Bi-Allelic Pathogenic Variations in MERTK Including Deletions Are Associated with an Early Onset Progressive Form of Retinitis Pigmentosa

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Editorial Policies and Ethical Considerations
2.2. Clinical Evaluation
2.3. Molecular Genetic Analysis
3. Results
3.1. Clinical Characteristics
3.2. Genetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef] [Green Version]
- Nash, B.M.; Wright, D.C.; Grigg, J.R.; Bennetts, B.; Jamieson, R.V. Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Transl. Pediatr. 2015, 4, 139–163. [Google Scholar] [CrossRef]
- Bertelsen, M.; Jensen, H.; Bregnhoj, J.F.; Rosenberg, T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiol. 2014, 21, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Li, Y.; Thompson, D.A.; Weir, J.; Orth, U.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Mohand-Said, S.; Boulanger-Scemama, E.; Zanlonghi, X.; Condroyer, C.; Demontant, V.; Boyard, F.; Antonio, A.; Mejecase, C.; El Shamieh, S.; et al. MERTK mutation update in inherited retinal diseases. Hum. Mutat. 2018, 39, 887–913. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Yasumura, D.; Matthes, M.T.; LaVail, M.M.; Vollrath, D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J. Biol. Chem. 2002, 277, 17016–17022. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.O.; Prieto, A.L.; Obin, M.S.; Abrams, T.A.; Burgess, B.L.; Heeb, M.J.; Agnew, B.J. Outer segment phagocytosis by cultured retinal pigment epithelial cells requires Gas6. Exp. Eye Res. 2001, 73, 509–520. [Google Scholar] [CrossRef]
- Law, A.L.; Parinot, C.; Chatagnon, J.; Gravez, B.; Sahel, J.A.; Bhattacharya, S.S.; Nandrot, E.F. Cleavage of Mer tyrosine kinase (MerTK) from the cell surface contributes to the regulation of retinal phagocytosis. J. Biol. Chem. 2015, 290, 4941–4952. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Cooper, D.N. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinform. 2012, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, E.; Duno, M.; Batbayli, M.; Vilhelmsen, K.; Rosenberg, T. A novel MERTK deletion is a common founder mutation in the Faroe Islands and is responsible for a high proportion of retinitis pigmentosa cases. Mol. Vis. 2011, 17, 1485–1492. [Google Scholar] [PubMed]
- Zampaglione, E.; Kinde, B.; Place, E.M.; Navarro-Gomez, D.; Maher, M.; Jamshidi, F.; Nassiri, S.; Mazzone, J.A.; Finn, C.; Schlegel, D.; et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 2020, 22, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schil, K.; Karlstetter, M.; Aslanidis, A.; Dannhausen, K.; Azam, M.; Qamar, R.; Leroy, B.P.; Depasse, F.; Langmann, T.; De Baere, E. Autosomal recessive retinitis pigmentosa with homozygous rhodopsin mutation E150K and non-coding cis-regulatory variants in CRX-binding regions of SAMD7. Sci. Rep. 2016, 6, 21307. [Google Scholar] [CrossRef] [Green Version]
- Bujakowska, K.M.; Fernandez-Godino, R.; Place, E.; Consugar, M.; Navarro-Gomez, D.; White, J.; Bedoukian, E.C.; Zhu, X.; Xie, H.M.; Gai, X.; et al. Copy-number variation is an important contributor to the genetic causality of inherited retinal degenerations. Genet. Med. 2017, 19, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1219. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ellard Sian, B.E.L.; Ian, B.; Natalie, F.; Clare, T.; Martina, O.; Diana, M.E.; Stephen, A.; Richard, S.; Deans Zandra, C.D.; Tracy, L.; et al. ACGS Best Practice Guidelines for Variant Classification 2019. Available online: https://www.acgs.uk.com/quality/best-practice-guidelines/#VariantGuidelines (accessed on 4 December 2020).
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Q. Statistical features of human exons and their flanking regions. Hum. Mol. Genet. 1998, 7, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, V.W.; Feng, Y.; Tian, X.; Li, F.Y.; Truong, C.; Wang, G.; Chiang, P.W.; Lewis, R.A.; Wong, L.J. Dependable and efficient clinical utility of target capture-based deep sequencing in molecular diagnosis of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6213–6223. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Campbell, C.; Barton, S.; Bhaskar, S.; Gupta, S.; Taylor, R.L.; Sergouniotis, P.I.; Horn, B.; Lamb, J.A.; Michaelides, M.; et al. Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur. J. Hum. Genet. 2017, 25, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7369–7375. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Horn, B.; Campbell, C.; Arno, G.; Barton, S.; Tate, C.; Bhaskar, S.; Sergouniotis, P.I.; Taylor, R.L.; Carss, K.J.; et al. Assessment of the incorporation of CNV surveillance into gene panel next-generation sequencing testing for inherited retinal diseases. J. Med. Genet. 2018, 55, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| ID | Sex (M/F) | Clinical Diagnosis | Decade First Symptoms | Clinical History | First OPH Exam, Year (Age) | Latest OPH Exam, Year (Age) | BCVA OD/OS First Visit (Age) | BCVA OD/OS Last Visit (Age) | Refractive Error OD/OS First Visit | Refractive Error OD/OS Last Visit | VF First Exam (Age) | VF Latest Exam (Age) | (Phph) | Colour Vision (Age) | Slit Lamp (Age) | Dark Adoptometry (Age) | ERG (Age) | Latest Fundoscopy (Age) | Latest OCT (Age) | Other Symptoms/Signs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
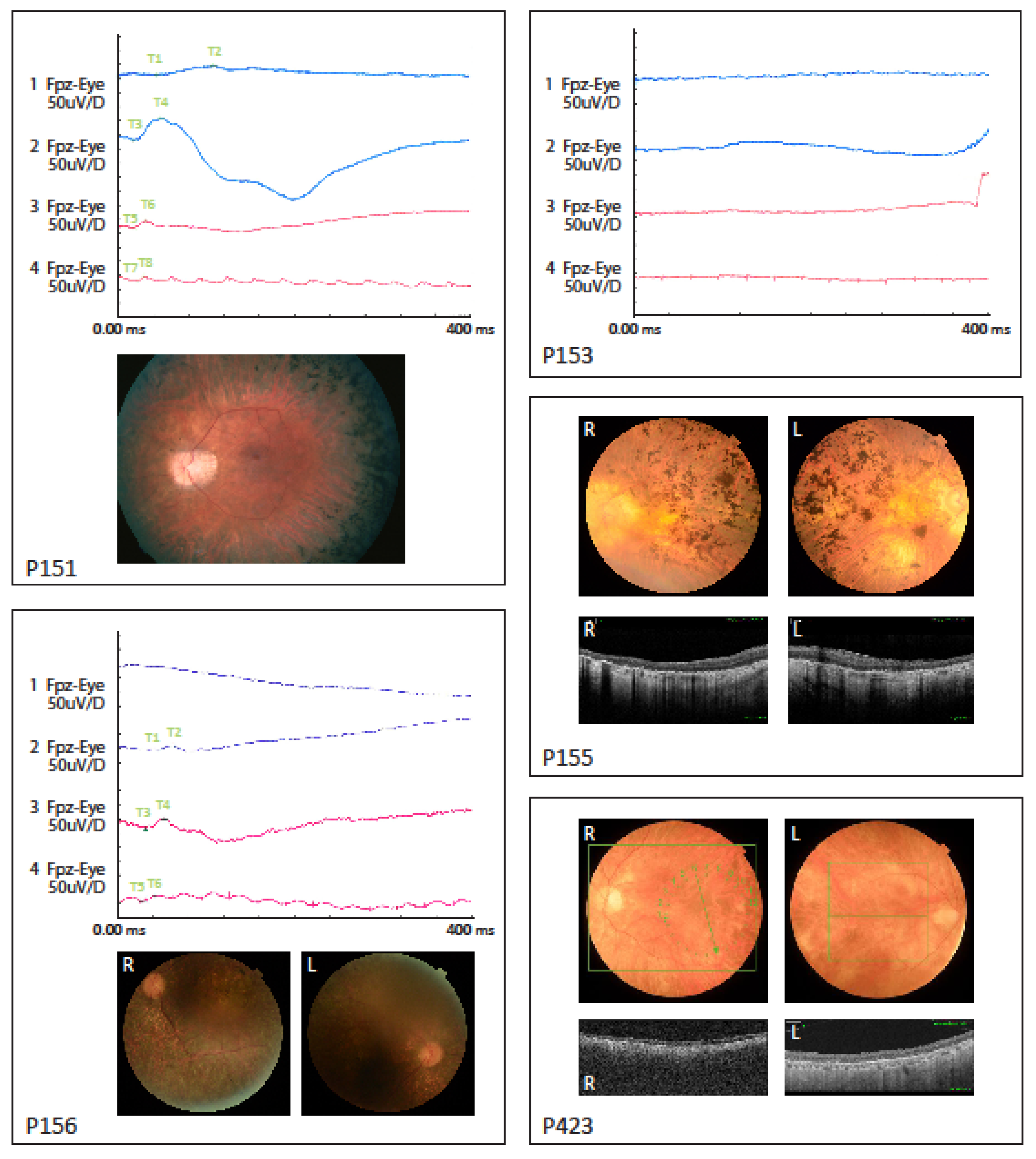
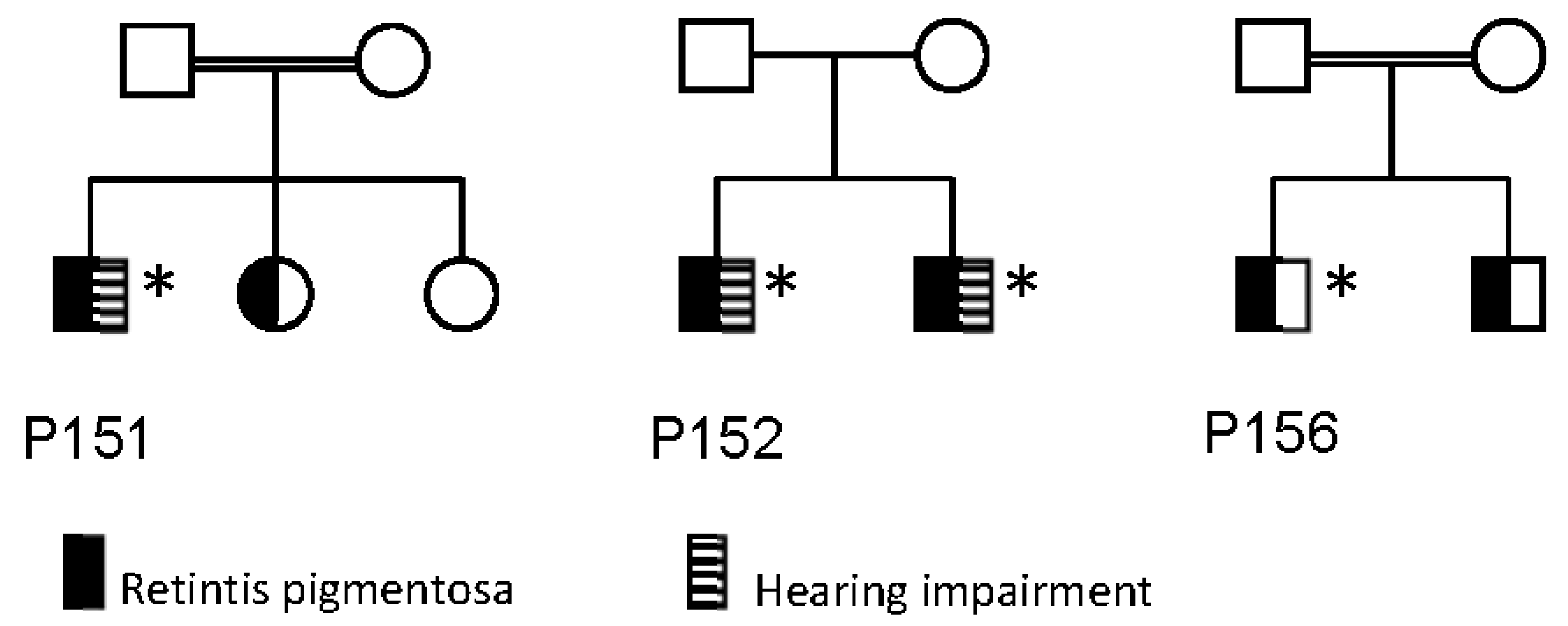
| 151 | M | RP | 1st | At age 7 poor night vision and peripheral visual field loss. Slow progression | 1964 (9) | 1991 (36) | 0.67/0.80 (9) | 0.6/0.6 (36) | −4.5−1.00 × 0/−4.5−1.00 × 0 | −8.5−2.00 × 170/−7.75−1.75 × 170 | Normal (ad modum Donders) (11) | Both eyes constricted to 20° (36) | 1 | ND | Normal (36) | Normal (36) | Reduced responses, rod-cone dysfunction (36) | Normal optic nerve, peripapillar atrophy, normal macula, attenuated retinal vessels, bone spicule hyperpigmentation in the periphery (36) | N/A | Mild hearing impairment |
| 152 | M | RP | 1st | First symptoms were night blindness and during early childhood progressive constrictive visual field and colour vision defects. Visual acuity 1/60 both eyes and a visual field of only a few degrees at the age of 24 years | 1971 (17) | 2016 (62) | 0.40/0.40 (17) | no LP (62) | −1.50−1.50 × 80 | −1.50−0.50 × 90 | Both eyes contricted to 5° (17) | No visual field (62) | 2 | Ishihara only errors both eyes (17) | Right eye: aphacic (phaco, 2016). Left eye: moderate nuclear and subcapsular cataract (62) | N/A | Undetectable (30) | Pale atrophic optic disc, attenuated vessels, severe generalized retinal degeneration with numerous hyperpigmentation both central and in the periphery (62) | Severe retinal degeneration and abnormal structure (62) | Hearing impairment, chronic lymphatic leukemia and hypertension |
| 153 | M | RP | 1st | First symptoms of night blindness and photophobia at age 7. Thereafter slow progression of visual acuity and visual field loss | 1984 (21) | 2003 (40) | 0.33/0.33 (21) | 0.10/0.10 (40) | +2.00−0.50 × 100 | +1.00−0.50 × 80 | Slightly constricted (ad modum Donders) (21) | Both eyes constricted to 5° (40) | 1 | N/A | Normal (40) | Delayed and final threshold elevated approximately 2 log units above normal level (30) | Undetectable (30) | Pale atrophic optic disc with peripapillary drusen. Attenuated vessels, severe generalized retinal degeneration with peripheral bone spicule hyperpigmentation (38) | N/A | No |
| 155 | M | RP | 1st | Since childhood decreased central vision and night vision. Slow progression of visual field loss | 1973 (17) | 2018 (61) | 0.10/0.10 (17) | LP/LP (61) | −5.00/−4.00 | +0.25−1.50 × 170/−0.25-1.25 × 172 | Both eyes: constricted to 1–2°, preserved temporal island (17) | Cannot cooperate to visual field test (61) | 1 | ND | Cataract operation in 2016, otherwise normal (61) | ND | ND | Waxy optic disc pallor, attenuated retinal vessels, pigment deposits in the macula, bone spicule hyperpigmentation in periphery (61) | Right eye: macular atrophy. Left eye: epiretinal membrane, macular atrophy and edema (61) | Hearing impairment since 2015 |
| 156 | M | RP | 2nd | Since early teens decreased central vision and poor night vision. Slow progression of visual field loss | 1993 (22) | 2017 (46) | 0.50/0.25 (22) | 0.04/0.03 (46) | −0.25−1.75 × 10/−0.25−2.00 × 10 | +1.00−1.50 × 120/+0.50−0.75 × 175 | Both eyes: constricted to 20° (22) | Both eyes constricted to 5° (46) | 1 | Affected (22) | Incipient cataract (46) | Monophasic curve with loss of rod mediated dark adaptation (22) | Reduced responses, rod-cone dysfunction (22) | Pale optic nerve, attenuated retinal vessels, macular atrophy, peripheral bone spicule hyperpigmentation (22) | No | |
| 423 | F | RP | 6th | Late onset with night blindness at the age of 53 years. Slow progression of visual field loss thereafter | 2005 (53) | 2015 (62) | 0.6/0.8 (53) | 0.30/0.50 (62) | −1.00−1.25 × 155/−0.5−0.5 × 0 | −0.50−0.50 × 0 (before cataract operation) | Normal (53) | Both eyes constricted to 5° and preserved inferior islands of 10 × 30°(62) | 1–2 | Slight dyschromatopsia (53) | Normal (53) | Monophasic curve with loss of rod mediated dark adaptation (53) | Rod response undetectable and cone response reduced and with prolonged implicit time (53) | Optic disc pallor and slightly attenuated vessels. Generalized light retinal coloring. No hyperpigmentation (53) | N/A | Thrombocytopenic purpura, rheumatoid arthritis and mixed connected tissue disease |
| ID | cDNA1 | Predicted Protein Change | AF in gnomAD | CADD Score | SSF/MES/NNS/GS | Class | ClinVar ID | Previously Reported |
|---|---|---|---|---|---|---|---|---|
| P151 | c.345C>G | p.(Cys115Trp) | 12/281826/0 | 22.9 | NA | VUS (PM2, PS4(mod), PP3) | RCV000787624.1 | [28,29] |
| c.345C>G | p.(Cys115Trp) | 12/281826/0 | 22.9 | NA | VUS (PM2, PS4(mod), PP3) | RCV000787624.1 | [28,29] | |
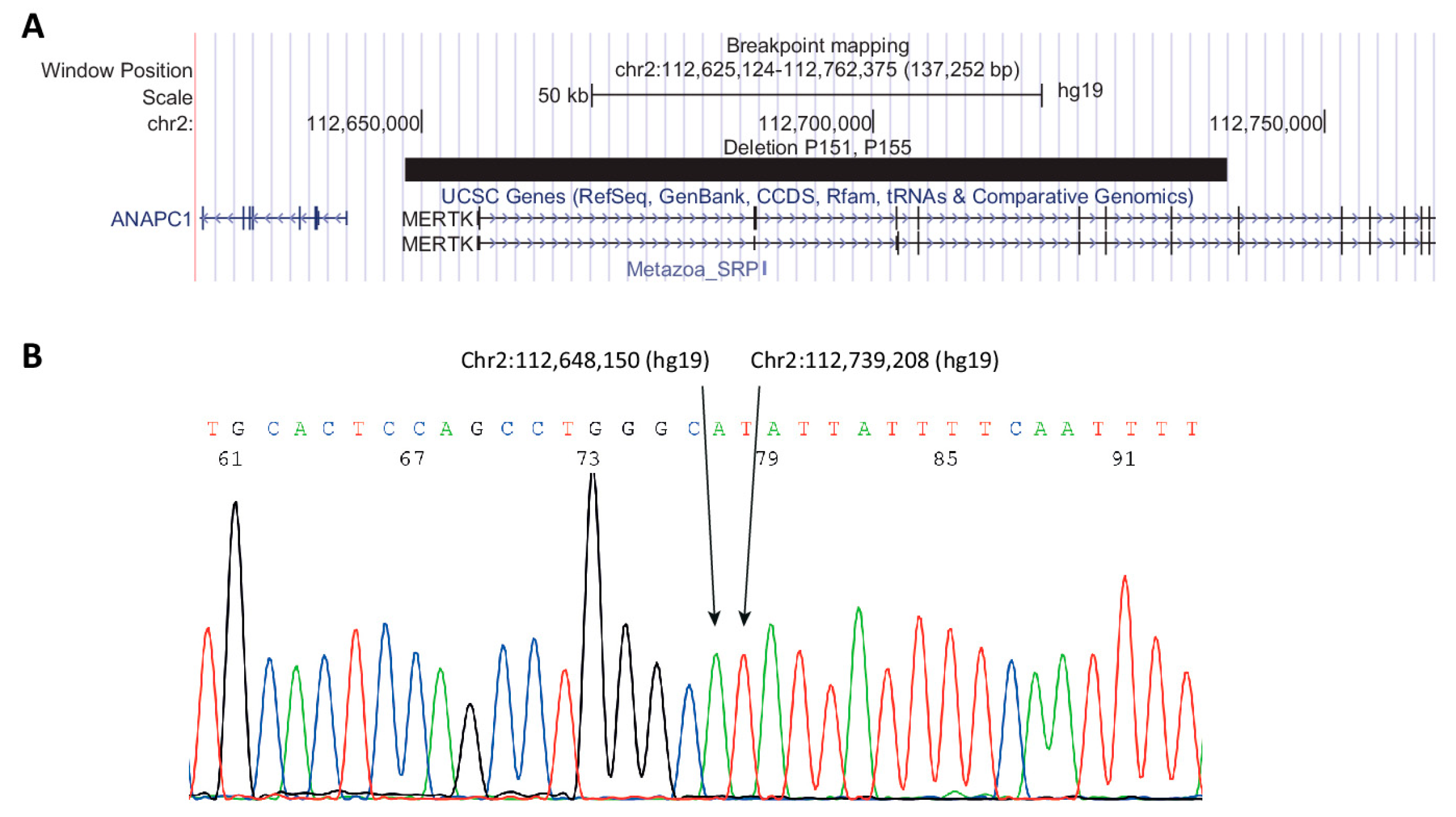
| P152 (+brother) | chr2:112,648,150-112,739,208del (hg19) | NA | 1/21694/0 | NA | NA | Pathogenic (PVS1, PM2, PP1) | VCV000636043.1 | [12,30] |
| chr2:112,648,150-112,739,208del (hg19) | NA | 1/21694/0 | NA | NA | Pathogenic (PVS1, PM2, PP1) | VCV000636043.1 | [12,30] | |
| P153 | c.960+1G>A | p.? | NP | NA | -/-/-/- | Likely pathogenic (PVS1, PM2) | RCV000787626.1 | [16] |
| c.960+1G>A | p.? | NP | NA | -/-/-/- | Likely pathogenic (PVS1, PM2) | RCV000787626.1 | [16] | |
| P155 | c.757+1G>A | p.? | NP | NA | -/-/-/- | Likely pathogenic (PVS1, PM2) | RCV000787625.1 | [16] |
| chr2:112,648,150-112,739,208del (hg19) | NA | 1/21694/0 | NA | -/-/-/- | Pathogenic (PVS1, PM2, PP1) | VCV000636043.1 | [12,30] | |
| P156 | c.1450G>A | p.(Gly484Ser), splice variant? | 3/251408/0 | 25.3 | NA | VUS (PM2, PS4(sup)) | RCV000132663.2 | [28,31] |
| c.1450G>A | p.(Gly484Ser), splice variant? | 3/251408/0 | 25.3 | NA | VUS (PM2, PS4(sup)) | RCV000132663.2 | [28,31] | |
| P423 | c.2060G>T;2305A>G | p.(Arg687Leu;Ile769Val) | NP; 92/282478/0 | 32/19.01 | NA | VUS (PM2, PP3) | RCV000787849.1/RCV787914.1 | Novel |
| c.2060G>T;2305A>G | p.(Arg687Leu;Ile769Val) | NP; 87/276840/0 | 32/19.01 | NA | VUS (PM2, PP3) | RCV000787849.1/RCV787914.1 | Novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jespersgaard, C.; Bertelsen, M.; Arif, F.; Gellert-Kristensen, H.G.; Fang, M.; Jensen, H.; Rosenberg, T.; Tümer, Z.; Møller, L.B.; Brøndum-Nielsen, K.; et al. Bi-Allelic Pathogenic Variations in MERTK Including Deletions Are Associated with an Early Onset Progressive Form of Retinitis Pigmentosa. Genes 2020, 11, 1517. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121517
Jespersgaard C, Bertelsen M, Arif F, Gellert-Kristensen HG, Fang M, Jensen H, Rosenberg T, Tümer Z, Møller LB, Brøndum-Nielsen K, et al. Bi-Allelic Pathogenic Variations in MERTK Including Deletions Are Associated with an Early Onset Progressive Form of Retinitis Pigmentosa. Genes. 2020; 11(12):1517. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121517
Chicago/Turabian StyleJespersgaard, Cathrine, Mette Bertelsen, Farah Arif, Helene Gry Gellert-Kristensen, Mingyan Fang, Hanne Jensen, Thomas Rosenberg, Zeynep Tümer, Lisbeth Birk Møller, Karen Brøndum-Nielsen, and et al. 2020. "Bi-Allelic Pathogenic Variations in MERTK Including Deletions Are Associated with an Early Onset Progressive Form of Retinitis Pigmentosa" Genes 11, no. 12: 1517. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11121517