
Genotyping of the Major SARS-CoV-2 Clade by Short-Amplicon High-Resolution Melting (SA-HRM) Analysis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Samples
2.2. cDNA Synthesis
2.3. Small-Amplicon High-Resolution Melting Analysis (SA-HRM)
2.4. Amplification-Refractory Mutation System (ARMS)
2.5. Sanger Sequencing
2.6. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Petrone, M.E.; Holmes, E.C. We shouldn’t worry when a virus mutates during disease outbreaks. Nat. Microbiol. 2020, 5, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495. [Google Scholar] [CrossRef]
- Arena, F.; Pollini, S.; Rossolini, G.M.; Margaglione, M. Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic. Int. J. Mol. Sci. 2021, 22, 1298. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; VanInsberghe, D.; Koelle, K. Insights from SARS-CoV-2 sequences. Science 2021, 371, 466–467. [Google Scholar] [CrossRef]
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- WHO. Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health, 8 January 2021; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Van Tan, L.; Man, D.N.H.; Hang, V.T.T.; Khanh, P.N.Q.; Xuan, T.C.; Phong, N.T.; Tu, T.N.H.; Hien, T.T.; Hung, L.M.; Truong, N.T. SARS-CoV-2 and co-infections detection in nasopharyngeal throat swabs of COVID-19 patients by metagenomics. J. Infect. 2020, 81, e175–e177. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Destras, G.; Gaymard, A.; Bouscambert-Duchamp, M.; Valette, M.; Escuret, V.; Frobert, E.; Billaud, G.; Trouillet-Assant, S.; Cheynet, V.; et al. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020, 26, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Sun, C.; Zheng, H.; Wang, L.; Zhang, H.; Zou, L.; Liu, Z.; Du, P.; Xu, X.; Liang, L.; et al. High-coverage SARS-CoV-2 genome sequences acquired by target capture sequencing. J. Med. Virol. 2020, 92, 2221–2226. [Google Scholar] [CrossRef]
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS ONE 2021, 16, e0247115. [Google Scholar] [CrossRef]
- Harilal, D.; Ramaswamy, S.; Loney, T.; Suwaidi, H.A.; Khansaheb, H.; Alkhaja, A.; Varghese, R.; Deesi, Z.; Nowotny, N.; Alsheikh-Ali, A.; et al. SARS-CoV-2 Whole Genome Amplification and Sequencing for Effective Population-Based Surveillance and Control of Viral Transmission. Clin. Chem. 2020, 66, 1450–1458. [Google Scholar] [CrossRef]
- Helmy, M.; Awad, M.; Mosa, K.A. Limited resources of genome sequencing in developing countries: Challenges and solutions. Appl. Transl. Genom. 2016, 9, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Liew, M.; Pryor, R.; Palais, R.; Meadows, C.; Erali, M.; Lyon, E.; Wittwer, C. Genotyping of Single-Nucleotide Polymorphisms by High-Resolution Melting of Small Amplicons. Clin. Chem. 2004, 50, 1156–1164. [Google Scholar] [CrossRef]
- López-Martínez, B.; Guzmán-Ortiz, A.L.; Nevárez-Ramírez, A.J.; Parra-Ortega, I.; Olivar-López, V.B.; Ángeles-Floriano, T.; Vilchis-Ordoñez, A.; Quezada, H. Saliva as a promising biofluid for SARS-CoV-2 detection during the early stages of infection. Bol. Med. Hosp. Infant. Mex. 2020, 77, 228–233. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Little, S. Amplification-Refractory Mutation System (ARMS) Analysis of Point Mutations. Curr. Protoc. Hum. Genet. 1995, 7, 9.8.1–9.8.12. [Google Scholar] [CrossRef] [PubMed]
- Walensky, R.P.; Walke, H.T.; Fauci, A.S. SARS-CoV-2 Variants of Concern in the United States—Challenges and Opportunities. JAMA 2021, 325, 1037–1038. [Google Scholar] [CrossRef] [PubMed]
- Toovey, O.T.R.; Harvey, K.N.; Bird, P.W.; Tang, J.W.-T.W.-T. Introduction of Brazilian SARS-CoV-2 484K.V2 related variants into the UK. J. Infect. 2021, S0163-4453(21)00047–5. [Google Scholar] [CrossRef] [PubMed]
- Wilfinger, W.W.; Mackey, K.; Chomczynski, P. Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. Biotechniques 1997, 22, 474–481. [Google Scholar] [CrossRef] [PubMed]
- El Bali, L.; Diman, A.; Bernard, A.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Comparative study of seven commercial kits for human DNA extraction from urine samples suitable for DNA biomarker-based public health studies. J. Biomol. Tech. JBT 2014, 25, 96. [Google Scholar]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3’→5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
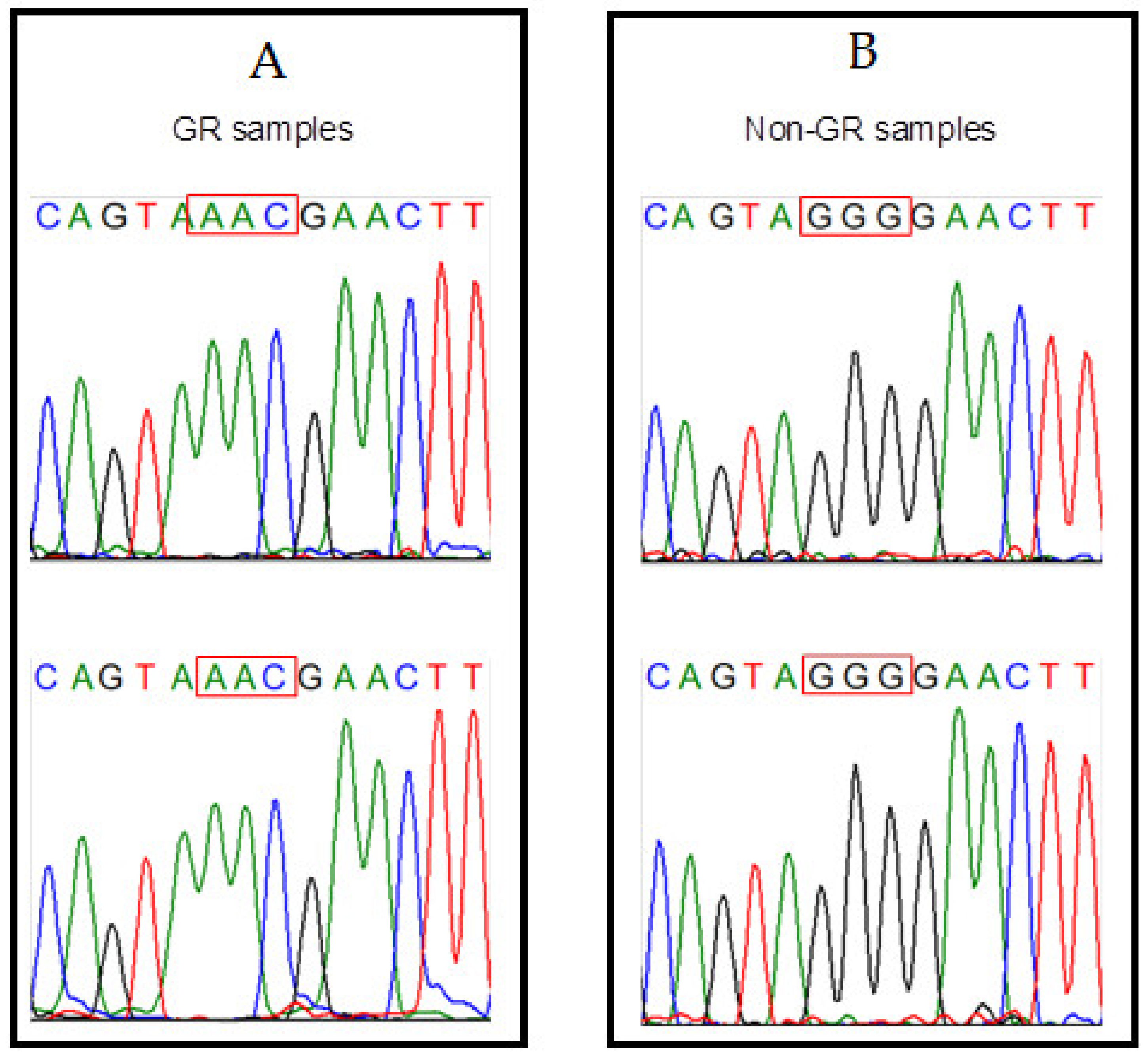{kind=link}
| Assay | Primer | Sequence (5′ → 3′) Position at SARS-CoV-2 Isolate Wuhan-Hu-1 * Position at SARS-CoV-2 N gene: NC_045512.2 ** | Annealing Temperature (°C) | Cycles | Product Size (bp) |
|---|---|---|---|---|---|
| SA-HRM | GR-SA-HRM-F | CAAGAAATTCAACTCCAGGCAG (nt28854 → 28875) * (nt581 → 602) ** | 61 | 29 | 54 |
| GR-SA-HRM-R | CAGCCATTCTAGCAGGAGAAGTT (nt28907 → 28885) * (nt634 → 612) ** | ||||
| ARMS | GR-ARMS-F | ACTCCAGGCAGCAGTAAAC (nt28865 → nt28883) * (nt592 → 610) ** | 55 | 40 | 649 |
| GR-ARMS-R | CACTGCTCATGGATTGTTG (nt29513 → 29495) * (nt1240 → 1222) ** | ||||
| non-GR-ARMS-F | CTACCTAGGAACTGGGCCAG (nt28606 → 28625) * (nt333 → 352) ** | 55 | 40 | 295 | |
| non-GR-ARMS-R | TCTAGCAGGAGAAGTTCCCC (nt28900 → 28881) * (nt627 → 608) ** | ||||
| Sanger | GR-Sanger-F | CAAAGACGGCATCATATGG (nt28651 → 28669) * (nt378 → 396) ** | 57 | 40 | 304 |
| GR-Sanger-R | CAATCTGTCAAGCAGCAGC (nt28954 → 28936) * (nt681 → 663) ** |
| Feature | Mean ± SD (n = 14) |
|---|---|
| Concentration (ng/μL) | 105.64 ± 23.34 |
| Purity (260/280) | 3.21 ± 0.31 |
| qPCR Ct E | 20.74 ± 6.92 |
| qPCR Ct RdRp | 20.07 ± 4.39 |
| qPCR Ct N | 21.10 ± 5.87 |
| Sample | Assay | |
|---|---|---|
| SA-HRM | ARMS | |
| SPL-1 | GR | GR |
| SPL-2 | GR | GR |
| SPL-3 | Non-GR | Non-GR |
| SPL-4 | Non-GR | - |
| SPL-5 | GR | GR |
| SPL-6 | Non-GR | Non-GR |
| SPL-7 | GR | GR |
| SPL-8 | GR | GR |
| SPL-9 | Non-GR | Non-GR |
| SPL-10 | GR | GR |
| SPL-11 | GR | GR |
| SPL-12 | GR | GR |
| SPL-13 | GR | - |
| SPL-14 | Non-GR | - |
| r (Relative Amplification) (n = 14) | Confidence Intervals (95%) | p-Value | ||
|---|---|---|---|---|
| Lower | Upper | |||
| Concentration (ng/μL) | 0.84 | 0.3777099 | 0.9450479 | 0.00331 |
| Purity (260/280) | −0.828 | −0.9449925 | −0.3772663 | 0.003323 |
| PCR Ct E | −0.586 | −0.7877534 | 0.3098872 | 0.2823 |
| PCR Ct RdRp | −0.351 | −0.8033255 | 0.3572016 | 0.3193 |
| PCR Ct N | −0.504 | −0.7711625 | 0.3476024 | 0.3394 |
| GR Samples | Non-GR Samples | p-Value | |
|---|---|---|---|
| Mean ± SD (n = 9) | Mean ± SD (n = 5) | ||
| Concentration (ng/μL) | 106.38 ± 25.67 | 104.29 ± 21.19 | 1 |
| Purity (260/280) | 3.16 ± 0.31 | 3.28 ± 0.34 | 0.5185 |
| PCR Ct E | 17.15 ± 2.25 | 27.22 ± 7.99 | 0.006993 |
| PCR Ct RdRp | 18.12 ± 2.03 | 24.46 ± 5.38 | 0.01119 |
| PCR Ct N | 18.05 ± 1.78 | 26.59 ± 6.87 | 0.006993 |
| Relative amplification | 1.60 ± 1.71 | 2.29 ± 3.19 | 0.9273 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Garcia, H.; Guzmán-Ortiz, A.L.; Angeles-Floriano, T.; Parra-Ortega, I.; López-Martínez, B.; Martínez-Saucedo, M.; Aquino-Jarquin, G.; Sánchez-Urbina, R.; Quezada, H.; Granados-Riveron, J.T. Genotyping of the Major SARS-CoV-2 Clade by Short-Amplicon High-Resolution Melting (SA-HRM) Analysis. Genes 2021, 12, 531. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040531
Diaz-Garcia H, Guzmán-Ortiz AL, Angeles-Floriano T, Parra-Ortega I, López-Martínez B, Martínez-Saucedo M, Aquino-Jarquin G, Sánchez-Urbina R, Quezada H, Granados-Riveron JT. Genotyping of the Major SARS-CoV-2 Clade by Short-Amplicon High-Resolution Melting (SA-HRM) Analysis. Genes. 2021; 12(4):531. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040531
Chicago/Turabian StyleDiaz-Garcia, Hector, Ana L. Guzmán-Ortiz, Tania Angeles-Floriano, Israel Parra-Ortega, Briceida López-Martínez, Mirna Martínez-Saucedo, Guillermo Aquino-Jarquin, Rocío Sánchez-Urbina, Hector Quezada, and Javier T. Granados-Riveron. 2021. "Genotyping of the Major SARS-CoV-2 Clade by Short-Amplicon High-Resolution Melting (SA-HRM) Analysis" Genes 12, no. 4: 531. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040531