast cancer, the region on chromosome 22 that contains CYP2D6 is commonly deleted [77]. Lastly, the process of formalin fixation damages somatic DNA, although repair processes have been developed [78].
6. Factors Impacting CYP2D6 Function beyond CYP2D6 Genetics
Adding to the complexity of CYP2D6 metabolism is the effect of non-genetic factors that contribute to metaboliser phenotype. As it stands, existing haplotypes alone may not reflect the full underlying genetic complexity of drug-metabolising CYPs due to, for example, rare variation, nor do they consider non-genetic factors. Epigenetic regulation, drug–drug interactions and some foodstuffs influence CYP2D6 activity. Of note, drugs that inhibit CYP2D6 function can lead to an individual having a less functionally active metaboliser phenotype than would be predicted by their genotype, a process termed ‘phenoconversion’ [79,80]. Examples of strong CYP2D6 inhibitors that increase the area under the concentration–time curve (AUC) of sensitive CYP2D6 substrates (e.g., dextromethorphan, nortriptyline, eliglustat) by ≥5-fold include fluoxetine, paroxetine and bupropion [81]. In addition, a range of food products and associated chemicals have also been identified as CYP2D6 inhibitors, including sesamin [82], curcumin and the botanical herb, goldenseal [83]. CYP2D6 phenotyping can evaluate phenoconversion, for example in dedicated drug-drug interaction studies incorporating a CYP2D6 probe drug (e.g., dextromethorphan) or other substrate of interest. Alternatively, endogenous metabolites have been suggested as alternate CYP2D6 probes, such as urinary M1 (m/z 444.3102) identified through global metabolomics [84], although further validation of endogenous metabolites should be sought. Several proposed methods have been suggested to counteract the issue of phenoconversion by CYP2D6 inhibition including prioritising phenotyping over genotyping where possible, utilising metabolic probes or even using endogenous compounds to determine phenotype. One suggested improvement is simply to evolve the nomenclature to incorporate phenoconversions, therefore separating this from ‘classic’ metabolism statuses (e.g., pUM instead of UM) [85]. This approach would theoretically reduce communication errors and provide additional information when capturing phenoconversion data.
Interestingly, there are no recognised drug inducers of CYP2D6, and so the UM phenotype appears predominantly genetic in origin. It appears that canonical xenobiotic-sensing receptors that upregulate expression of CYP3A4 and other CYPs, including pregnane X receptor (PXR) and constitutive androstane receptor (CAR), have little effect on CYP2D6 [86]. Nevertheless, the expression of CYP2D6 transcripts is controlled by different transcriptional regulators and growing evidence suggests they contribute to part of the observed CYP2D6 variability [43]. For example, the transcription factor, hepatocyte nuclear factor 4α (HNF4α), is a global regulator of genes involved in liver-specific functions, and it has been shown in a systematic study of over 450 human livers that the mRNA levels of HNF4α correlate with the microsomal activity of multiple drug-metabolising CYPs, including CYP2D6 [87]. Moreover, a rich pharmacokinetic study that administered a single dose of tolterodine (a CYP2D6 substrate) to 30 healthy Korean individuals determined that a genetic variant within HNF4A, G60D, was significantly associated with tolterodine exposure independent of CYP2D6 genotype, and accounted for approximately a quarter of the variation explained by CYP2D6 [88]. It has also been observed that pregnancy induces CYP2D6 activity [43]. Although mechanistic understanding of this is incomplete, mouse models (e.g., CYP2D6-humanised mice) and cell lines (e.g., HepG2 cells) have led to the identification of the novel transcription factors, small heterodimer partner (SHP) and Krüppel-like factor 9 (KLF9), that appear to act as a corepressor and coactivator of HNF4α-mediated transactivation of the CYP2D6 promoter, respectively [43]. Thus, in pregnancy, it appears that expression of the corepressor SHP is reduced, whilst levels of the coactivator KLF9 are increased, plausibly leading to CYP2D6 induction [89,90].
7. CYP2D6 Clinical Impact
Clinical Guidelines and Pharmacogenomics Implementation
The clinical guidelines developed by both CPIC and DPWG for CYP2D6-substrate drugs have overall a high degree of similarity in their determination of CYP2D6 metabolism status and therapeutic recommendations [91]. Moreover, recent efforts to address inconsistencies between these guidelines have further harmonised CYP2D6 metabolism status [39,60,92]. Nevertheless, the advice between the guideline writing groups differs for a few drugs, including fluvoxamine and paroxetine (Table 1). It is also apparent that few drugs have had guidelines developed for them from all of the aforementioned pharmacogenomic guideline-writing committees, reflecting the international collaborative approach of the relatively small pharmacogenomics research field, the effort and expertise required to develop clinical guidance, and the likely influence of different patterns of prescribing between countries and regions.
As stated earlier, actionable pharmacogenomic guidance has been developed for 26 drugs involving CYP2D6 to date. The majority of these drugs are established drugs that already have a license. An exception is the glucosylceramide synthase inhibitor, eliglustat, which is a new drug indicated in Gaucher disease type 1, and was licensed with a requirement for companion CYP2D6 testing, as stated in its product label [93]. The evidence underpinning CYP2D6 clinical guidelines is generally based on the totality of published, predominantly observational, studies, due to an absence of large randomized control trials. Despite the limitation of this lack of gold-standard evidence, the amalgamation of congruent pharmacokinetic studies, clinical observational studies and case reports within clinical guidance provides a pragmatic approach to sensibly interpret and facilitate the use of the existing knowledge base. It can be contended that this evidence base is equivalent to the evidence underpinning dose/drug modifications based on renal or liver function biochemical testing, which is routinely carried out for many drugs in clinical practice. Overall, the question over the evidential threshold needed for dose/drug optimisation strategies has yet to be fully addressed; however intuitively, it seems reasonable to suggest that, given limited resources for research, it should not always require the same level of evidence as required for tests that carry greater clinical consequences (e.g., tests that lead to a new diagnosis, or, for example, high-risk BRCA mutations that can lead to prophylactic surgery).
Nevertheless, to inform the evidence base for pharmacogenomics implementation, one initiative being conducted is the PREPARE study. PREPARE is a large pan-European block randomised prospective pharmacogenomics intervention study, coordinated by the Ubiquitous Pharmacogenomics consortium, that has recruited ~7000 patients. These patients started one of 42 drugs, for which a DPWG guideline is available, and have been followed up for adverse events for 12 weeks [19]. In the control arm, patients were dosed according to standard care. In the intervention arm, patients have been genotyped for 44 variants in 12 genes, including CYP2D6, and DPWG therapeutic recommendations have been made available based on these genotype results to the patient’s healthcare team within one week of starting the drug. It is expected that the primary results of PREPARE will be available in 2021 and, whilst PREPARE is not powered to answer whether individual drug–gene pairs should be implemented, it will address whether or not an overarching pharmacogenomics approach is clinically and/or cost-effective [19].
Recommendations in CYP2D6 guidelines include: starting with the usual dose of the drug (e.g., in EMs), altering its dose (e.g., in IMs), selecting an alternative drug (e.g., in PM/UMs), or considering increased patient monitoring and therapeutic drug monitoring if available (e.g., in PMs). In a few guidelines (e.g., atomoxetine, tamoxifen), the IM category is further subdivided and specific recommendations are provided according to the presence/absence of the CYP2D6*10 allele (AS 0.25). This metaboliser status classification system has been similarly used in the pharmacogenomic guidelines for other important CYP pharmacogenes (e.g., CYP2B6, CYP2C9, CYP2C19, CYP3A5), albeit usually without activity scoring because of the lower complexity of genetic variation compared to CYP2D6. However, these other CYP pharmacogenes can have different gene-specific considerations, such as an additional rapid metaboliser (RM) category for CYP2C19, and no UM group for CYP3A5.
A nationwide study in the Netherlands of over 45 commonly prescribed drugs paired with genotype data found that, overall, the implementation of pharmacogenomics using DPWG guidelines could lead to 5.4% of prescriptions being altered (e.g., dose adjustments or selection of alternative drugs) [94]. For drugs linked to CYP2D6, the proportion of patients with an actionable recommendation varied by drug according to which metaboliser statuses were deemed actionable. Thus, for example, only 5% of aripiprazole prescriptions were associated with a recommendation (reduced maximum dose) because only the PM category is considered actionable in DPWG guidance for aripiprazole. However, DPWG guidance recommends optional (for IM or PM patients) or definite (for UM patients) prescription alterations for 47% of tramadol prescriptions. Several other drugs, such as flecainide, venlafaxine and imipramine, were similarly recommended a drug or dosage change based on CYP2D6 genotype in >45% of cases [94]. The following subsections highlight CYP2D6 pharmacogenomics in relation to two case-studies: codeine and tamoxifen.
8. CYP2D6-Drug Case Studies
8.1. Codeine
Codeine is an opiate prodrug used primarily as an analgesic agent and to treat diarrhoea and cough. However, codeine can have a range of adverse effects from relatively mild nausea, constipation and headaches, through to drowsiness and serious respiratory depression. Metabolism of codeine into active morphine is dependent on CYP2D6, and so is strongly influenced by CYP2D6 genotype (Figure 4). Codeine displays a reduced binding potential to mu opioid receptors compared to morphine, resulting in milder analgesic effects. Codeine is also considered to be the safer of the two compounds [95,96]. If an individual is a CYP2D6 PM, there is an increased likelihood of an insufficient analgesic effect [97]. Conversely, several importantly case reports have identified severe respiratory depression in UM children following tonsillectomies [97,98,99]. Mothers who are CYP2D6 UMs also appear to have increased morphine present in their breast milk, and this has been associated with severe adverse events in breastfed infants [100].
8.2. Tamoxifen
Tamoxifen is a selective oestrogen receptor modulator (SERM) frequently used to reduce the incidence of breast cancer recurrence in pre- and perimenopausal women with oestrogen receptor-positive breast cancer. Although generally well tolerated, it can cause hot flushes, fatigue and nausea amongst other adverse effects. Tamoxifen metabolism is extensive and complex (Figure 5). Nevertheless, an important secondary metabolite, endoxifen, has almost 100-fold greater anti-oestrogenic potency than parent tamoxifen [101]. The major route of endoxifen formation is demethylation via CYP3A4/5 followed by oxidation by CYP2D6 [101]. CYP2D6 IM/PMs have been associated with reduced endoxifen plasma concentration [102], although the evidence relating CYP2D6 to clinical outcomes indicative of reduced tamoxifen efficacy, such as breast cancer recurrence and mortality, are inconsistent [101]. Whilst an association has been shown in some studies [103,104], two large studies dispute an effect [104,105]. Notably, tumour tissue was used for genotyping in both these studies which, as mentioned previously, is a major limitation and their findings may not match the genotype of somatic tissue for the same patients. Furthermore, a recent review suggested that a link between CYP2D6 genotype and tamoxifen adherence was uncovered in these large studies [106] and other studies linked a specific CYP2D6 haplotype to outcome in tamoxifen-treated patients [107]. Accordingly, clinical guidance for tamoxifen-CYP2D6 has been developed by CPIC and DPWG as well as other societies, which generally recommends alternative treatment, such as an aromatase inhibitor, in CYP2D6 IMs and PMs [80,108].
Other common CYP2D6-mediated drugs such as amitriptyline have also been found to have strong links between CYP2D6 status and increased risk of ADRs [110]. Overall, the impact of CYP2D6 genotypes will vary across medical specialities depending on the prevalence of drug prescriptions, which metaboliser phenotypes are associated with altered response for a given drug, and the frequency and severity of a drug’s adverse events.
9. Novel Approaches to CYP2D6 Phenotyping
Many CYP2D6 variants, including rare variants and complex combinations of haplotypes, have been identified but not assigned a metabolic function. One in vitro state-of-the-art approach to comprehensively determine the empirical functionality of missense variants is saturation mutagenesis with massively parallel functional assays. In this approach, a mutagenesis library of missense variants is constructed that represent all possible amino acid substitutions within a protein and their in vitro functionality is determined. This high-throughput functional screening approach has been successfully applied to NUDT15 and could be conceivably applied to CYP2D6 [111]. Analysis of large datasets is another approach to assign functional effects to these undetermined haplotypes, and is being increasingly carried out to interrogate CYP2D6 [112,113]. One example is the use of sequence data and phenotypic data from >450,000 UK biobank participants [112]. Analysis of these data has been used to elucidate haplotypes, but it was unable to capture structural variations. Technologies such as Nanopore utilise long read sequencing, which is able to sequence, identify and capture structural variants in CYP2D6 [114].
In recent years, numerous promising approaches have been employed to predict CYP2D6 function, utilising machine learning and deep learning. One interesting example leveraged the observation that CYP2D6 is amongst the most highly expressed CYPs in the brain and so used machine learning to identify imaging patterns within brain perfusion images from functional magnetic resonance imaging (fMRI) associated with CYP2D6 metaboliser groups. This technique was able to detect UM individuals with sensitivity and specificity values of 85–87%, and PM individuals with sensitivity and specificity values of 71–79% [115]. Another recent study utilised a deep learning approach to predict the function of CYP2D6 genotypes, including those with rare variants, and then derive a subsequent classification model that predicts whether a CYP2D6 haplotype has a normal or reduced function, and was shown to have a high degree of accuracy [116]. Lastly, a neural network model was recently developed and trained on long-read CYP2D6 sequencing data in conjunction with information on the rate of CYP2D6-mediated tamoxifen metabolism from 561 patients with breast cancer. This model linked CYP2D6 variation to the tamoxifen metabolic ratio on a continuous scale, rather than categorising it into the conventional discrete metaboliser phenotype groups. Importantly, it could account for 79% of observed interindividual variation in CYP2D6 activity, compared to 54% with the conventional approach. Furthermore, this model could assign enzyme activity to haplotypes containing uncharacterised combinations of variants, and was validated in independent cohorts. Thus, this approach highlights the potential of combining fully phased gene sequence data with machine learning approaches and continuous scale readout to improve pharmacogenotype–phenotype translation [117].
10. Veterinary Pharmacogenomics and CYP2D6 Orthologues
Beyond human pharmacogenomics, interindividual variability in drug response in veterinary practice is also being increasingly explored. However, the scale and implementation of veterinary pharmacogenomics remains limited to date. The primary example of veterinary pharmacogenomics is the identification of an ABCB1 (also known as MDR1 and encodes P-glycoprotein) allele that reduces the function of P-gp and has been associated with severe ADRs following administration of the anti-parasite drug, ivermectin, in homozygous individuals [118,119]. This variant is very common in specific canine breeds (e.g., collies and English shepherd dogs) due to selective breeding to historically desired traits, and highlights the requirement for veterinary pharmacogenomic research to go beyond broad species genetics and to target genetics at breed [118,120] and eventually individual level.
To date, the role of CYP2D6 orthologues in veterinary pharmacogenomics has been poorly elucidated. However, a few examples of CYP2D6 orthologue genotypes influencing drug metabolism have been studied. Canine and equine orthologues of human CYP2D6 are CYP2D15 and CYP2D50, respectively [118,121,122]. Four haplotypes have been identified in canine CYP2D15 (*1, *2, V1 and WT2), all of which have been shown to metabolise celecoxib at different rates in beagles and, additionally, the rate of metabolism of bufuralol was shown to be reduced in the WT2 haplotype [121]. CYP2D15 also has a role in the metabolism of desipramine, dextromethorphan and imipramine [123]. Moreover, the equine orthologue, CYP2D50, has been found to harbour SNPs and deletions associated with varying rates of tramadol metabolism, allowing the categorisation of horses into CYP2D50 PMs and EMs, whereas one horse displayed UM status [122].
11. Conclusions
CYP2D6 is a very important pharmacogene with dozens of common haplotypes. These haplotypes can significantly influence the metabolism and, thus, systemic exposure of CYP2D6 substrates, leading to variable drug response. As a result, clinical pharmacogenomic guidelines have been developed for at least 48 drugs, which collectively span a wide range of therapeutic indications. Moreover, as approximately 20% of commonly used drugs are metabolised by CYP2D6, the ramifications of variable CYP2D6 activity on drug response is likely to be notable in clinical practice. Thus, incorporating CYP2D6 metaboliser status into routine care is expected to reduce ADRs, save healthcare system resources and improve the efficiency of healthcare sectors. However, there is not yet consensus on which CYP2D6 variants should be routinely tested for clinical use. Moreover, a fast turnaround, cost-effective, non-labour intensive, accurate and reliable method for analysing CYP2D6 with the potential for rapid upscaling is required for effective implementation of CYP2D6 pharmacogenomics in the clinic. However, CYP2D6 presents a number of technical challenges to mass testing in a healthcare setting. In particular, structural variation in CYP2D6 can be challenging to identify and characterise, complicates the design of CYP2D6-specific amplification and detection methods, and can even mask potentially actionable diplotypes. Nevertheless, several technologies and solutions exist that can identify CYP2D6 structural variants, although they are, on the whole, relatively expensive, labour-intensive and/or time-consuming for mass testing. The continued efforts to develop new pharmacogenomics guidelines for different drugs will further aid adoption of CYP2D6 testing in healthcare settings. Finally, novel machine learning approaches that can comprehensively parse CYP2D6 genotype-phenotype relationships hold significant promise for application to whole genome sequencing initiatives. However, there remains much to do to translate current understanding of CYP2D6 into tangible benefit for patients in the clinic.
Author Contributions
All authors listed have contributed significantly to the knowledge, research and structuring of this review. Literature review and compiling of data was conducted by C.T., R.M.T., V.Y. and M.P.; Images and tables were created by C.T., I.C. and P.M.; Biotechnology and novel CYP2D6 approaches section was written by I.C. and P.M. All authors have read and agreed to the published version of the manuscript.
Funding
This review was written as part of a KTP project funded by UKRI through Innovate UK, the Welsh Government and MC Diagnostics. C. Taylor was funded by the Innovate UK Knowledge Transfer Partnership. R. M. Turner was supported by a Postdoctoral Research Fellowship from Health Education England Genomics Education Programme (HEE GEP). The views expressed in this publication are those of the authors and not necessarily those of HEE GEP.
Conflicts of Interest
Christopher Taylor, Ian Crosby and Peter Maguire work with MC Diagnostics which operates as a commercial genetic testing kit manufacturer. The remaining authors have not conflict of interest to declare.
References
- Munro, A.W.; Lindsay, J.G. Bacterial cytochromes P-450. Mol. Microbiol. 1996, 20, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Satta, Y. Substrate-dependent evolution of cytochrome P450: Rapid turnover of the detoxification-type and conservation of the biosynthesis-type. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome Biol. 2000, 1, reviews3003.1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Šrejber, M.; Navrátilová, V.; Paloncýová, M.; Bazgier, V.; Berka, K.; Anzenbacher, P.; Otyepka, M. Membrane-attached mammalian cytochromes P450: An overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J. Inorg. Biochem. 2018, 183, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Otyepka, M.; Skopalík, J.; Anzenbacherová, E.; Anzenbacher, P. What common structural features and variations of mammalian P450s are known to date? Biochim. Biophys. Acta-Gen. Subj. 2007, 1770, 376–389. [Google Scholar] [CrossRef]
- Hasemann, C.A.; Kurumbail, R.G.; Boddupalli, S.S.; Peterson, J.A.; Deisenhofer, J. Structure and function of cytochromes P450: A comparative analysis of three crystal structures. Structure 1995, 3, 41–62. [Google Scholar] [CrossRef]
- Porter, T.D. New insights into the role of cytochrome P450 reductase (POR) in microsomal redox biology. Acta Pharm. Sin. B 2012, 2, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.F.; Li, Z.H.; Liu, J.Y.; Liu, T.T.; Wang, P.; Fang, Y.; Zhou, J.; Cui, M.Z.; Gao, N.; Tian, X.; et al. Correlation of cytochrome P450 oxidoreductase expression with the expression of 10 isoforms of cytochrome P450 in human liver. Drug Metab. Dispos. 2016, 44, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Gopisankar, M.G. CYP2D6 pharmacogenomics. Egypt. J. Med. Hum. Genet. 2017, 18, 309–313. [Google Scholar] [CrossRef]
- Spear, B.; Heath-Chiozzi, M.; Huff, J. Clinical application of pharmacogenetics. Trends Mol. Med. 2001, 7, 201–204. [Google Scholar] [CrossRef]
- Pirmohamed, M.; James, S.; Meakin, S.; Green, C.; Scott, A.K.; Walley, T.J.; Farrar, K.; Park, B.K.; Breckenridge, A.M. Adverse drug reactions as cause of admission to hospital: Prospective analysis of 18 820 patients. Br. Med. J. 2004, 329, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvy, J.C.; De Bruin, M.L.; Koopmanschap, M.A. Epidemiology of Adverse Drug Reactions in Europe: A Review of Recent Observational Studies. Drug Saf. 2015, 38, 437–453. [Google Scholar] [CrossRef] [Green Version]
- Davies, E.C.; Green, C.F.; Taylor, S.; Williamson, P.R.; Mottram, D.R. Adverse Drug Reactions in Hospital In-Patients: A Prospective Analysis of 3695 Patient-Episodes. PLoS ONE 2009, 4, e4439. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.C.S.; De Oliveira, C. Drug-drug interactions and adverse drug reactions in polypharmacy among older adults: An integrative review. Rev. Lat. Am. Enferm. 2016, 24, 1–17. [Google Scholar] [CrossRef]
- Ferner, R.E.; McGettigan, P. Adverse drug reactions. BMJ 2018, 363. [Google Scholar] [CrossRef]
- NICE Costing Statement: Medicines Optimisation: Implementing the NICE Guideline on Medicines Optimisation (NG5). Available online: https://www.nice.org.uk/guidance/ng5/resources/costing-statement-6916717 (accessed on 12 September 2020).
- Nebert, D.W.; Zhang, G.; Vesell, E.S. From human genetics and genomics to pharmacogenetics and pharmacogenomics: Past lessons, future directions. Drug Metab. Rev. 2008, 40, 187–224. [Google Scholar] [CrossRef] [Green Version]
- Van der Wouden, C.H.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; Dávila-Fajardo, C.L.; Deneer, V.H.; Dolžan, V.; Ingelman-Sundberg, M.; Jönsson, S.; Karlsson, M.O.; et al. Implementing Pharmacogenomics in Europe: Design and Implementation Strategy of the Ubiquitous Pharmacogenomics Consortium. Clin. Pharmacol. Ther. 2017, 101, 341–358. [Google Scholar] [CrossRef]
- Daly, A.K. Pharmacogenetics: A general review on progress to date. Br. Med. Bull. 2017, 124, 65–79. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Cardiovascular pharmacogenomics: Expectations and practical benefits. Clin. Pharmacol. Ther. 2014, 95, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Table of Pharmacogenomic Biomarkers in Drug Labeling|FDA. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 28 August 2020).
- PharmGKB Clinical Guideline Annotations. Available online: https://www.pharmgkb.org/guidelineAnnotations (accessed on 15 September 2020).
- Dutch Pharmacogenetic Working Group. DPWG Recommendations; Dutch Pharmacogenetic Working Group: Den Haag, The Netherlands, 2020. [Google Scholar]
- The Pharmacogenetics Implementation Consortium Guidelines—CPIC. Available online: https://cpicpgx.org/guidelines/ (accessed on 13 October 2020).
- Canadian Pharmacogenomics Network for Drug Safety Pharmacogenomics—Canadian Pharmacogenomics Network for Drug Safety. Available online: http://cpnds.ubc.ca/faqs/pharmacogenomics (accessed on 13 October 2020).
- Pharmacogénétiqu, the R.N. de the Réseau National de Pharmacogénétiqu Recommendations. Available online: http://www.pharmacogenetics.fr/7.html (accessed on 13 October 2020).
- Mahgoub, A.; Dring, L.G.; Idle, J.R.; Lancaster, R.; Smith, R.L. Polymorphic Hydroxylation of Debrisoquin in Man. Lancet 1977, 310, 584–586. [Google Scholar] [CrossRef]
- Eichelbaum, M.; Ingelman-Sundberg, M.; Evans, W.E. Pharmacogenetics and individualized drug therapy. Annu. Rev. Med. 2005, 57, 119–156. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, F.; Dauchy, S.; Diry, M.; Sazdovitch, V.; Cloarec, O.; Mellottée, L.; Bièche, I.; Ingelman-Sundberg, M.; Flinois, J.P.; De Waziers, I.; et al. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. 2009, 37, 1528–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantione, K.J.; Cadet, P.; Zhu, W.; Kream, R.M.; Sheehan, M.; Fricchione, G.L.; Goumon, Y.; Esch, T.; Stefano, G.B. Endogenous morphine signaling via nitric oxide regulates the expression of CYP2D6 and COMT: Autocrine/paracrine feedback inhibition. Addict. Biol. 2008, 13, 118–123. [Google Scholar] [CrossRef]
- Funae, Y.; Kishimoto, W.; Cho, T.; Niwa, T.; Hiroi, T. CYP2D in the Brain. Drug Metab. Pharmacokinet. 2003, 18, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cao, J. Finite-time synchronization of coupled neural networks via discontinuous controllers. Cogn. Neurodyn. 2011, 5, 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, J.; Dong, G.; Yue, J. The endogenous substrates of brain CYP2D. Eur. J. Pharmacol. 2014, 724, 211–218. [Google Scholar] [CrossRef]
- Williams, I.S.; Gatchie, L.; Bharate, S.B.; Chaudhuri, B. Biotransformation, using recombinant CYP450-expressing baker’s yeast cells, identifies a novel cyp2d6.10 a122v variant which is a superior Metabolizer of codeine to morphine than the wild-type enzyme. ACS Omega 2018, 3, 8903–8912. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Botton, M.R.; Scott, E.R.; Scott, S.A. Sequencing the CYP2D6 gene: From variant allele discovery to clinical pharmacogenetic testing. Pharmacogenomics 2017, 18, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, J.P.; Peter, A.P.; Shaman, J.A. Consequences of CYP2D6 copy-number variation for pharmacogenomics in psychiatry. Front. Psychiatry 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gaedigk, A. Complexities of CYP2D6 gene analysis and interpretation. Int. Rev. Psychiatry 2013, 25, 534–553. [Google Scholar] [CrossRef]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Muller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; LLerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beoris, M.; Wilson, J.A.; Garces, J.A.; Lukowiak, A.A. CYP2D6 copy number distribution in the US population. Pharm. Genom. 2016, 26, 96–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleeman, N.; Dundar, Y.; Dickson, R.; Jorgensen, A.; Pushpakom, S.; McLeod, C.; Pirmohamed, M.; Walley, T. Cytochrome P450 testing for prescribing antipsychotics in adults with schizophrenia: Systematic review and meta-analyses. Pharm. J. 2011, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PharmGKB CYP2D6—Drug Label Annotations. Available online: https://www.pharmgkb.org/gene/PA128/labelAnnotation (accessed on 15 September 2020).
- Pan, X.; Ning, M.; Jeong, H. Transcriptional regulation of CYP2D6 expression. Drug Metab. Dispos. 2017, 45, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.; Li, M.S.; Sundaram, S.K.; Tomlinson, B.; Cheung, P.Y.; Tzang, C.H. CYP2D6 allele frequencies, copy number variants, and tandems in the population of Hong Kong. J. Clin. Lab. Anal. 2019, 33. [Google Scholar] [CrossRef] [Green Version]
- Pharmacogene Variation Consortium PharmVar CYP2D6 Alleles. Available online: https://www.pharmvar.org/gene/CYP2D6 (accessed on 12 October 2020).
- Nofziger, C.; Turner, A.J.; Sangkuhl, K.; Whirl-Carrillo, M.; Agúndez, J.A.G.; Black, J.L.; Dunnenberger, H.M.; Ruano, G.; Kennedy, M.A.; Phillips, M.S.; et al. PharmVar GeneFocus: CYP2D6. Clin. Pharmacol. Ther. 2020, 107, 154–170. [Google Scholar] [CrossRef] [Green Version]
- PharmVar Structural Variation CYP2D6. Available online: https://www.pharmvar.org/gene-support/Variation_CYP2D6.pdf (accessed on 9 September 2020).
- Ramírez, B.; Niño-Orrego, M.J.; Cárdenas, D.; Ariza, K.E.; Quintero, K.; Contreras Bravo, N.C.; Tamayo-Agudelo, C.; González, M.A.; Laissue, P.; Fonseca Mendoza, D.J. Copy number variation profiling in pharmacogenetics CYP-450 and GST genes in Colombian population. BMC Med. Genom. 2019, 12, 110. [Google Scholar] [CrossRef]
- Gaedigk, A.; Fuhr, U.; Johnson, C.; Bérard, L.A.; Bradford, D.; Leeder, J.S. CYP2D7-2D6 hybrid tandems: Identification of novel CYP2D6 duplication arrangements and implications for phenotype prediction. Pharmacogenomics 2010, 11, 43–53. [Google Scholar] [CrossRef]
- Petrović, J.; Pešić, V.; Lauschke, V.M. Frequencies of clinically important CYP2C19 and CYP2D6 alleles are graded across Europe. Eur. J. Hum. Genet. 2020, 28, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Del Tredici, A.L.; Malhotra, A.; Dedek, M.; Espin, F.; Roach, D.; Zhu, G.; Voland, J.; Moreno, T.A. Frequency of CYP2D6 alleles including structural variants in the United States. Front. Pharmacol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.K.; Idle, J.R.; Fairbrother, K.S.; Andreassen, O.A.; London, S.J.; Steen, V.M. Characterization and PCR-based detection of two different hybrid CYP2D7P/CYP2D6 alleles associated with the poor metabolizer phenotype. Pharmacogenetics 1996, 6, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Kramer, W.E.; Walker, D.L.; O’Kane, D.J.; Mrazek, D.A.; Fisher, P.K.; Dukek, B.A.; Bruflat, J.K.; Black, J.L. CYP2D6: Novel genomic structures and alleles. Pharm. Genom. 2009, 19, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, J.L.; Walker, D.L.; O’Kane, D.J.; Harmandayan, M. Frequency of undetected CYP2D6 hybrid genes in clinical samples: Impact on phenotype prediction. Drug Metab. Dispos. 2012, 40, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaedigk, A.; Bradford, L.D.A.; Alander, S.W.; Leeder, J.S. CYP2D6*36 gene arrangements within the CYP2D6 locus: Association of CYP2D6*36 with poor metabolizer status. Drug Metab. Dispos. 2006, 34, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyotani, K.; Shimizu, M.; Kumai, T.; Kamataki, T.; Kobayashi, S.; Yamazaki, H. Limited effects of frequent CYP2D6*36-*10 tandem duplication allele on in vivo dextromethorphan metabolism in a Japanese population. Eur. J. Clin. Pharmacol. 2010, 66, 1065–1068. [Google Scholar] [CrossRef]
- Wang, A.; Savas, U.; Hsu, M.H.; Stout, C.D.; Johnson, E.F. Crystal structure of human cytochrome P450 2D6 with prinomastat bound. J. Biol. Chem. 2012, 287, 10834–10843. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yang, L.P.; Zhang, X.Z.; Huang, S.Q.; Bartlam, M.; Zhou, S.F. New insights into the structural characteristics and functional relevance of the human cytochrome P450 2D6 enzyme. Drug Metab. Rev. 2009, 41, 573–643. [Google Scholar] [CrossRef]
- Sehnal, D.; Rose, A.; Koca, J.; Burley, S.; Velankar, S. Mol*: Towards a common library and tools for web molecular graphics. In Proceedings of the Workshop on Molecular Graphics and Visual Analysis of Molecular Data, Brno, Czech Republic, 4 June 2018; pp. 29–33. [Google Scholar]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2020, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Gaedigk, A.; Sangkuhl, K.; Whirl-Carrillo, M.; Klein, T.; Steven Leeder, J. Prediction of CYP2D6 phenotype from genotype across world populations. Genet. Med. 2017, 19, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Parveen, F.; Faridi, R.M.; Das, V.; Tripathi, G.; Agrawal, S. Genetic association of phase I and phase II detoxification genes with recurrent miscarriages among North Indian women. Mol. Hum. Reprod. 2010, 16, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Hicks, J.K.; Sangkuhl, K.; Swen, J.J.; Ellingrod, V.L.; M€ Uller, D.J.; Shimoda, K.; Bishop, J.R.; Kharasch, E.D.; Skaar, T.C.; Gaedigk, A.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline (CPIC) for CYP2D6 and CYP2C19 Genotypes and Dosing of Tricyclic Antidepressants: 2016 Update. Clin. Pharmacol. Ther. 2017, 102, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousman, C.A.; Jaksa, P.; Pantelis, C. Systematic evaluation of commercial pharmacogenetic testing in psychiatry: A focus on CYP2D6 and CYP2C19 allele coverage and results reporting. Pharm. Genom. 2017, 27, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Dorado, P.; López-Torres, E.; Peñas-Lledó, E.M.; Martínez-Antón, J.; Llerena, A. Neurological toxicity after phenytoin infusion in a pediatric patient with epilepsy: Influence of CYP2C9, CYP2C19 and ABCB1 genetic polymorphisms. Pharm. J. 2013, 13, 359–361. [Google Scholar] [CrossRef]
- Puaprasert, K.; Chu, C.; Saralamba, N.; Day, N.P.J.; Nosten, F.; White, N.J.; Dondorp, A.M.; Imwong, M. Real time PCR detection of common CYP2D6 genetic variants and its application in a Karen population. Malar. J. 2018, 17, 1–14. [Google Scholar] [CrossRef]
- Siqueira, J.F.; Fouad, A.F.; Rôças, I.N. Pyrosequencing as a tool for better understanding of human microbiomes. J. Oral Microbiol. 2012, 4, 10743. [Google Scholar] [CrossRef]
- Scantamburlo, G.; Tziolia, K.; Zopf, M.; Bernardinelli, E.; Soyal, S.M.; Civello, D.A.; Vanoni, S.; Dossena, S.; Patsch, W.; Patrinos, G.P.; et al. Allele Drop Out Conferred by a Frequent CYP2D6 Genetic Variation for Commonly Used CYP2D6*3 Genotyping Assays. Cell. Physiol. Biochem. 2017, 43, 2297–2309. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, A.; Flockhart, D.A.; Hosono, N.; Kubo, M.; Nakamura, Y.; Skaar, T.C. Differential quantification of CYP2D6 gene copy number by four different quantitative real-time PCR assays. Pharm. Genom. 2010, 20, 451–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamenski, G.; Ayazseven, S.; Berndt, A.; Fink, W.; Kamenski, L.; Zehetmayer, S.; Pühringer, H. Clinical Relevance of CYP2D6 Polymorphisms in Patients of an Austrian Medical Practice: A Family Practice-Based Observational Study. Drugs Real World Outcomes 2020, 7, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Heller, T.; Kirchheiner, J.; Armstrong, V.W.; Luthe, H.; Tzvetkov, M.; Brockm??ller, J.; Oellerich, M. AmpliChip CYP450 GeneChip: A new gene chip that allows rapid and accurate CYP2D6 genotyping. Ther. Drug Monit. 2006, 28, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Bank, P.C.D.; Swen, J.J.; Guchelaar, H.J.; Van Der Straaten, T. GenoChip CYP2D6 macroarray as a method to genotype for CYP2D6 variants: Results of a validation study in a Caucasian population. Pharmacogenomics 2015, 16, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Wheeler, M.M.; Patterson, K.; McGee, S.; Dalton, R.; Woodahl, E.L.; Gaedigk, A.; Thummel, K.E.; Nickerson, D.A. Stargazer: A software tool for calling star alleles from next-generation sequencing data using CYP2D6 as a model. Genet. Med. 2019, 21, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Twist, G.P.; Gaedigk, A.; Miller, N.A.; Farrow, E.G.; Willig, L.K.; Dinwiddie, D.L.; Petrikin, J.E.; Soden, S.E.; Herd, S.; Gibson, M.; et al. Constellation: A tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. NPJ Genom. Med. 2016, 1. [Google Scholar] [CrossRef]
- Chen, X.; Shen, F.; Gonzaludo, N.; Malhotra, A.; Rogert, C.; Taft, R.J.; Bentley, D.R.; Eberle, M.A.; Eberle, M. Accurate CYP2D6 genotyping using whole genome sequencing data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ratain, M.J.; Nakamura, Y.; Cox, N.J. CYP2D6 genotype and tamoxifen activity: Understanding interstudy variability in methodological quality. Clin. Pharmacol. Ther. 2013, 94, 185–187. [Google Scholar] [CrossRef] [Green Version]
- Hosein, A.N.; Song, S.; McCart Reed, A.E.; Jayanthan, J.; Reid, L.E.; Kutasovic, J.R.; Cummings, M.C.; Waddell, N.; Lakhani, S.R.; Chenevix-Trench, G.; et al. Evaluating the repair of DNA derived from formalin-fixed paraffin-embedded tissues prior to genomic profiling by SNP-CGH analysis. Lab. Investig. 2013, 93, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Gaedigk, A.; Dinh, J.C.; Jeong, H.; Prasad, B.; Leeder, J.S. Ten years’ experience with the CYP2D6 activity score: A perspective on future investigations to improve clinical predictions for precision therapeutics. J. Pers. Med. 2018, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Westergaard, N.; Nielsen, R.S.; Jørgensen, S.; Vermehren, C. Drug use in Denmark for drugs having pharmacogenomics (PGx) based dosing guidelines from CPIC or DPWG for CYP2D6 and CYP2C19 drug–gene pairs: Perspectives for introducing PGx test to polypharmacy patients. J. Pers. Med. 2020, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers|FDA. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 29 July 2020).
- Yasuda, K.; Ikushiro, S.; Kamakura, M.; Ohta, M.; Sakaki, T. Metabolism of sesamin by cytochrome P450 in human liver microsomes. Drug Metab. Dispos. 2010, 38, 2117–2123. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Sato, Y.; Kumagai, T.; Yoshinari, K.; Nagata, K. Effect of health foods on cytochrome P450-mediated drug metabolism. J. Pharm. Health Care Sci. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Tay-Sontheimer, J.; Shireman, L.M.; Beyer, R.P.; Senn, T.; Witten, D.; Pearce, R.E.; Gaedigk, A.; Gana Fomban, C.L.; Lutz, J.D.; Isoherranen, N.; et al. Detection of an endogenous urinary biomarker associated with CYP2D6 activity using global metabolomics. Pharmacogenomics 2014, 15, 1947–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R.; Smith, R.L. Addressing phenoconversion: The Achilles’ heel of personalized medicine. Br. J. Clin. Pharmacol. 2015, 79, 222–240. [Google Scholar] [CrossRef] [Green Version]
- Tolson, A.H.; Wang, H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 2010, 62, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Yeo, C.W.; Lee, S.S.; Oh, M.K.; Ghim, J.L.; Shon, J.H.; Kim, H.S.; Kim, E.Y.; Kim, D.H.; Shin, J.G. Effect of HNF4α genetic polymorphism G60D on the pharmacokinetics of CYP2D6 substrate tolterodine in healthy Korean individuals. Pharm. Genom. 2013, 23, 175–179. [Google Scholar] [CrossRef]
- Koh, K.H.; Pan, X.; Shen, H.W.; Arnold, S.L.M.; Yu, A.M.; Gonzalez, F.J.; Isoherranen, N.; Jeong, H. Altered expression of small Heterodimer Partner Governs Cytochrome P450 (CYP) 2D6 induction during Pregnancy in CYP2D6-humanized mice. J. Biol. Chem. 2014, 289, 3105–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.H.; Pan, X.; Zhang, W.; McLachlan, A.; Urrutia, R.; Jeong, H. Krüppel-like factor 9 promotes hepatic cytochrome P450 2D6 expression during pregnancy in CYP2D6-humanized mice. Mol. Pharmacol. 2014, 86, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, P.C.D.; Caudle, K.E.; Swen, J.J.; Gammal, R.S.; Whirl-Carrillo, M.; Klein, T.E.; Relling, M.V.; Guchelaar, H.J. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin. Pharmacol. Ther. 2018, 103, 599–618. [Google Scholar] [CrossRef]
- Caudle, K.E.; Thorn, C.F.; Klein, T.E.; Swen, J.J.; McLeod, H.L.; Diasio, R.B.; Schwab, M. Clinical pharmacogenetics implementation consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin. Pharmacol. Ther. 2013, 94, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Egan, A. Highlights of Prescribing Information—CERDELGATM (Eliglustat) Capsules. Available online: www.fda.gov/medwatch (accessed on 10 October 2020).
- Bank, P.C.D.; Swen, J.J.; Guchelaar, H.J. Estimated nationwide impact of implementing a preemptive pharmacogenetic panel approach to guide drug prescribing in primary care in The Netherlands. BMC Med. 2019, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignat, C.; Wille, U.; Ziegler, A. Affinity profiles of morphine, codeine, dihydrocodeine and their glucuronides at opioid receptor subtypes. Life Sci. 1995, 56, 793–799. [Google Scholar] [CrossRef]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. Codeine and Morphine Pathway. Available online: http://journals.lww.com/01213011-200907000-00009 (accessed on 24 October 2020).
- Dean, L. Codeine Therapy and CYP2D6 Genotype. In Medical Genetics Summaries; National Center for Biotechnology Information: Bethesda, MD, USA, 2012; pp. 1–9. [Google Scholar]
- Boyle, K.L.; Rosenbaum, C.D. Oxycodone overdose in the pediatric population: Case files of the University of Massachusetts Medical Toxicology Fellowship. J. Med. Toxicol. 2014, 10, 280–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racoosin, J.A.; Roberson, D.W.; Pacanowski, M.A.; Nielsen, D.R. New evidence about an old drug—Risk with codeine after adenotonsillectomy. N. Engl. J. Med. 2013, 368, 2155–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Use of Codeine and Tramadol Products in Breastfeeding Women—Questions and Answers|FDA. Available online: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/use-codeine-and-tramadol-products-breastfeeding-women-questions-and-answers (accessed on 15 September 2020).
- Goetz, M.P.; Sangkuhl, K.; Guchelaar, H.-J.; Schwab, M.; Province, M.; Whirl-Carrillo, M.; Symmans, W.F.; Mcleod, H.L.; Ratain, M.J.; Zembutsu, H.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and Tamoxifen Therapy. Clin. Pharmacol. Ther. 2018, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez de Dueñas, E.; Ochoa Aranda, E.; Blancas Lopez-Barajas, I.; Ferrer Magdalena, T.; Bandrés Moya, F.; Chicharro García, L.M.; Gómez Capilla, J.A.; Zafra Ceres, M.; de Haro, T.; Romero Llorens, R.; et al. Adjusting the dose of tamoxifen in patients with early breast cancer and CYP2D6 poor metabolizer phenotype. Breast 2014, 23, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Lee, H.J.; Lee, K.S.; Lee, E.S.; Jang, I.J.; Ro, J. Clinical implications of CYP2D6 genotypes predictive of tamoxifen pharmacokinetics in metastatic breast cancer. J. Clin. Oncol. 2007, 25, 3837–3845. [Google Scholar] [CrossRef]
- Rae, J.M.; Drury, S.; Hayes, D.F.; Stearns, V.; Thibert, J.N.; Haynes, B.P.; Salter, J.; Sestak, I.; Cuzick, J.; Dowsett, M. CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J. Natl. Cancer Inst. 2012, 104, 452–460. [Google Scholar] [CrossRef]
- Regan, M.M.; Leyland-Jones, B.; Bouzyk, M.; Pagani, O.; Tang, W.; Kammler, R.; Dell’Orto, P.; Biasi, M.O.; Thürlimann, B.; Lyng, M.B.; et al. CYP2D6 Genotype and Tamoxifen Response in Postmenopausal Women with Endocrine-Responsive Breast Cancer: The Breast International Group 1-98 Trial. J. Natl. Cancer 2008, 37, 1–9. [Google Scholar] [CrossRef]
- Chan, C.W.H.; Law, B.M.H.; So, W.K.W.; Chow, K.M.; Waye, M.M.Y. Pharmacogenomics of breast cancer: Highlighting CYP2D6 and tamoxifen. J. Cancer Res. Clin. Oncol. 2020, 146, 1395–1404. [Google Scholar] [CrossRef]
- Wei, X.; Sun, H.; Zhuang, J.; Weng, X.; Zheng, B.; Lin, Q.; Zhang, G.; Cai, J. Cost-effectiveness analysis of CYP2D6*10 pharmacogenetic testing to guide the adjuvant endocrine therapy for postmenopausal women with estrogen receptor positive early breast cancer in China. Clin. Drug Investig. 2020, 40, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Kamal, A.; Ames, M.M. Tamoxifen pharmacogenomics: The role of CYP2D6 as a predictor of drug response. Clin. Pharmacol. Ther. 2008, 83, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D.J.; Thorn, C.F.; Desta, Z.; Flockhart, D.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Tamoxifen pathway, pharmacokinetics. Pharm. Genom. 2013, 23, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Hicks, J.K.; Swen, J.J.; Thorn, C.F.; Sangkuhl, K.; Kharasch, E.D.; Ellingrod, V.L.; Skaar, T.C.; Müller, D.J.; Gaedigk, A.; Stingl, J.C. Clinical pharmacogenetics implementation consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin. Pharmacol. Ther. 2013, 93, 402–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suiter, C.C.; Moriyama, T.; Matreyek, K.A.; Yang, W.; Scaletti, E.R.; Nishii, R.; Yang, W.; Hoshitsuki, K.; Singh, M.; Trehan, A.; et al. Massive parallel variant characterization identifies NUDT15 alleles associated with thiopurine toxicity. bioRxiv 2020, 1–42. [Google Scholar] [CrossRef]
- Mcinnes, G.; Dalton, R.; Sangkuhl, K.; Whirl-Carrillo, M.; Lee, S.-B.; Tsao, P.S.; Gaedigk, A.; Altman, R.B.; Woodahl, E.L. Transfer learning enables prediction of CYP2D6 haplotype function. bioRxiv 2020, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Lauschke, V.M.; Ingelman-Sundberg, M. Emerging strategies to bridge the gap between pharmacogenomic research and its clinical implementation. NPJ Genom. Med. 2020, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Liau, Y.; Maggo, S.; Miller, A.L.; Pearson, J.F.; Kennedy, M.A.; Cree, S.L. Nanopore sequencing of the pharmacogene CYP2D6 allows simultaneous haplotyping and detection of duplications. Pharmacogenomics 2019, 20, 1033–1047. [Google Scholar] [CrossRef]
- Napolitano, G.; Stingl, J.C.; Schmid, M.; Viviani, R. Predicting CYP2D6 phenotype from resting brain perfusion images by gradient boosting. Psychiatry Res. Neuroimaging 2017, 259, 16–24. [Google Scholar] [CrossRef]
- McInnes, G.; Dalton, R.; Sangkuhl, K.; Whirl-Carrillo, M.; Lee, S.; Altman, R.B.; Woodahl, E.L. Hubble2D6: A deep learning approach for predicting drug metabolic activity. bioRxiv 2019, 684357. [Google Scholar] [CrossRef] [Green Version]
- Van Der Lee, M.; Allard, W.G.; Vossen, R.H.A.M.; Baak-Pablo, R.F.; Menafra, R.; Deiman, B.A.L.M.; Deenen, M.J.; Neven, P.; Johansson, I.; Gastaldello, S.; et al. A unifying model to predict variable drug response for personalised medicine. bioRxiv 2020, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Campion, D.P.; Dowell, F.J. Translating pharmacogenetics and pharmacogenomics to the clinic: Progress in human and veterinary medicine. Front. Vet. Sci. 2019, 6, 1–11. [Google Scholar] [CrossRef]
- Mealey, K.L. Adverse drug reactions in herding-breed dogs: The role of P-glycoprotein. Compend. Contin. Educ. Pract. Vet. 2006, 28, 23–33. [Google Scholar]
- Fleischer, S.; Sharkey, M.; Mealey, K.; Ostrander, E.A.; Martinez, M. Pharmacogenetic and metabolic differences between dog breeds: Their impact on canine medicine and the use of the dog as a preclinical animal model. AAPS J. 2008, 10, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, S.K.; Engel, L.; Reitz, B.; Bolten, S.; Burton, E.G.; Maziasz, T.J.; Yan, B.; Schoenhard, G.L. Evidence for polymorphism in the canine metabolism of the cyclooxygenase 2 inhibitor, celecoxib. Drug Metab. Dispos. 1999, 27, 1133–1142. [Google Scholar]
- Corado, C.R.; McKemie, D.S.; Young, A.; Knych, H.K. Evidence for polymorphism in the cytochrome P450 2D50 gene in horses. J. Vet. Pharmacol. Ther. 2016, 39, 245–254. [Google Scholar] [CrossRef]
- Court, M.H. Canine Cytochrome P-450 Pharmacogenetics. Vet. Clin. N. Am. Small Anim. Pract. 2013, 43, 1027–1038. [Google Scholar]
Figure 1.
Venn Diagram showing recognised (non-HLA) pharmacogenes according to their association with drugs used in different therapeutic areas. CYP2D6 overlaps with four distinct therapeutic areas. BCHE: Butyrylcholinesterase. CFTR: Cystic fibrosis transmembrane conductance regulator. CYP2B6, CYP2C9, CYP2C19, CYP3A5, CYP4F2: Members of cytochrome P450 superfamily. DPYD: Dihydropyrimidine dehydrogenase. F5: Factor 5. G6PD: Glucose-6-phosphate dehydrogenase. IFNL3: Interferon lambda 3. ITPA: Inosine triphosphate pyrophosphohydrolase. NAT2: N-Acetyltransferase 2. NUDT15: Nudix hydrolase 15. POLG: DNA polymerase subunit γ. RARG: Retinoic acid receptor γ. SLCO1B1: Solute carrier organic anion transporter family member 1B1. SLC28A3: Solute carrier family 28 member 3. TPMT: Thiopurine S-methyltransferase. UGT1A1: UDP-glucuronosyltransferases. VKORC1: Vitamin K epoxide reductase complex.
Figure 1.
Venn Diagram showing recognised (non-HLA) pharmacogenes according to their association with drugs used in different therapeutic areas. CYP2D6 overlaps with four distinct therapeutic areas. BCHE: Butyrylcholinesterase. CFTR: Cystic fibrosis transmembrane conductance regulator. CYP2B6, CYP2C9, CYP2C19, CYP3A5, CYP4F2: Members of cytochrome P450 superfamily. DPYD: Dihydropyrimidine dehydrogenase. F5: Factor 5. G6PD: Glucose-6-phosphate dehydrogenase. IFNL3: Interferon lambda 3. ITPA: Inosine triphosphate pyrophosphohydrolase. NAT2: N-Acetyltransferase 2. NUDT15: Nudix hydrolase 15. POLG: DNA polymerase subunit γ. RARG: Retinoic acid receptor γ. SLCO1B1: Solute carrier organic anion transporter family member 1B1. SLC28A3: Solute carrier family 28 member 3. TPMT: Thiopurine S-methyltransferase. UGT1A1: UDP-glucuronosyltransferases. VKORC1: Vitamin K epoxide reductase complex.

Figure 2.
Summary of main CYP2D6 structural variant types. Adapted from PharmVar CYP2D6 Structural Variation document [47]. (A) shows the wildtype layout of CYP2D6, CYP2D7 and CYP2D8. (B) shows examples of structural variants involving CYP2D6. For simplicity, REP6, REP7 and downstream elements have been omitted in (B).
Figure 2.
Summary of main CYP2D6 structural variant types. Adapted from PharmVar CYP2D6 Structural Variation document [47]. (A) shows the wildtype layout of CYP2D6, CYP2D7 and CYP2D8. (B) shows examples of structural variants involving CYP2D6. For simplicity, REP6, REP7 and downstream elements have been omitted in (B).

Figure 3.
The 3D structure of CYP2D6 from different angles, annotated with the locations of notable star allele-defining variants (neon green). Images (A–C) show CYP2D6 from different angles. The images on the left and right show the molecular surface of CYP2D6 and ribbon model of CYP2D6, respectively. The central heme group is highlighted in the ribbon models. Molecular structures were generated and annotated using Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) software [59].
Figure 3.
The 3D structure of CYP2D6 from different angles, annotated with the locations of notable star allele-defining variants (neon green). Images (A–C) show CYP2D6 from different angles. The images on the left and right show the molecular surface of CYP2D6 and ribbon model of CYP2D6, respectively. The central heme group is highlighted in the ribbon models. Molecular structures were generated and annotated using Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) software [59].

Figure 4.
This figure illustrates the CYP2D6-mediated conversion of codeine to morphine. This pathway results in the metabolism of 0–15% of codeine. Other metabolites codeine-6-glucuronide and norcodeine are metabolized by UGT2B4 and UGT2B7, and CYP3A4, respectively, accounting for remaining codeine metabolism. [96]. Secondary metabolites such as morphine-6-glucuronide are not shown for simplicity.
Figure 4.
This figure illustrates the CYP2D6-mediated conversion of codeine to morphine. This pathway results in the metabolism of 0–15% of codeine. Other metabolites codeine-6-glucuronide and norcodeine are metabolized by UGT2B4 and UGT2B7, and CYP3A4, respectively, accounting for remaining codeine metabolism. [96]. Secondary metabolites such as morphine-6-glucuronide are not shown for simplicity.

Figure 5.
This figure provides a simplified overview focusing on the role of CYP2D6 in the formation of endoxifen. Over 90% of tamoxifen metabolism is CYP3A-mediated N-demethylation to the primary metabolite, N-desmethyltamoxifen, which can be oxidized further by CYP2D6 to endoxifen. Approximately 7% of tamoxifen metabolism is CYP2D6-mediated hydroxylation to 4-hydroxytamoxifen. There is high interindividual variation in endoxifen exposure, which is a significantly more potent anti-oestrogen compared to parent tamoxifen [101,109].
Figure 5.
This figure provides a simplified overview focusing on the role of CYP2D6 in the formation of endoxifen. Over 90% of tamoxifen metabolism is CYP3A-mediated N-demethylation to the primary metabolite, N-desmethyltamoxifen, which can be oxidized further by CYP2D6 to endoxifen. Approximately 7% of tamoxifen metabolism is CYP2D6-mediated hydroxylation to 4-hydroxytamoxifen. There is high interindividual variation in endoxifen exposure, which is a significantly more potent anti-oestrogen compared to parent tamoxifen [101,109].


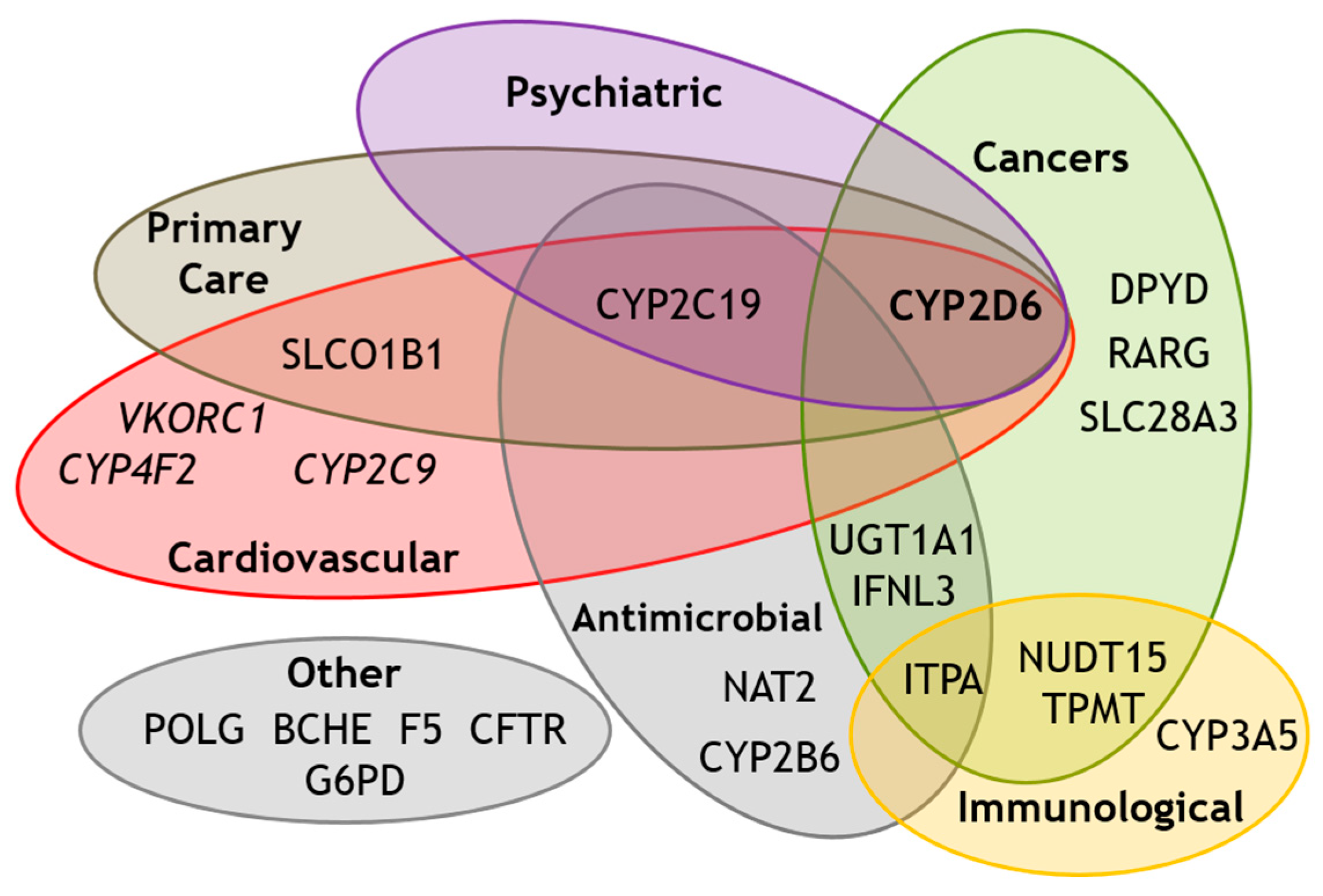{kind=link}
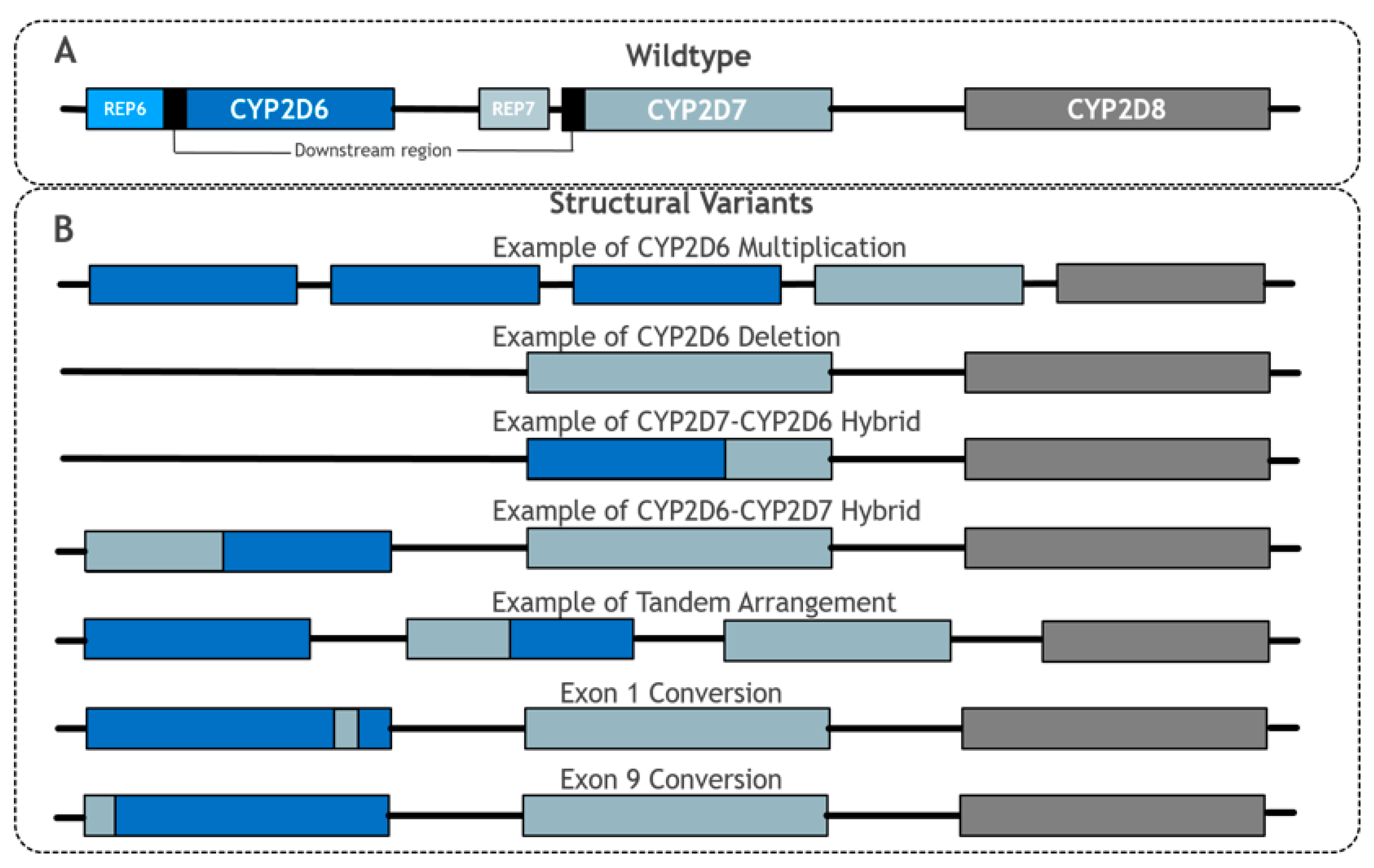{kind=link}
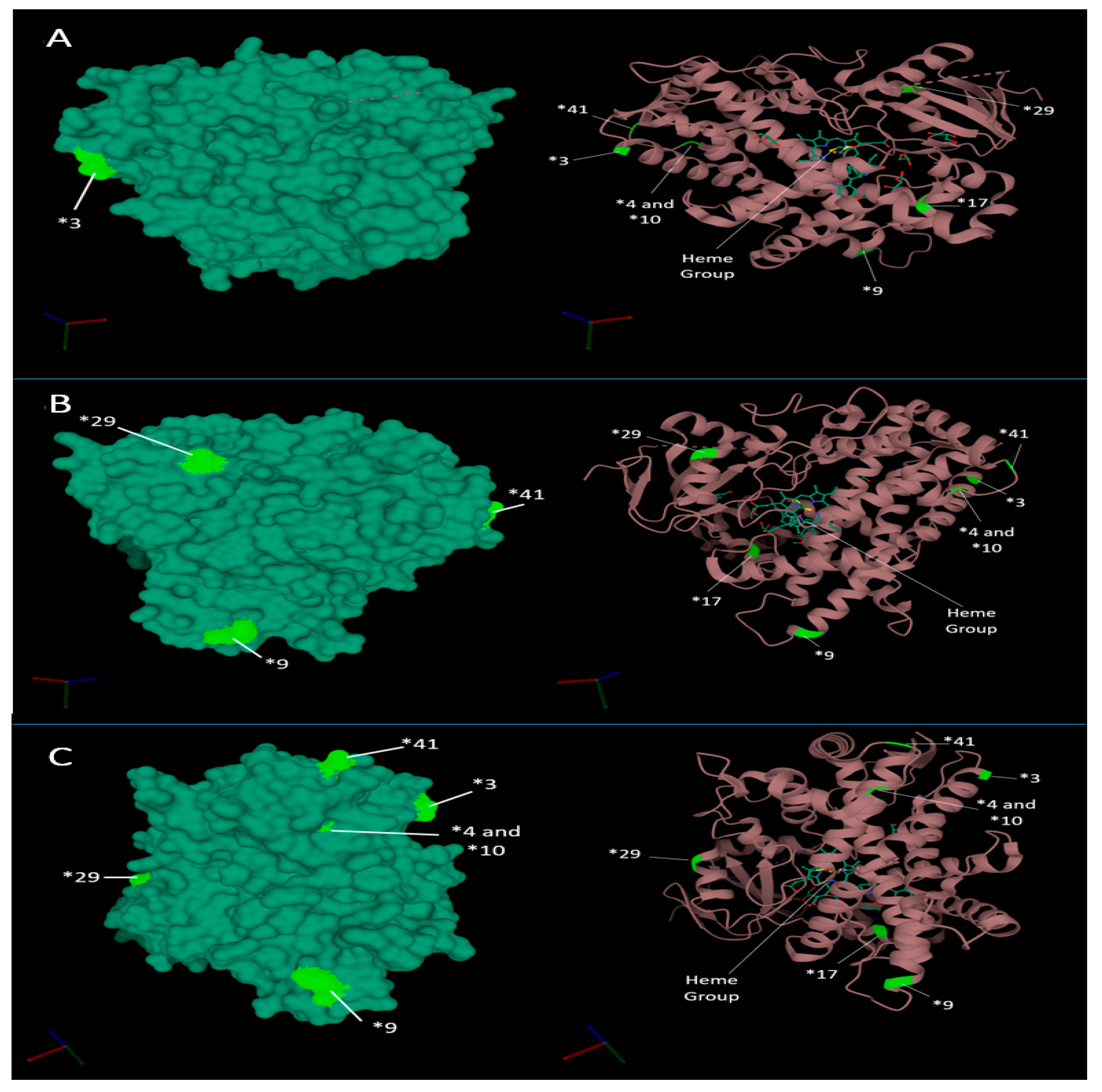{kind=link}
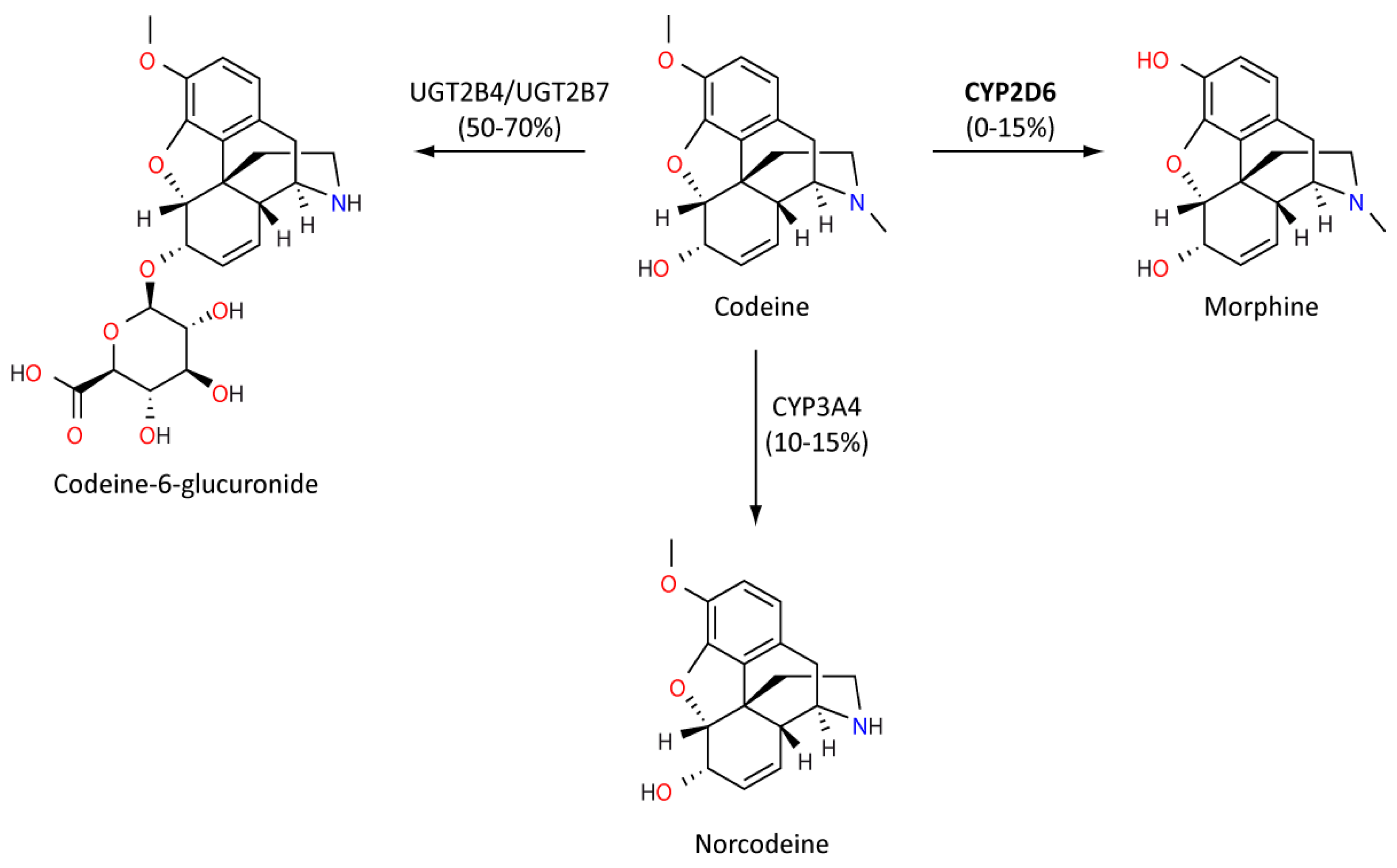{kind=link}
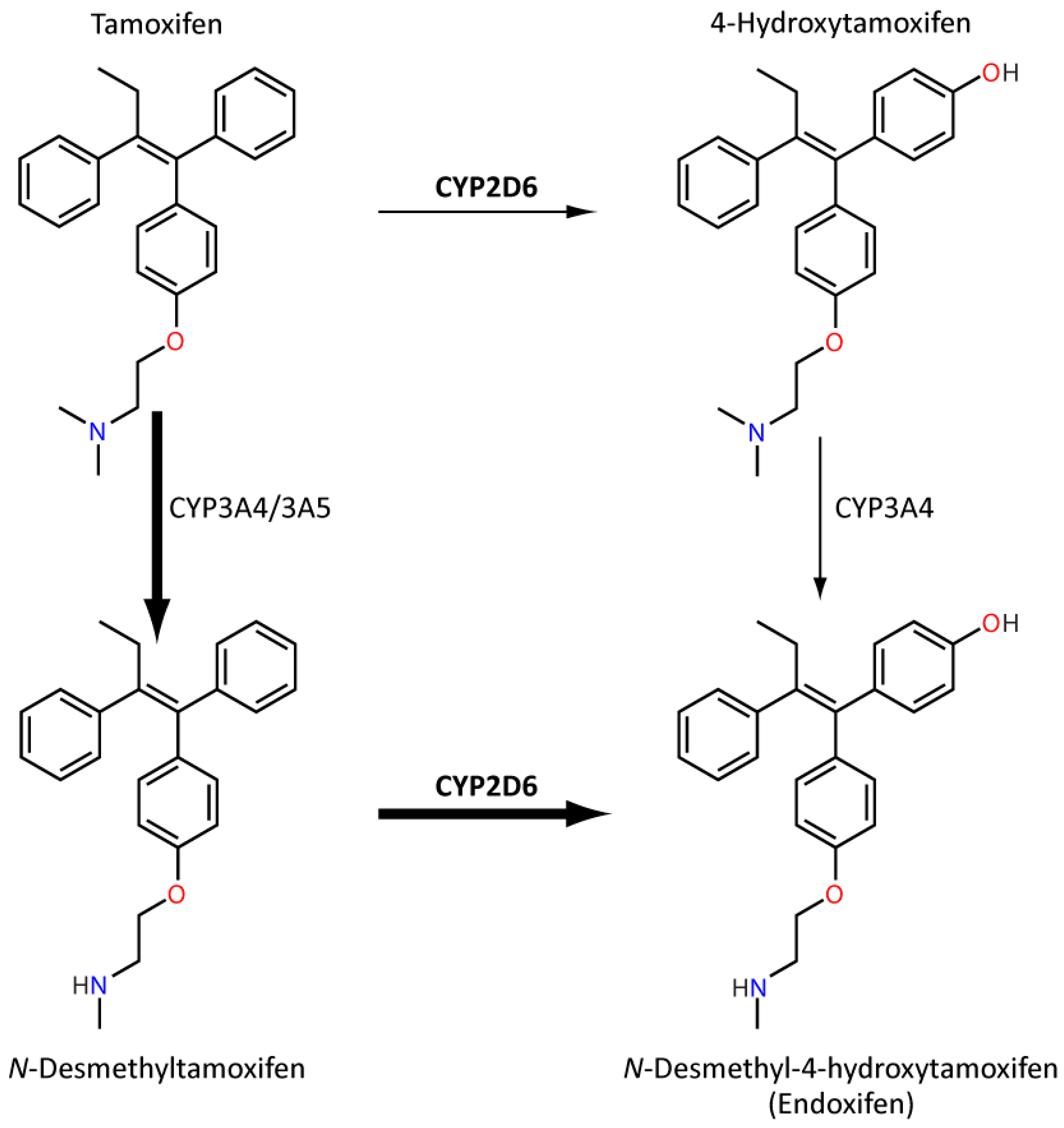{kind=link}
Table 1.
Available CYP2D6-Drug Clinical Guidelines.
| Drug | Drug Class | Guideline Committee Recommendations | PharmGKB Evidence Level | ||
|---|---|---|---|---|---|
| CPIC | DPWG | Other | |||
| Amiodarone | Antiarrhythmic | - | No Recommendation | - | - |
| Amitriptyline | Antidepressant | CD or AD | CD or AD | - | 1A |
| Aripiprazole | Antipsychotic | - | CD | - | 3 |
| Atenolol | β blocker | - | No Recommendation | - | - |
| Atomoxetine | ADHD treatment | CD | CD or AD | - | 1A |
| Bisoprolol | β blocker | - | No Recommendation | - | - |
| Brexpiprazole | Antipsychotic | - | CD | - | - |
| Carvedilol | β blocker | - | No Recommendation | - | 3 |
| Citalopram | Antidepressant | - | No Recommendation | - | 3 |
| Clomipramine | Antidepressant | CD or AD | CD or AD | - | 1A |
| Clonidine | Antihypertensive | - | No Recommendation | - | - |
| Clozapine | Antipsychotic | - | No Recommendation | - | - |
| Codeine | Analgesic | AD | CD or AD | CPNDs: AD | 1A |
| Desipramine | Antidepressant | CD or AD | - | - | 1A |
| Disopyramide | Antiarrhythmic | - | No Recommendation | - | - |
| Doxepin | Antidepressant | CD or AD | CD or AD | - | 1A |
| Duloxetine | Antidepressant | - | No Recommendation | - | - |
| Eliglustat | Gaucher’s disease | - | CD with CM or AD | - | - |
| Escitalopram | Antidepressant | - | No Recommendation | - | 3 |
| Flecainide | Antiarrhythmic | - | CD | - | 2A |
| Fluoxetine | Antidepressant | - | No Recommendation | - | 3 |
| Flupenthixol | Antipsychotic | - | No Recommendation | - | - |
| Fluphenazine | Antipsychotic | - | No Recommendation | - | - |
| Fluvoxamine | Antidepressant | CD or AD | No Recommendation | - | 1A |
| Gefitinib | Cancer treatment | - | No Recommendation | - | 3 |
| Haloperidol | Antipsychotic | - | CD or AD | - | 3 |
| Imipramine | Antidepressant | CD or AD | CD | - | 1A |
| Methylphenidate | ADHD treatment | - | No Recommendation | - | 4 |
| Metoprolol | β blocker | - | CD or AD | - | 2A |
| Mirtazapine | Antidepressant | - | No Recommendation | - | 3 |
| Nortriptyline | Antidepressant | CD or AD | CD or AD | - | 1A |
| Olanzapine | Antipsychotic | - | No Recommendation | - | 3 |
| Ondansetron | Antiemetic | AD | - | - | 1A |
| Oxycodone | Analgesic | - | No Recommendation | - | 2A |
| Paroxetine | Antidepressant | CD or AD | AD | - | 1A |
| Pimozide | Antipsychotic | - | CD | - | 4 |
| Propafenone | Antiarrhythmic | - | CD or AD | - | 2A |
| Quetiapine | Antipsychotic | - | No Recommendation | - | 4 |
| Quinidine | Antiarrhythmic | - | No Recommendation | - | - |
| Risperidone | Antipsychotic | - | CD or AD | - | 1B |
| Sertraline | Antidepressant | - | No Recommendation | - | 3 |
| Sotalol | Antiarrhythmic | - | No Recommendation | - | - |
| Tamoxifen | Cancer treatment | CD or AD | CD or AD | CPNDS: AD, RNPGx: No Recommendation | 1A |
| Tramadol | Analgesic | - | CD or AD | - | 1B |
| Trimipramine | Antidepressant | CD or AD | - | - | 1A |
| Tropisetron | Antiemetic | AD | - | - | 1A |
| Venlafaxine | Antidepressant | - | CD or AD | - | 2A |
| Zuclopenthixol | Antipsychotic | - | CD or AD | - | 3 |
Data extracted from PharmGKB Clinical Guideline Annotations; guidance is currently available for 48 specific drugs [23]. CPIC: Clinical Pharmacogenetics Implementation Consortium. DPWG: Dutch Pharmacogenetics Working Group. CPNDS: Canadian Pharmacogenomics Network for Drug Safety. RNPGx: French National Network of Pharmacogenetics. CD: Change Dosage based on metabolism status. AD: Alternative Drug recommended based on metabolism status. CM: Co-medication. ADHD: attention deficit hyperactivity disorder. It should be noted that Pharmgkb describes evidence between drug and variants and in some cases different variants within a gene are assigned differing levels of evidence. Although guidelines have been generated, the level of evidence can vary. Thus, PharmGKB assigns CYP2D6 variant-drug annotations a level based on the strength of evidence. Class 1A and 1B annotations indicate strong evidence based on multiple studies and/or is based on the variant–drug combination having been integrated into pharmacogenetic guidelines or major healthcare system. Class 2A and 2B indicated moderate evidence with an effect seen in multiple studies. Class 3 indicates low evidence, typically a single, highly significant finding of association. Class 4 indicates only preliminary evidence of association.
Table 2.
Selection of common CYP2D6 variant frequencies in major population groups.
| CYP2D6 Allele | Frequency | Predicted Function | |||
|---|---|---|---|---|---|
| AFR | EUR | EAS | SAS | ||
| *xN | 0.07 | 0.03 | 0.02 | 0.02 | Allele Specific |
| *1xN | 0.03 | 0.01 | 0.01 | 0.01 | Increased Metaboliser |
| *2 | 0.27 | 0.34 | 0.14 | 0.30 | Normal Metaboliser |
| *3 | 0.00 | 0.04 | 0.00 | 0.00 | Decreased Metaboliser |
| *4 | 0.12 | 0.15 | 0.00 | 0.09 | None Functional |
| *4xN | 0.02 | 0.00 | 0.00 | 0.00 | None Functional |
| *5 | 0.04 | 0.03 | 0.07 | 0.03 | None Functional |
| *6 | 0.00 | 0.02 | 0.00 | 0.00 | None Functional |
| *9 | 0.00 | 0.02 | 0.00 | 0.01 | Decreased Metaboliser |
| *10/114(A) | 0.03 | 0.00 | 0.59 | 0.20 | Decreased Metaboliser |
| *17 | 0.20 | 0.00 | 0.00 | 0.00 | Decreased Metaboliser |
| *29 | 0.09 | 0.00 | 0.00 | 0.00 | Decreased Metaboliser |
| *33 | 0.00 | 0.01 | 0.00 | 0.00 | Normal Metaboliser |
| *35 | 0.00 | 0.06 | 0.00 | 0.00 | Normal Metaboliser |
| *36 | 0.00 | 0.00 | 0.01 | 0.01 | Decreased Metaboliser |
| *41 | 0.03 | 0.03 | 0.03 | 0.08 | Decreased Metaboliser |
AFR: African Population, EUR: European Population, EAS: East Asian population, SAS: South Asian population. Frequencies for AFR, EUR and EAS populations derived from [63] and SAS from [61]. Predicted function was extracted via the Clinical Pharmacogenetics Implementation Consortium (CPIC) tricyclic antidepressants guideline [64].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Taylor, C.; Crosby, I.; Yip, V.; Maguire, P.; Pirmohamed, M.; Turner, R.M. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes 2020, 11, 1295. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111295
AMA Style
Taylor C, Crosby I, Yip V, Maguire P, Pirmohamed M, Turner RM. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes. 2020; 11(11):1295. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111295
Chicago/Turabian StyleTaylor, Christopher, Ian Crosby, Vincent Yip, Peter Maguire, Munir Pirmohamed, and Richard M. Turner. 2020. "A Review of the Important Role of CYP2D6 in Pharmacogenomics" Genes 11, no. 11: 1295. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111295
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.