Seascape Genomics of the Sugar Kelp Saccharina latissima along the North Eastern Atlantic Latitudinal Gradient

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials & Methods
2.1. Sampling, DNA Extraction, and Genotyping
2.2. Genetic Diversity and Analysis of Population Structure
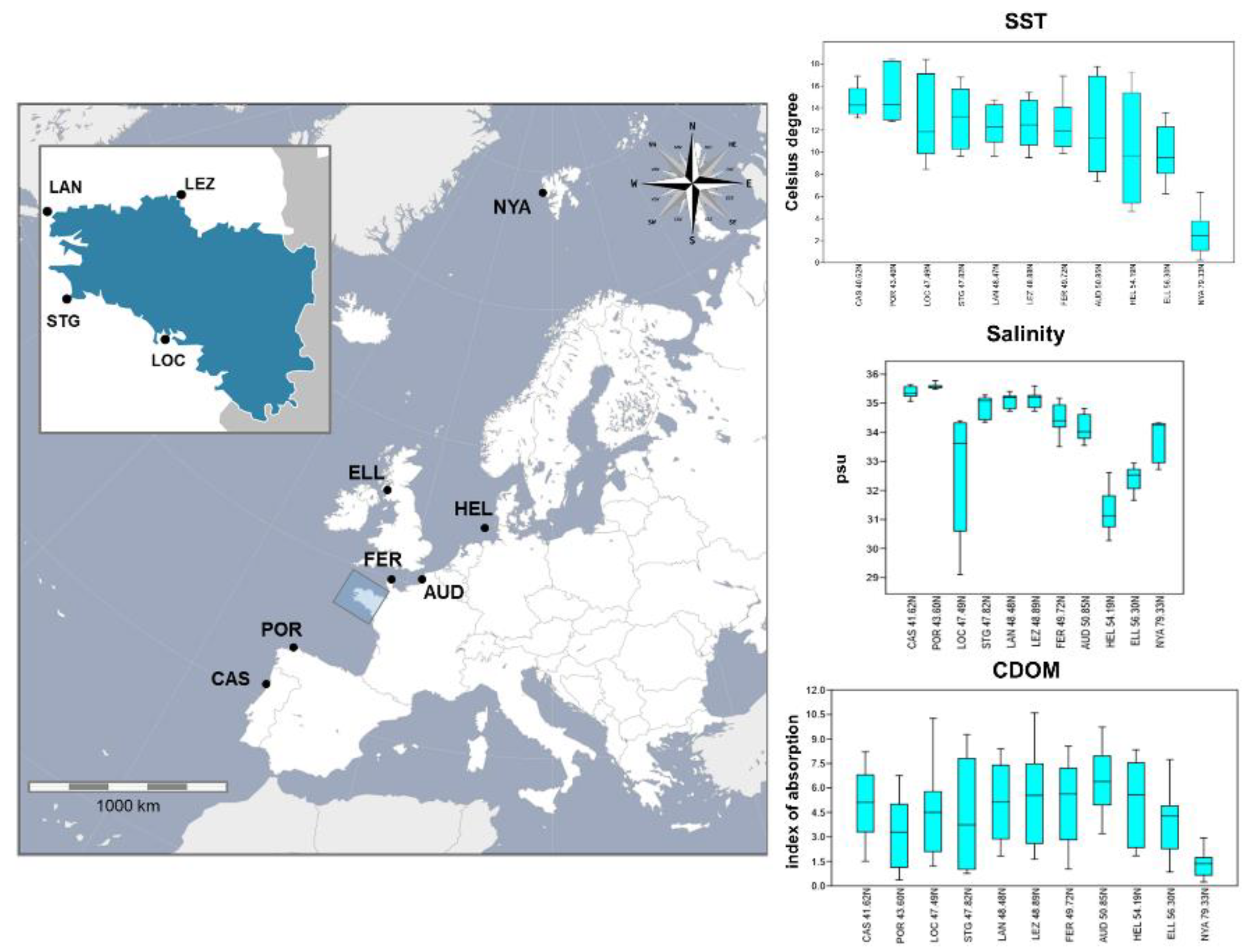
2.3. Description of the Environmental Variables
2.4. Test for Detection of Candidate Outlier Loci
2.5. Environment-Genome Multivariate Correlation Tests
3. Results
3.1. ddRAD-seq Genotyping Effort and Locus and SNP Marker Filtering
3.2. Genetic Diversity
3.3. Deviation from Random Mating
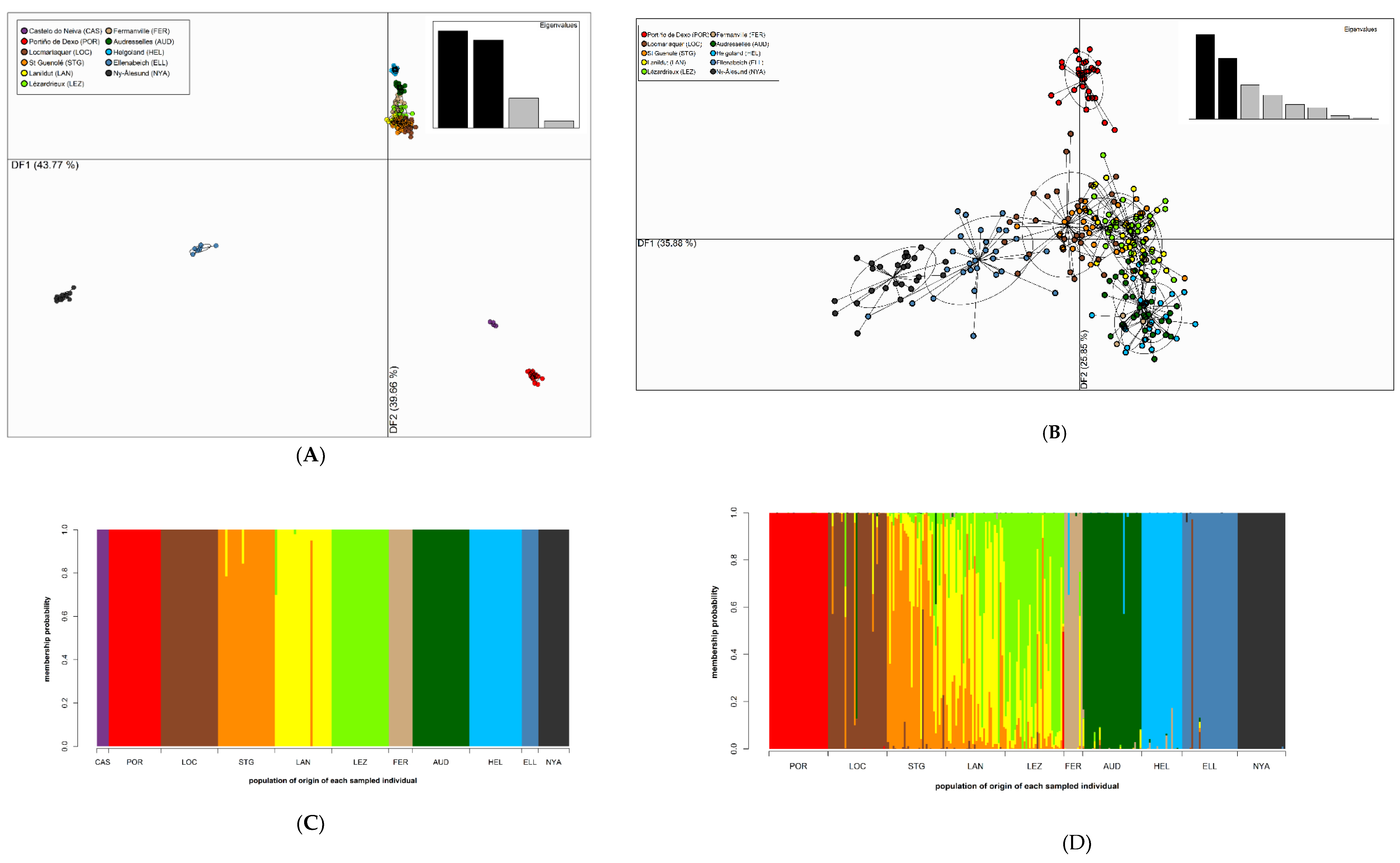
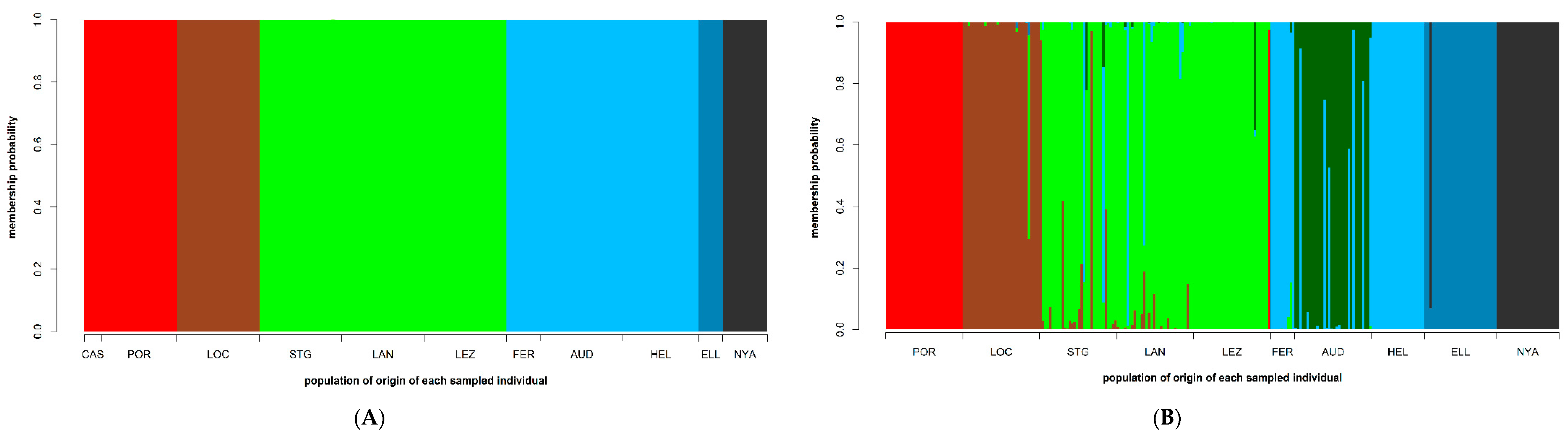
3.4. Population Structure
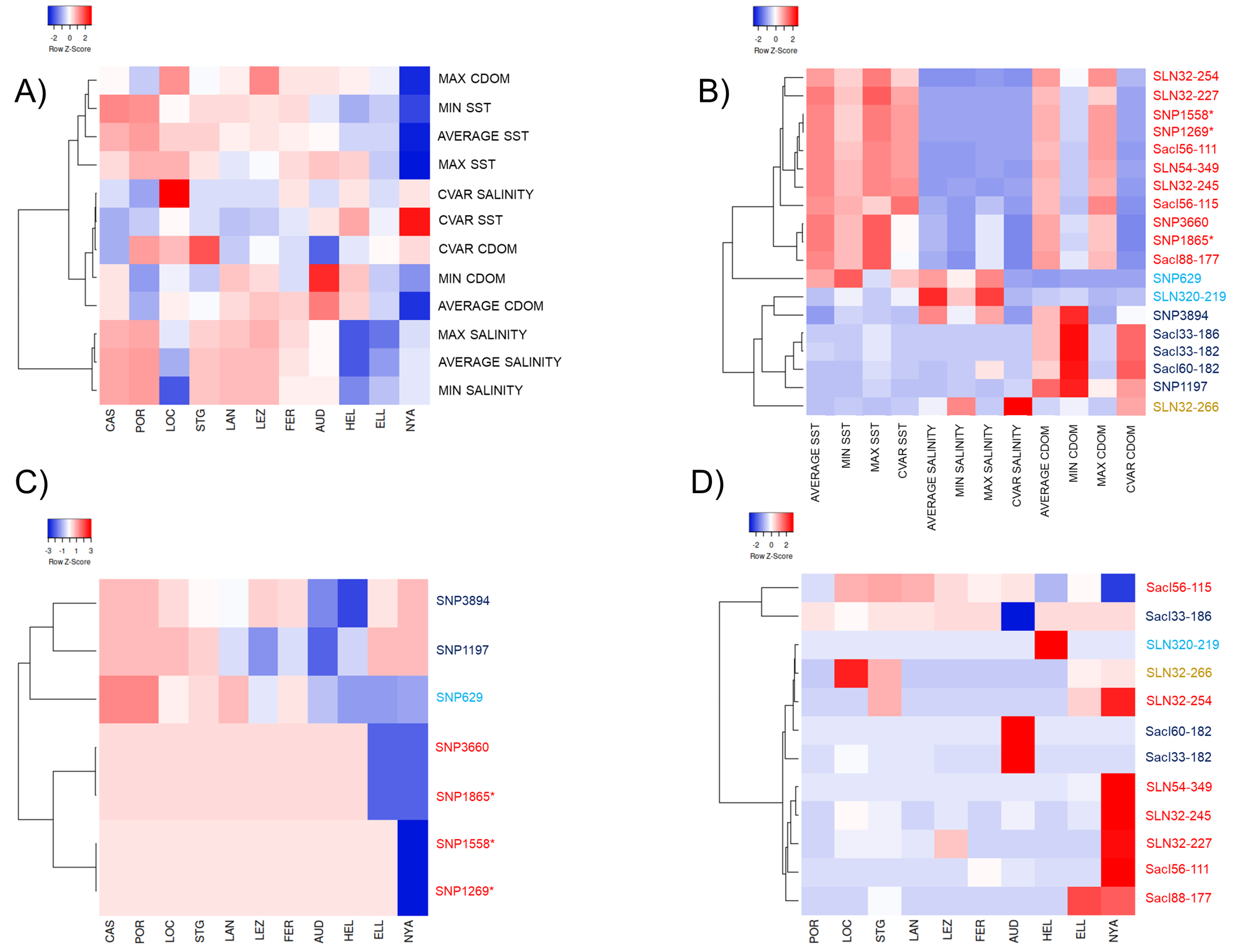
3.5. Test for Detection of Candidate Outlier Loci and Correlations with the Environmental Parameters
4. Discussion
4.1. Patterns of Variation in Within-Locality Genetic Diversity
4.2. Low Connectivity Levels between Disjunct S. latissima Sampling Sites
4.3. Adaptive Functional Responses to the Seascape in S. latissima
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data
References
- Frankel, O.H.; Soulé, M.E. (Eds.) Conservation and Evolution; Cambridge University Press: Cambridge, UK, 1981; pp. 1–327. [Google Scholar]
- Holsinger, K.E.; Vitt, P. The future of conservation biology, what’s a geneticist to do? In The Ecological Basis of Conservation; Pickett, S.T.A., Ostfeld, R.S., Shachak, M., Likens, G.E., Eds.; Springer: Boston, MA, USA, 1997; pp. 202–216. [Google Scholar]
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Excoffier, L.; Foll, M.; Petit, R.J. Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 481–501. [Google Scholar] [CrossRef]
- Hampe, A.; Petit, R.J. Conserving biodiversity under climate change, the rear edge matters. Ecol. Lett. 2005, 8, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Provan, J.; Maggs, C.A. Unique genetic variation at a species' rear edge is under threat from global climate change. Proc. R. Soc. B 2012, 279, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Hirao, A.S.; Watanabe, M.; Tsuyuzaki, S.; Shimono, A.; Li, X.; Masuzawa, T.; Wada, N. Genetic diversity within populations of an arctic–alpine species declines with decreasing latitude across the Northern Hemisphere. J. Biogeogr. 2017, 44, 2740–2751. [Google Scholar] [CrossRef]
- Ninua, L.; Tarkhnishvili, D.; Gvazava, E. Phylogeography and taxonomic status of trout and salmon from the Ponto-Caspian drainages, with inferences on European brown trout evolution and taxonomy. Ecol. Evol. 2018, 8, 2645–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horreo, J.L.; Griffiths, A.M.; Machado-Schiaffino, G.; Stevens, J.R.; Garcia-Vazquez, E. Northern areas as refugia for temperate species under current climate warming, Atlantic salmon (Salmo salar L.) as a model in northern Europe. J. Fish Biol. 2019, 95, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmesan, C. Ecological and Evolutionary Responses to Recent Climate Change. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 637–669. [Google Scholar] [CrossRef] [Green Version]
- Ruthrof, K.X.; Breshears, D.D.; Fontaine, J.B.; Froend, R.H.; Matusick, G.; Kala, J.; Miller, B.P.; Mitchell, P.J.; Wilson, S.K.; van Keulen, M.; et al. Subcontinental heat-wave triggers terrestrial and marine, multi-taxa responses. Sci. Rep. 2018, 8, 13094. [Google Scholar] [CrossRef]
- Straub, S.C.; Wernberg, T.; Thomsen, M.S.; Moore, P.J.; Burrows, M.T.; Harvey, B.P.; Smale, D.A. Resistance, extinction, and everything in between—The diverse responses of seaweeds to marine heatwaves. Front. Mar. Sci. 2019, 6, 763. [Google Scholar] [CrossRef]
- Wernberg, T.; Coleman, M.A.; Bennett, S.; Thomsen, M.S.; Tuya, F.; Kelaher, B.P. Genetic diversity and kelp forest vulnerability to climatic stress. Sci. Rep. 2018, 8, 1851. [Google Scholar] [CrossRef] [PubMed]
- Assis, J.; Araújo, M.B.; Serrão, E.A. Projected climate changes threaten ancient refugia of kelp forests in the North Atlantic. Glob. Chang. Biol. 2018, e55–e66. [Google Scholar] [CrossRef] [PubMed]
- Luikart, G.; England, P.R.; Tallmon, D.A.; Jordan, S.; Taberlet, P. The power and promise of population genomics, from genotyping to genome typing. Nat Rev Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Allendorf, F.; Hohenlohe, P.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697–709. [Google Scholar] [CrossRef]
- Haasl, R.J.; Payseur, B.A. Fifteen years of genome wide scans for selection, trends, lessons and unaddressed genetic sources of complication. Mol. Ecol. 2016, 25, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Frichot, E.; Schoville, S.; De Villemereuil, P.; Gaggiotti, O.E.; François, O. Detecting adaptive evolution based on association with ecological gradients: Orientation matters! Heredity 2015, 115, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nature 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Amish, S.J.; Ali, O.; Peacock, M.; Miller, M.; Robinson, M.; Smith, S.; Luikart, G.; Neville, H. Assessing thermal adaptation using family-based association and FST-outlier tests in a threatened trout. Mol. Ecol. 2019, 28, 2573–2593. [Google Scholar] [CrossRef]
- Steneck, R.S.; Graham, M.H.; Bourque, B.J.; Corbett, D.; Erlandson, J.M.; Estes, J.A.; Tegner, M.J. Kelp forest ecosystems, biodiversity, stability, resilience and future. Environ. Conserv. 2002, 29, 436–459. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, I.; Wiencke, C.; Bischof, K.; Buchholz, C.M.; Buck, B.H.; Eggert, A.; Feuerpfeil, P.; Hanelt, D.; Jacobsen, S.; Karez, R.; et al. The genus Laminaria sensu lato, recent insights and developments. Eur. J. Phycol. 2008, 43, 1–86. [Google Scholar] [CrossRef]
- Oppliger, L.V.; Correa, J.A.; Engelen, A.H.; Tellier, F.; Vieira, V.; Faugeron, S.; Valero, M.; Gomez, G.; Destombe, C. Temperature effects on gametophyte life–history traits and geographic distribution of two cryptic kelp species. PLoS ONE 2012, 7, e39289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, C.M.M.; Li, H.; Bischof, K.; Bartsch, I.; Valentin, K.U.; Corre, E.; Collén, J.; Harms, L.; Glöckner, G.; Heinrich, S. Is geographical variation driving the transcriptomic responses to multiple stressors in the kelp Saccharina latissima? BMC Plant Biol. 2019, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Smale, D.A. Impacts of ocean warming on kelp forest ecosystems. New Phytol. 2020, 225, 1447–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggs, C.; Castilho, R.; Foltz, D.; Hensler, C.; Jolly, M.; Kelly, J.; Olsen, J.; Perez, E.; Stam, W.; Vainola, R.; et al. Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 2008, 89, S108–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assis, J.; Coelho, N.C.; Alberto, F.; Valero, M.; Raimondi, P.; Reed, D.; Serrao, E.A. High and distinct range–edge genetic diversity despite local bottlenecks. PLoS ONE 2013, 8, e68646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringloe, T.T.; Verbruggen, H.; Saunders, G.W. Unique biodiversity in Arctic marine forests is shaped by diverse recolonization pathways and far northern glacial refugia. Proc. Nat. Acad. Sci. USA 2020, 202002753. [Google Scholar] [CrossRef]
- Robuchon, M.; Le Gall, L.; Mauger, S.; Valero, M. Contrasting genetic diversity patterns in two sister kelp species co-distributed along the coast of Brittany, France. Mol. Ecol. 2014, 23, 2669–2685. [Google Scholar] [CrossRef] [Green Version]
- Brennan, G.; Kregting, L.; Beatty, G.E.; Cole, C.; Elsäßer, B.; Savidge, G.; Provan, J. Understanding macroalgal dispersal in a complex hydrodynamic environment, a combined population genetic and physical modelling approach. J. R. Soc. Interface 2014, 11, 20140197. [Google Scholar] [CrossRef]
- Neiva, J.; Serrão, E.A.; Paulino, C.; Gouveia, L.; Want, A.; Tamigneaux, É.; Ballenghien, M.; Mauger, S.; Fouqueau, L.; Engel-Gautier, C.; et al. Genetic structure of amphi-Atlantic Laminaria digitata (Laminariales, Phaeophyceae) reveals a unique range-edge gene pool and suggests post-glacial colonization of the NW Atlantic. Eur. J. Phycol. 2020, 54, 1–12. [Google Scholar]
- King, N.G.; McKeown, N.J.; Smale, D.A.; Bradbury, S.; Stamp, T.; Jüterbock, A.; Egilsdóttir, H.; Groves, E.A.; Moore, P.J. Hierarchical genetic structuring in the cool boreal kelp, Laminaria digitata, implications for conservation and management. ICES J. Mar. Sci. 2020, 77, 1906–1913. [Google Scholar] [CrossRef]
- Evankow, A.; Christie, H.; Hancke, K.; Brysting, A.K.; Junge, C.; Fredriksen, S.; Thaulow, J. Genetic heterogeneity of two bioeconomically important kelp species along the Norwegian coast. Conserv. Genet. 2019, 20, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Schoenrock, K.M.; O’Connor, A.M.; Mauger, S.; Valero, M.; Neiva, J.; Serrão, E.Á.; Krueger–Hadfield, S.A. Genetic diversity of a marine foundation species, Laminaria hyperborea (Gunnerus) Foslie, along the coast of Ireland. Eur. J. Phycol. 2020, 55, 310–326. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Paulino, C.; Neiva, J.; Krause–Jensen, D.; Bruhn, A.; Serrao, E.A. Genetic diversity of Saccharina latissima (Phaeophyceae) along a salinity gradient in the North Sea–Baltic Sea transition zone. J. Phycol. 2016, 52, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Breton, T.S.; Nettleton, J.C.; O’Connell, B.; Bertocci, M. Fine–scale population genetic structure of sugar kelp, Saccharina latissima (Laminariales, Phaeophyceae), in eastern Maine, USA. Phycologia 2018, 57, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzinski, J.; Mauger, S.; Cock, J.M.; Valero, M. Characterization of newly developed expressed sequence tag–derived microsatellite markers revealed low genetic diversity within and low connectivity between European Saccharina latissima populations. J. Appl. Phycol. 2016, 28, 3057–3070. [Google Scholar] [CrossRef] [Green Version]
- Luttikhuizen, P.C.; van den Heuvel, F.H.M.; Rebours, C.; Witte, H.J.; van Bleijswijk, J.D.L.; Timmermans, K. Strong population structure but no equilibrium yet. Genetic connectivity and phylogeography in the kelp Saccharina latissima (Laminariales, Phaeophyta). Ecol. Evol. 2018, 8, 4265–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesner, D.; Fouqueau, L.; Valero, M.; Roleda, M.Y.; Pearson, G.A.; Bischof, K.; Valentin, K.; Bartsch, I. Heat stress responses and population genetics of the kelp Laminaria digitata (Phaeophyceae) across latitudes reveal differentiation among North Atlantic populations. Ecol. Evol. 2020, 10, 9144–9177. [Google Scholar] [CrossRef]
- Gurgel, C.F.D.; Camacho, O.; Minne, A.J.; Wernberg, T.; Coleman, M.A. Marine heatwave drives cryptic loss of genetic diversity in underwater forests. Curr. Biol. 2020, 30, 1199–1206. [Google Scholar] [CrossRef]
- Bolton, J.; Lüning, K. Optimal growth and maximal survival temperatures of Atlantic Laminaria species (Phaeophyta) in culture. Mar. Biol. 1982, 66, 89–94. [Google Scholar] [CrossRef]
- Gerard, V.A. Ecotypic differentiation in light–related traits of the kelp Laminaria saccharina. Mar Biol. 1988, 97, 25–36. [Google Scholar] [CrossRef]
- Lee, R.K.S. (Ed.) A Catalogue of the Marine Algae of the Canadian Arctic; National Museums of Canada: Ottawa, ON, Canada, 1980; Volume 9. [Google Scholar]
- Bekkby, T.; Moy, F.E. Developing spatial models of sugar kelp (Saccharina latissima) potential distribution under natural conditions and areas of its disappearance in Skagerrak. Estuar. Coast Shelf Sci. 2011, 95, 477–483. [Google Scholar] [CrossRef]
- Neiva, J.; Paulino, C.; Nielsen, M.M.; Krause-Jensen, D.; Saunders, G.W.; Assis, J.; Bárbara, I.; Tamigneaux, É.; Gouveia, L.; Aires, T. Glacial vicariance drives phylogeographic diversification in the amphi–boreal kelp Saccharina latissima. Sci. Rep. 2018, 8, 1112. [Google Scholar] [CrossRef] [PubMed]
- Gerard, V.A.; Du Bois, K. Temperature ecotypes near the southern boundary of the kelp Laminaria saccharina. Mar. Biol. 1988, 97, 575–580. [Google Scholar] [CrossRef]
- Muller, R.; Wiencke, C.; Bischof, K. Interactive effects of UV radiation and temperature on microstages of Laminariales (Phaeophyceae) from the Arctic and North Sea. Clim. Res. 2008, 37, 203–213. [Google Scholar] [CrossRef]
- Olischlager, M.; Iniguez, C.; Koch, K.; Wiencke, C.; Gordillo, F.J.L. Increased pCO(2) and temperature reveal ecotypic differences in growth and photosynthetic performance of temperate and Arctic populations of Saccharina latissima. Planta 2017, 245, 119–136. [Google Scholar] [CrossRef]
- Paulino, C.; Neiva, J.; Coelho, N.C.; Aires, T.; Marba, N.; Krause–Jensen, D.; Serrao, E.A. Characterization of 12 polymorphic microsatellite markers in the sugar kelp Saccharina latissima. J. Appl. Phycol. 2016, 28, 3071–3074. [Google Scholar] [CrossRef] [Green Version]
- Guzinski, J.; Ballenghien, M.; Daguin-Thiébaut, C.; Lévêque, L.; Viard, F. Population genomics of the introduced and cultivated Pacific kelp Undaria pinnatifida. Marinas—Not farms—Drive regional connectivity and establishment in natural rocky reefs. Evol. Appl. 2018, 11, 1582–1597. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks, an analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, T.; Lamothe, M.; Devloo-Delva, F.; Grewe, P. Radiator: RADseq Data Exploration, Manipulation and Visualization using R. R package version 1.1.8. 2020. Available online: https://thierrygosselin.github.io/radiator/ (accessed on 25 February 2020).
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [Green Version]
- Mauger, S.; Couceiro, L.; Valero, M. A simple and cost–effective method to synthesize an internal size standard amenable to use with a 5–dye system. Prime Res. Biotechnol. 2012, 2, 40–46. [Google Scholar]
- Goudet, J.; Jombart, T. Hierfstat, Estimation and Tests of Hierarchical F–Statistics. 2015; R package version 0.04. [Google Scholar]
- Adamack, A.T.; Gruber, B. PopGenReport, simplifying basic population genetic analyses in R. Methods Ecol. Evol. 2014, 5, 384–387. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007, a complete re–implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate, a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5, a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet, a R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Beugin, M.P.; Gayet, T.; Pontier, D.; Devillard, S.; Jombart, T. A fast likelihood solution to the genetic clustering problem. Methods Ecol. Evol. 2018, 9, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX 4.05, Logiciel Sous Windows TM Pour la Génétique des Populations. Laboratoire Génome, Populations, Interactions; CNRS UMR 5000; Université de Montpellier II: Montpellier, France, 2004. [Google Scholar]
- White, N.; Marshall, C.E. Saccharina latissima Sugar kelp. In Marine Life Information Network: Biology and Sensitivity Key Information Reviews; Tyler-Walters, H., Hiscock, K., Eds.; Marine Biological Association of the United Kingdom: Plymouth, UK, 2007. [Google Scholar]
- Guallar, C.; Flos, J. Linking phytoplankton primary production and chromophoric dissolved organic matter in the sea. Progr. Oceanogr. 2019, 176, 102116. [Google Scholar] [CrossRef]
- Beaumont, M.A.; Nichols, R.A. Evaluating loci for use in the genetic analysis of population structure. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1996, 263, 1619–1626. [Google Scholar]
- Günther, T.; Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef] [Green Version]
- De Mita, S.; Thuillet, A.C.; Gay, L.; Ahmadi, N.; Manel, S.; Ronfort, J.; Vigouroux, Y. Detecting selection along environmental gradients, analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol. Ecol. 2013, 22, 1383–1399. [Google Scholar] [CrossRef]
- Lotterhos, K.E.; Whitlock, M.C. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 2015, 24, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, P.; Du, X.; Crawford, D.L.; Oleksiak, M.F. Evolutionary toxicogenomics of the striped killifish (Fundulus majalis) in the New Bedford Harbor (Massachusetts, USA). Int. J. Mol. Sci. 2019, 20, 1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, Ø.; Harper, D.A.T.; Paul, D.R. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol Electron. 2001, 4, 9. [Google Scholar]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper, web–enabled heat mapping for all. Nucleic Acids Resour. 2016, 44, 147–153. [Google Scholar] [CrossRef]
- Andersen, G.S.; Pedersen, M.F.; Nielsen, S.L. Temperature acclimation and heat tolerance of photosynthesis in Norwegian Saccharina latissima (Laminariales, Phaeophyceae). J. Phycol. 2013, 49, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Sunde, J.; Yıldırım, Y.; Tibblin, P.; Forsman, A. Comparing the performance of microsatellites and radseq in population genetic studies, analysis of data for pike (Esox lucius) and a synthesis of previous studies. Front. Genet. 2020, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Lemopoulos, A.; Prokkola, J.M.; Uusi-Heikkilä, S.; Vasemägi, A.; Huusko, A.; Hyvärinen, P.; Koljonen, M.L.; Koskiniemi, J.; Vainikka, A. Comparing RADseq and microsatellites for estimating genetic diversity and relatedness—Implications for brown trout conservation. Ecol. Evol. 2019, 9, 2106–2120. [Google Scholar] [CrossRef]
- D’Aloia, C.C.; Andrés, J.A.; Bogdanowicz, S.M.; McCune, A.R.; Harrison, R.G.; Buston, P.M. Unraveling hierarchical genetic structure in a marine metapopulation, A comparison of three high-throughput genotyping approaches. Mol. Ecol. 2020, 29, 2189–2203. [Google Scholar] [CrossRef]
- Mao, X.; Augyte, S.; Huang, M.; Hare, M.P.; Bailey, D.; Umanzor, S.; Marty-Rivera, M.; Robbins, K.R.; Yarish, C.; Lindell, S. Population genetics of sugar kelp throughout the northeastern united states using genome-wide markers. Front. Mar. Sci. 2020, 7, 694. [Google Scholar] [CrossRef]
- Gagnaire, P.A.; Broquet, T.; Aurelle, D.; Viard, F.; Souissi, A.; Bonhomme, F.; Arnaud-Haond, S.; Bierne, N. Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evol. Appl. 2015, 8, 769–786. [Google Scholar] [CrossRef] [Green Version]
- Stevison, L.S.; McGaugh, S.E. It's time to stop sweeping recombination rate under the genome scan rug. Mol. Ecol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, K.A.; D’Aloia, C.C.; Crandall, E.; Iacchei, M.; Liggins, L.; Puritz, J.B.; von der Heyden, S.; Toonen, R.J. A decade of seascape genetics, contributions to basic and applied marine connectivity. Mar. Ecol. Progr. Ser. 2016, 554, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Liggins, L.; Treml, E.A.; Riginos, C. Seascape genomics, contextualizing adaptive and neutral genomic variation in the ocean environment. In Population Genomics: Marine Organisms; Oleksiak, M., Rajora, O., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Trubenová, B.; Krejca, M.S.; Lehre, P.K.; Kötzing, T. Surfing on the seascape, Adaptation in a changing environment. Evolution 2019, 73, 1356–1374. [Google Scholar] [CrossRef] [PubMed]
- Nepper-Davidsen, J.; Andersen, D.T.; Pedersen, M.F. Exposure to simulated heatwave scenarios causes long–term reductions in performance in Saccharina latissima. Mar. Ecol. Prog. Ser. 2019, 630, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Smith, M.D.; Fitzpatrick, L.; Schnell, D.J. In vivo analysis of the role of atTic20 in protein import in to chloroplasts. Plant Cell 2002, 14, 641–654. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kikuchi, S.; Oishi, M.; Oishi, M.; Nakai, M. In vivo studies on the roles of two closely related Arabidopsis Tic20 proteins, AtTic20–I and AtTic20–IV. Plant Cell Physiol. 2011, 52, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.L.; Chen, Z.H.; Li, Q.Y.; Zhang, J.; Liu, S.; Duan, D.L. High-density SNP-based QTL mapping and candidate gene screening for yield–related blade length and width in Saccharina japonica Laminariales, Phaeophyta). Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Mohanty, S.; Tripathy, B.C. Role of temperature stress on chloroplast biogenesis and protein import in pea. Plant Physiol. 2009, 150, 1050–1061. [Google Scholar] [CrossRef]
- Henkel, S.K.; Hofmann, G.E. Differing patterns of hsp70 gene expression in invasive and native kelp species: Evidence for acclimation-induced variation. J. Appl. Phycol. 2008, 20, 915–924. [Google Scholar] [CrossRef]
- Tomaneck, L. Variation in the heat shock response and its implication for predicting the effect of global climate change on species' biogeographical distribution ranges and metabolic costs. J. Exp. Biol. 2010, 213, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cui, C.; Li, Y.; Wu, H. A genome screen for the development of sex-specific DNA markers in Saccharina japonica. J. Appl. Phycol. 2018, 30, 1239–1246. [Google Scholar] [CrossRef]
- Lipinska, A.P.; Ahmed, S.; Peters, A.F.; Faugeron, S.; Cock, J.M.; Coelho, S.M. Development of pcr-based markers to determine the sex of kelps. PLoS ONE 2015, 10, e0140535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, S.M.; Rijstenbil, J.W.; Brown, M.T. Impacts of anthropogenic stresses on the early development stages of seaweeds. J. Aquat. Ecosyst. Stress Recovery 2000, 7, 317–333. [Google Scholar] [CrossRef]
- Fredersdorf, J.; Müller, R.; Becker, S.; Wiencke, C.; Bischof, K. Interactive effects of radiation, temperature and salinity on different life history stages of the Arctic kelp Alaria esculenta (Phaeophyceae). Oecologia 2009, 160, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Kim, J.H.; Park, S.K.; Yu, O.H.; Kim, Y.S.; Choi, H.G. Diverse responses of sporophytic photochemical efficiency and gametophytic growth for two edible kelps, Saccharina japonica and Undaria pinnatifida, to ocean acidification and warming. Mar. Poll. Bull. 2019, 142, 315–320. [Google Scholar] [CrossRef]
- Monteiro, C.M.M.; Heinrich, S.; Bartsch, I.; Valentin, K.U.; Corre, E.; Collén, J.; Harms, L.; Glöckner, G.; Bischof, K. Temperature modulates sex–biased gene expression in the gametophytes of the kelp Saccharina latissima. Front. Mar. Sci. 2019, 6, 769. [Google Scholar] [CrossRef] [Green Version]
- Booker, T.R.; Yeaman, S.; Whitlock, M.C. Variation in recombination rate affects detection of FST outliers under neutrality. bioRxiv 2020, 937813. [Google Scholar]
- Inoue, A.; Satoh, A.; Morishita, M.; Tokunaga, Y.; Miyakawa, T.; Tanokura, M.; Ojima, T. Functional heterologous expression and characterization of mannuronan C5–epimerase from the brown alga Saccharina japonica. Algal Res. 2016, 16, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Michel, G.; Tonon, T.; Scornet, D.; Cock, J.M.; Kloareg, B. Central and storage carbon metabolism of the brown alga Ectocarpus siliculosus, insights into the origin and evolution of storage carbohydrates in eukaryotes. New Phytol. 2010, 188, 67–81. [Google Scholar] [CrossRef]
- Deniaud-Bouёt, E.; Kervarec, N.; Michel, G.; Tonon, T.; Kloareg, B.; Hervé, C. Chemical and enzymatic fractionation of cell walls from Fucales, insights into the structure of the extracellular matrix of brown algae. Ann. Bot. 2014, 114, 1203–1216. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.; Zhang, P.; Lu, C.; Li, S.; Chen, Z.; Wang, X.; Duan, D. Transcriptome sequencing of Saccharina japonica sporophytes during whole developmental periods reveals regulatory networks underlying alginate and mannitol biosynthesis. BMC Genom. 2019, 20, 975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, J.D.; Corradino, G.; Ziervogel, K.; Schnetzer, A.; Osburn, C.L. Formation of chromophoric dissolved organic matter by bacterial degradation of phytoplankton–derived aggregates. Front. Mar. Sci. 2018, 4, 430. [Google Scholar] [CrossRef]
- Schiener, P.; Black, K.D.; Stanley, M.S.; Green, D.H. The seasonal variation in the chemical composition of the kelp species Laminaria digitata, Laminaria hyperborea, Saccharina latissima and Alaria esculenta. J. Appl. Phycol. 2015, 27, 363–373. [Google Scholar] [CrossRef]
- Küpper, F.C.; Carpenter, L.J.; McFiggans, G.B.; Palmer, C.J.; Waite, T.J.; Boneberg, E.-M.; Woitsch, S.; Weiller, M.; Abela, R.; Grolimund, D.; et al. Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 6954–6958. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.; Liu, T.; Yin, H.; Xu, X.; Zhu, W.; Wang, Y.; Wang, C.; Lv, H. The brown alga Saccharina japonica features distinct vanadium–dependent bromoperoxidases and iodoperoxidases. bioRxiv 2018, 243410. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wang, X.; Yao, J.; Fu, W.; Duan, D. Development of expressed sequence tag–derived microsatellite markers for Saccharina (Laminaria) japonica. J. Appl. Phycol. 2010, 22, 109–111. [Google Scholar] [CrossRef]
- Liu, F.; Wang, F.; Duan, D. EST–SSR markers derived from Laminaria digitata and its transferable application in Saccharina japonica. J. Appl. Phycol. 2012, 24, 501–505. [Google Scholar] [CrossRef]
- Heinrich, S.; Valentin, K.; Frickenhaus, S.; John, U.; Wiencke, C. Transcriptomic analysis of acclimation to temperature and light stress in Saccharina latissima (Phaeophyceae). PLoS ONE 2012, 7, e44342. [Google Scholar] [CrossRef] [Green Version]
- Strasburg, J.L.; Sherman, N.A.; Wright, K.M.; Moyle, L.C.; Willis, J.H.; Rieseberg, L.H. What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, D.; Smithson, A.; Krauss, S.L. Signatures of diversifying selection at EST–SSR loci and association with climate in natural Eucalyptus populations. Mol. Ecol. 2013, 2, 5112–5129. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, M.; Li, F.; Weng, Q.; Zhou, C.; Li, M.; Li, J.; Huang, H.; Mo, X.; Gan, S. Genome scans for divergent selection in natural populations of the widespread hardwood species Eucalyptus grandis (Myrtaceae) using microsatellites. Sci. Rep. 2016, 6, 34941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Sun, Y.; Huang, H.W.; Cannon, C.H. Footprints of divergent selection in natural populations of Castanopsis fargesii (Fagaceae). Heredity 2014, 113, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Zheng, X.; Chen, L.; Gao, H.; Yang, H.; Long, P.; Rong, J.; Lu, B.; Li, J.; Luo, L. Genetic Differentiation Revealed by Selective Loci of Drought–Responding EST–SSRs between Upland and Lowland Rice in China. PLoS ONE 2014, 9, e106352. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Araújo, R.; Rebours, C.; Jacquemin, B.; Holdt, S.L.; Charrier, B. Development and objectives of the PHYCOMORPH European Guidelines for the Sustainable Aquaculture of Seaweeds (PEGASUS). Bot. Mar. 2020, 63, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Valero, M.; Guillemin, M.L.; Destombe, C.; Jacquemin, B.; Gachon, C.M.; Badis, Y.; Buschmann, A.H.; Camus, C.; Faugeron, S. Perspectives on domestication research for sustainable seaweed aquaculture. Persp. Phycol. 2017, 4, 33–46. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Code | N SNP | HO (SE) SNP | HE (SE) SNP | FIS SNP | Ar (SE) SNP | Pr Allele % SNP | N SSR | HO (SE) SSR | HE (SE) SSR | FIS SSR | Ar (SE) SSR | Pr Allele % SSR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAS | 5 | 0.037 (0.002) | 0.034 (0.002) | −0.077 | 1.086 (0.004) | 8.3 | NA | NA | NA | NA | NA | NA |
| POR | 22 | 0.038 (0.002) | 0.036 (0.002) | −0.049 * | 1.098 (0.004) | 12.5 | 32 | 0.223 (0.069) | 0.228 (0.071) | 0.022 | 2.313 (0.412) | 16.2 |
| LOC | 24 | 0.047 (0.002) | 0.051 (0.002) | 0.074 | 1.156 (0.005) | 13.8 | 32 | 0.221 (0.075) | 0.231 (0.079) | 0.047 | 2.518 (0.535) | 2.7 |
| STG | 24 | 0.050 (0.002) | 0.054 (0.002) | 0.076 | 1.173 (0.005) | 10.3 | 32 | 0.316 (0.080) | 0.316 (0.080) | −0.001 | 3.218 (0.700) | 5.4 |
| LAN | 24 | 0.051 (0.002) | 0.052 (0.002) | 0.022 | 1.162 (0.005) | 5.2 | 32 | 0.297 (0.083) | 0.289 (0.081) | −0.026 | 3.230 (0.696) | 29.7 |
| LEZ | 24 | 0.046 (0.002) | 0.051 (0.002) | 0.095 * | 1.149 (0.005) | 5.1 | 32 | 0.305 (0.072) | 0.330 (0.077) | 0.077 | 3.128 (0.633) | 0 |
| FER | 10 | 0.044 (0.002) | 0.045 (0.002) | 0.019 | 1.131 (0.005) | 8.8 | 10 | 0.211 (0.066) | 0.216 (0.068) | 0.023 | 2.228 (0.442) | 2.7 |
| AUD | 24 | 0.027 (0.002) | 0.027 (0.002) | 0.004 * | 1.074 (0.004) | 2.6 | 32 | 0.250 (0.067) | 0.249 (0.066) | −0.004 | 2.328 (0.440) | 8.1 |
| HEL | 22 | 0.016 (0.001) | 0.014 (0.001) | −0.130 * | 1.036 (0.003) | 0.6 | 22 | 0.192 (0.053) | 0.181 (0.051) | −0.062 | 1.812 (0.247) | 8.1 |
| ELL | 7 | 0.055 (0.002) | 0.064 (0.002) | 0.145 | 1.177 (0.006) | 11.6 | 30 | 0.334 (0.068) | 0.382 (0.078) | 0.125 | 3.250 (0.626) | 8.1 |
| NYA | 13 | 0.056 (0.002) | 0.060 (0.002) | 0.059 * | 1.165 (0.005) | 21.1 | 26 | 0.340 (0.064) | 0.367 (0.066) | 0.074 | 2.775 (0.429) | 18.9 |
| Locus/Allele | Species Associated with Genbank Hits | Functional Trait/Genome Location | NCBI Accession |
|---|---|---|---|
| SNP1269 (SNP) | Saccharina japonica | tic20-like protein gene | KY411551.1 |
| KY411556.1 | |||
| KY411554.1 | |||
| SNP1558 (SNP) | Saccharina japonica | heat shock protein 70 (hsp70) gene | JF507714.1 |
| SNP1691 (SNP) | Saccharina japonica | c5epi gene for mannuronan C-5 epimerase | LC053765.1 |
| SNP1865 (SNP) | Saccharina japonica | female-specific marker Msj68/58/2 genomic sequence | MF850255.1 |
| SNP2361 (SNP) | Saccharina japonica | carbonic anhydrase (CA) gene | KY041784.1 |
| SNP3030 (SNP) | Chlamydotis macqueenii, Clupea harengus, Picoides pubescens | solute carrier family 2, facilitated glucose transporter member 1 | XM_009907977.1 |
| XM_012835870.2 | |||
| XM_010120537.1 | |||
| SNP3705 (SNP) | Saccharina japonica | heat shock protein 70 (hsp70) gene | JF507714.1 |
| SLN35 (Microsatellite) | Saccharina latissima | genomic microsatellite locus | KT723018 |
| Locus/Allele | Species Associated with Genbank Hits | Functional Trait/Genome Location | Environmental Correlations | NCBI Accession |
|---|---|---|---|---|
| SNP629 (SNP) | Saccharina japonica | phosphomannose isomerase 1 (PMI1) gene | Min-SST (+) (R2 = 0.76) | KF937207.1 |
| Average-Salinity (+) (R2 = 0.57) | ||||
| Max-Salinity (+) (R2 = 0.64) | ||||
| SNP1197 (SNP) | Saccharina japonica | phosphomannose isomerase 1 (PMI1) gene | Min-CDOM (−) (R2 = 0.66) | KF937207.1 |
| cvar-CDOM (+) (R2 = 0.38) | ||||
| SNP1269 (SNP) * | Saccharina japonica | tic20-like protein gene | Average-SST (+) (R2 = 0.77) | KY411551.1 |
| Min-SST (+) (R2 = 0.53) | ||||
| Max-SST (+) (R2 = 0.81) | ||||
| cvar-SST (−) (R2 = 0.70) | ||||
| Average-CDOM (+) (R2 = 0.64) | ||||
| Max-CDOM (+) (R2 = 0.71) | ||||
| SNP1558 (SNP) * | Saccharina japonica | heat shock protein 70 (hsp70) gene | Average-SST (+) (R2 = 0.77) | JF507714.1 |
| Min-SST (+) (R2 = 0.53) | ||||
| Max-SST (+) (R2 = 0.81) | ||||
| cvar-SST (−) (R2 = 0.70) | ||||
| Average-CDOM (+) (R2 = 0.64) | ||||
| Max-CDOM (+) (R2 = 0.71) | ||||
| SNP1865 (SNP) * | Saccharina japonica | female-specific marker Msj68/58/2 genomic sequence | Average-SST (+) (R2 = 0.61) | MF850255.1 |
| Max-SST (+) (R2 = 0.68) | ||||
| Average-CDOM (+) (R2 = 0.51) | ||||
| SNP3660 (SNP) | Saccharina japonica | c5epi gene for mannuronan C-5 epimerase | Average-SST (+) (R2 = 0.61) | LC053765.1 |
| Max-SST (+) (R2 = 0.68) | ||||
| SNP3894 (SNP) | Saccharina japonica | vanadium-dependent iodine peroxidase gene | Min-CDOM (−) (R2 = 0.47) | MG195955.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzinski, J.; Ruggeri, P.; Ballenghien, M.; Mauger, S.; Jacquemin, B.; Jollivet, C.; Coudret, J.; Jaugeon, L.; Destombe, C.; Valero, M. Seascape Genomics of the Sugar Kelp Saccharina latissima along the North Eastern Atlantic Latitudinal Gradient. Genes 2020, 11, 1503. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121503
Guzinski J, Ruggeri P, Ballenghien M, Mauger S, Jacquemin B, Jollivet C, Coudret J, Jaugeon L, Destombe C, Valero M. Seascape Genomics of the Sugar Kelp Saccharina latissima along the North Eastern Atlantic Latitudinal Gradient. Genes. 2020; 11(12):1503. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121503
Chicago/Turabian StyleGuzinski, Jaromir, Paolo Ruggeri, Marion Ballenghien, Stephane Mauger, Bertrand Jacquemin, Chloe Jollivet, Jerome Coudret, Lucie Jaugeon, Christophe Destombe, and Myriam Valero. 2020. "Seascape Genomics of the Sugar Kelp Saccharina latissima along the North Eastern Atlantic Latitudinal Gradient" Genes 11, no. 12: 1503. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121503