
Computational and Transcriptomic Analysis Unraveled OsMATE34 as a Putative Anthocyanin Transporter in Black Rice (Oryza sativa L.) Caryopsis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Anthocyanin Measurement
2.3. In Silico Analysis of OsMATE in Rice
2.3.1. Identification of Putative MATE Genes in Rice and other Plant and Their Sequence-Structure Analysis
2.3.2. Phylogenetic Analysis and Gene Structure
2.4. Transcriptome Analysis
2.4.1. RNA Isolation, Library Preparation, and Illumina Hiseq Xten/Nova seq 6000 Sequencing for Transcriptome Analysis
2.4.2. Data Filtering and Mapping of Reads
2.4.3. Differential Expression Functional Enrichment Analysis
2.5. Expression Analysis of Putative Anthocyanin OsMATE Genes by Reverse Transcription Quantitative PCR (RT-qPCR)
3. Results
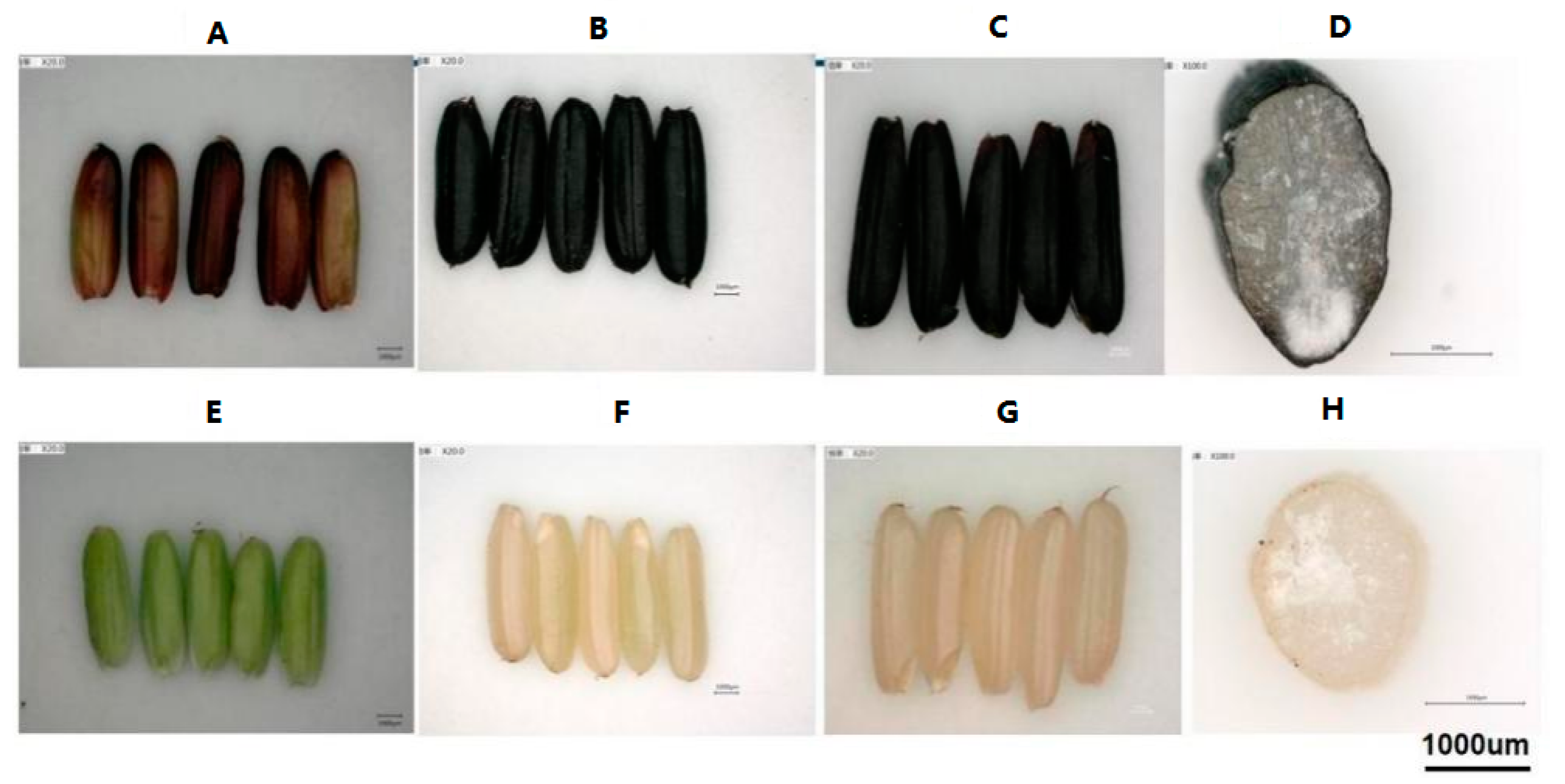
3.1. Anthocyanin Quantification at Different Developmental Stages
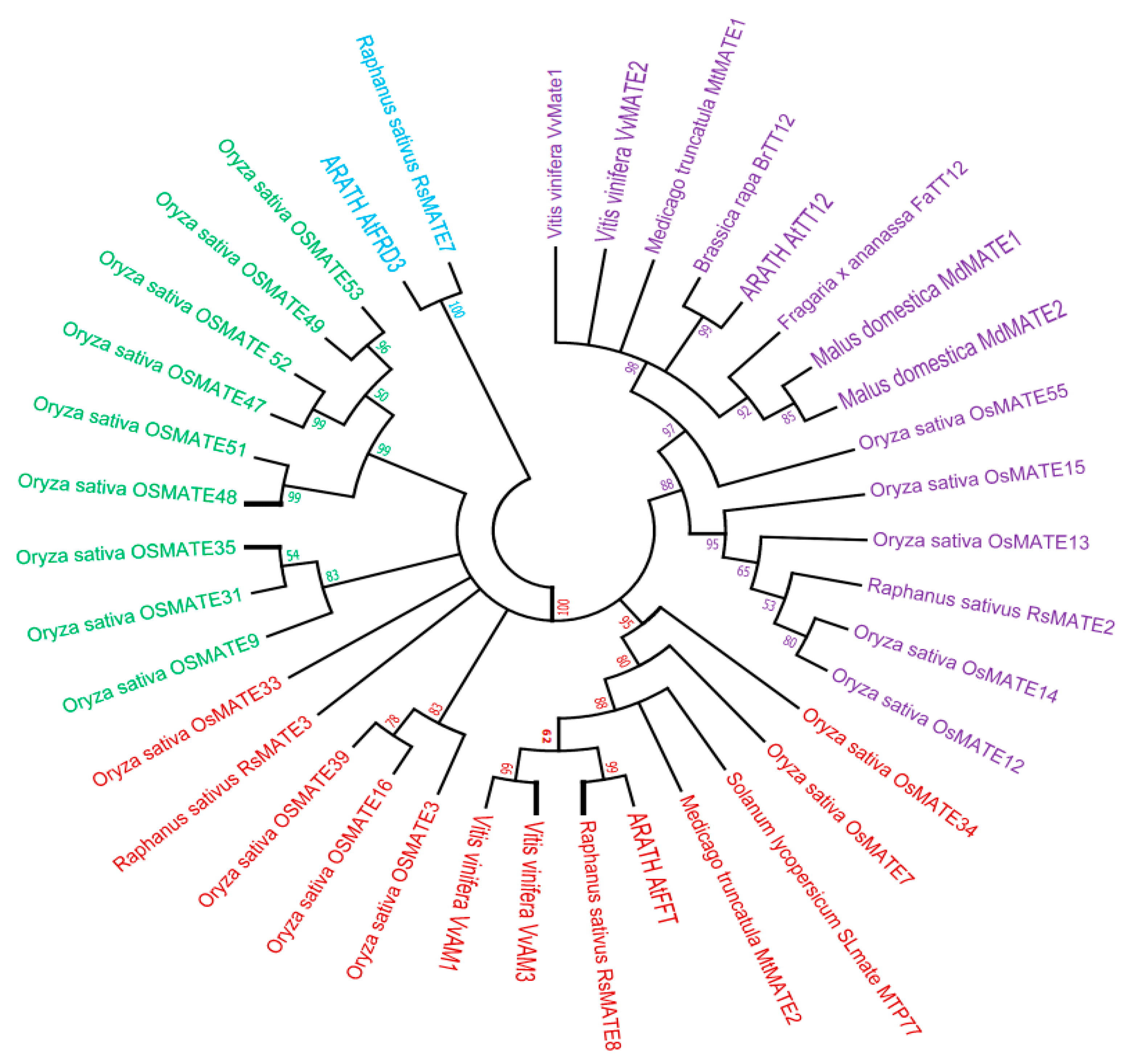
3.2. Phylogenetic Tree Analysis of MATE Proteins and Deduction of Putative OsMATE Gene for Anthocyanin Transportation
3.3. Molecular Characteristics and Structure of Putative Anthocyanin MATE Protein
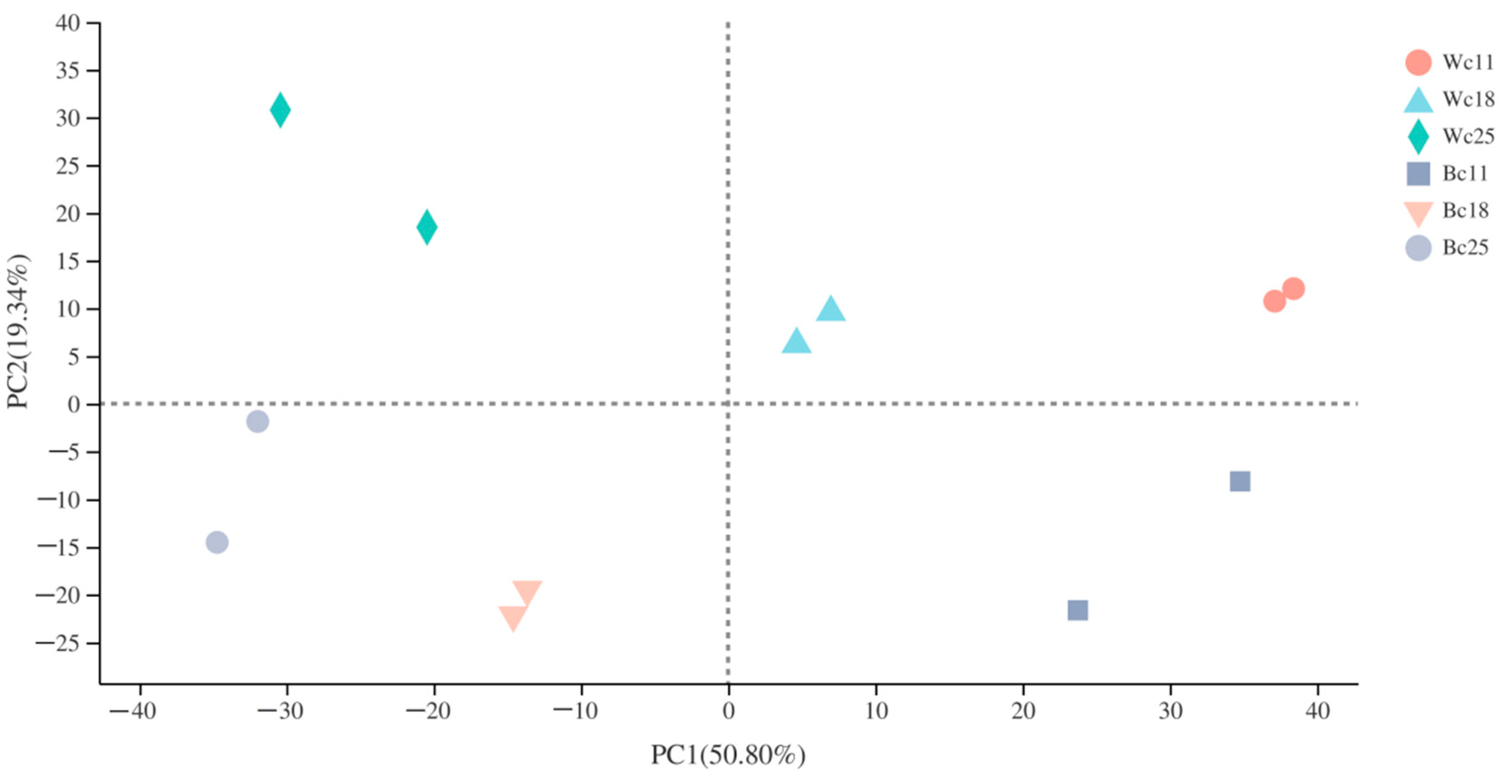
3.4. Transcriptome Sequencing and De Novo Assembly
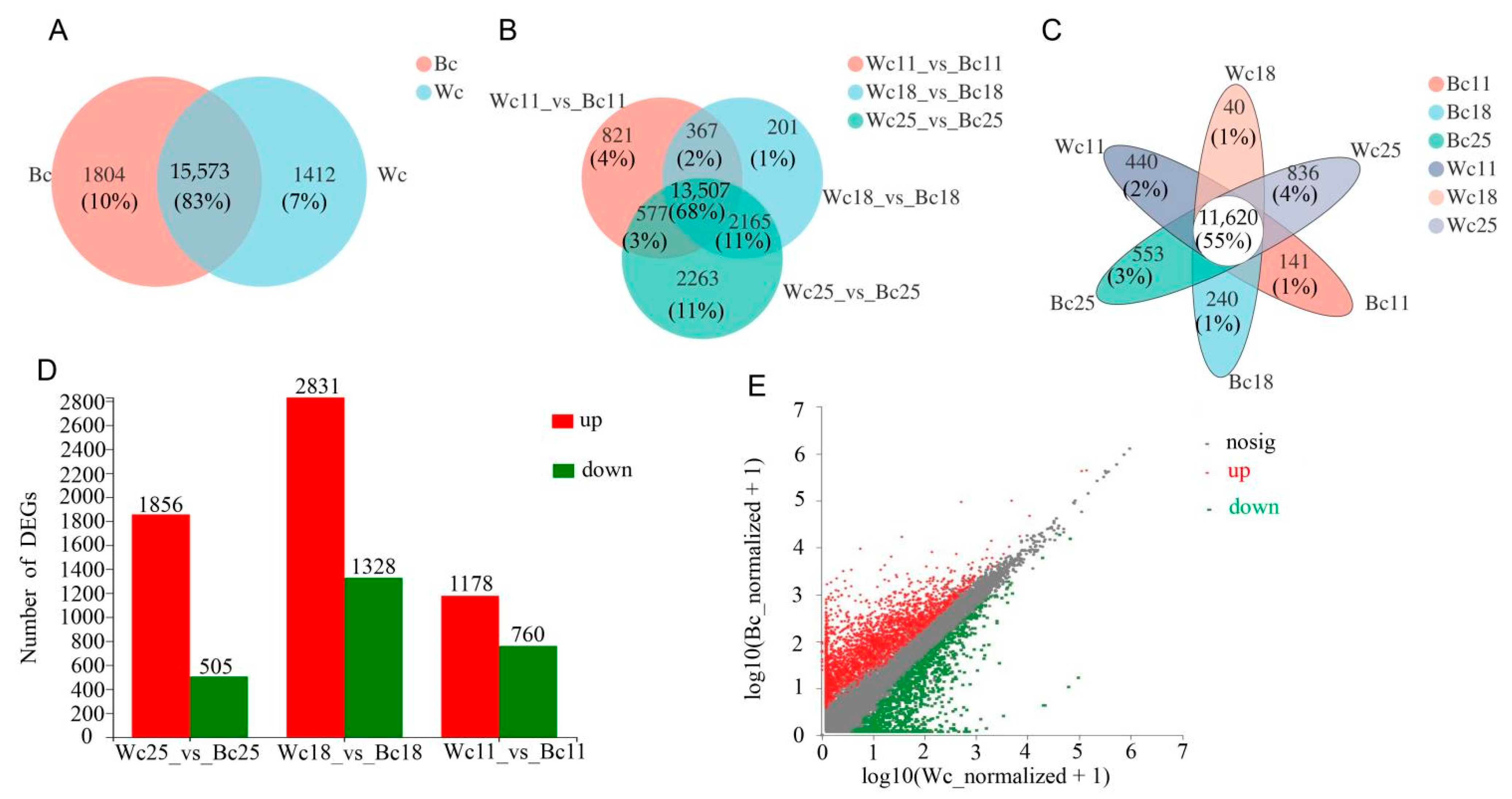
3.5. Identification of Differentially Expressed Genes (DEGs)
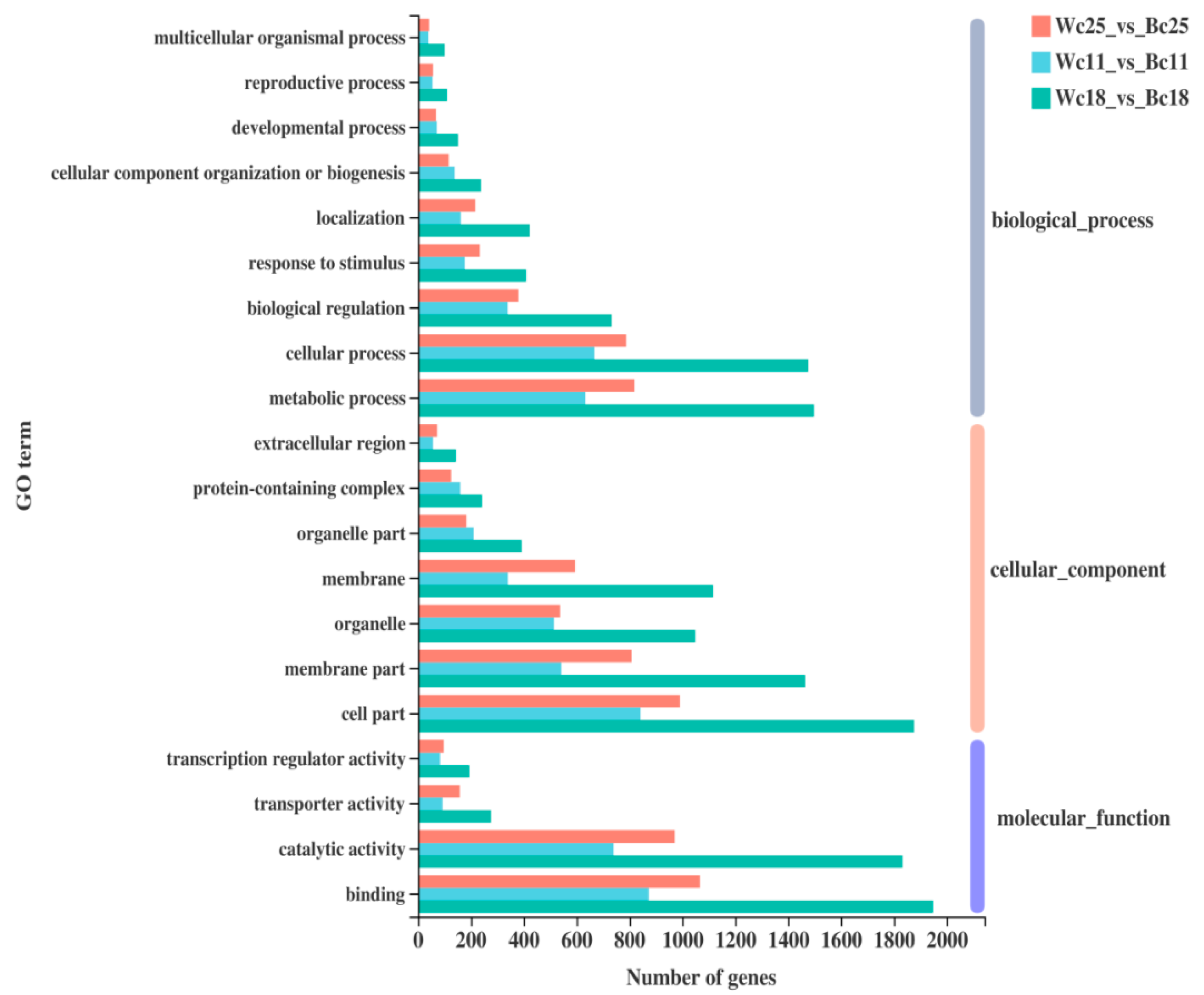
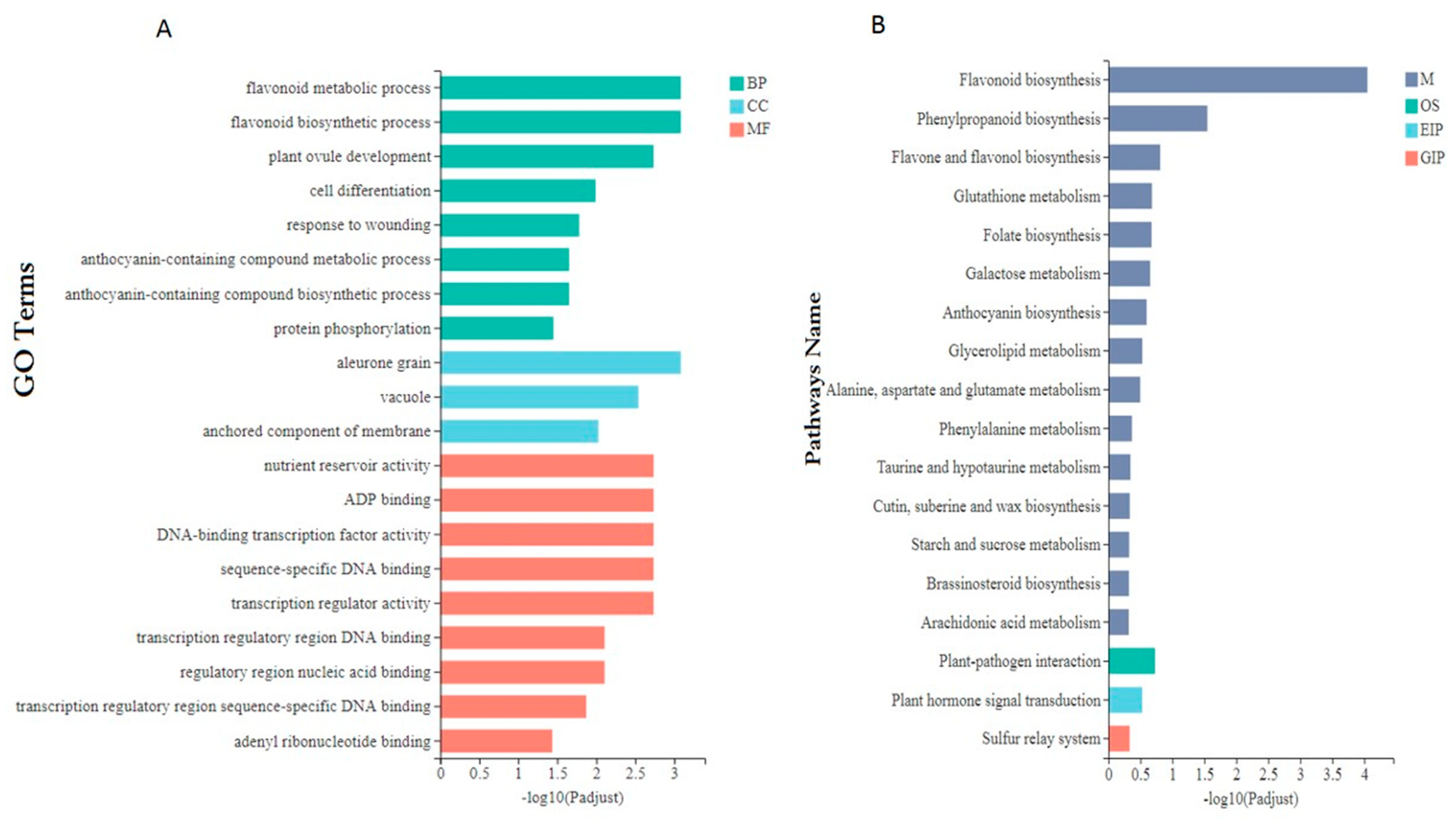
3.6. Functional Annotation and Enrichment Analysis of DEGs
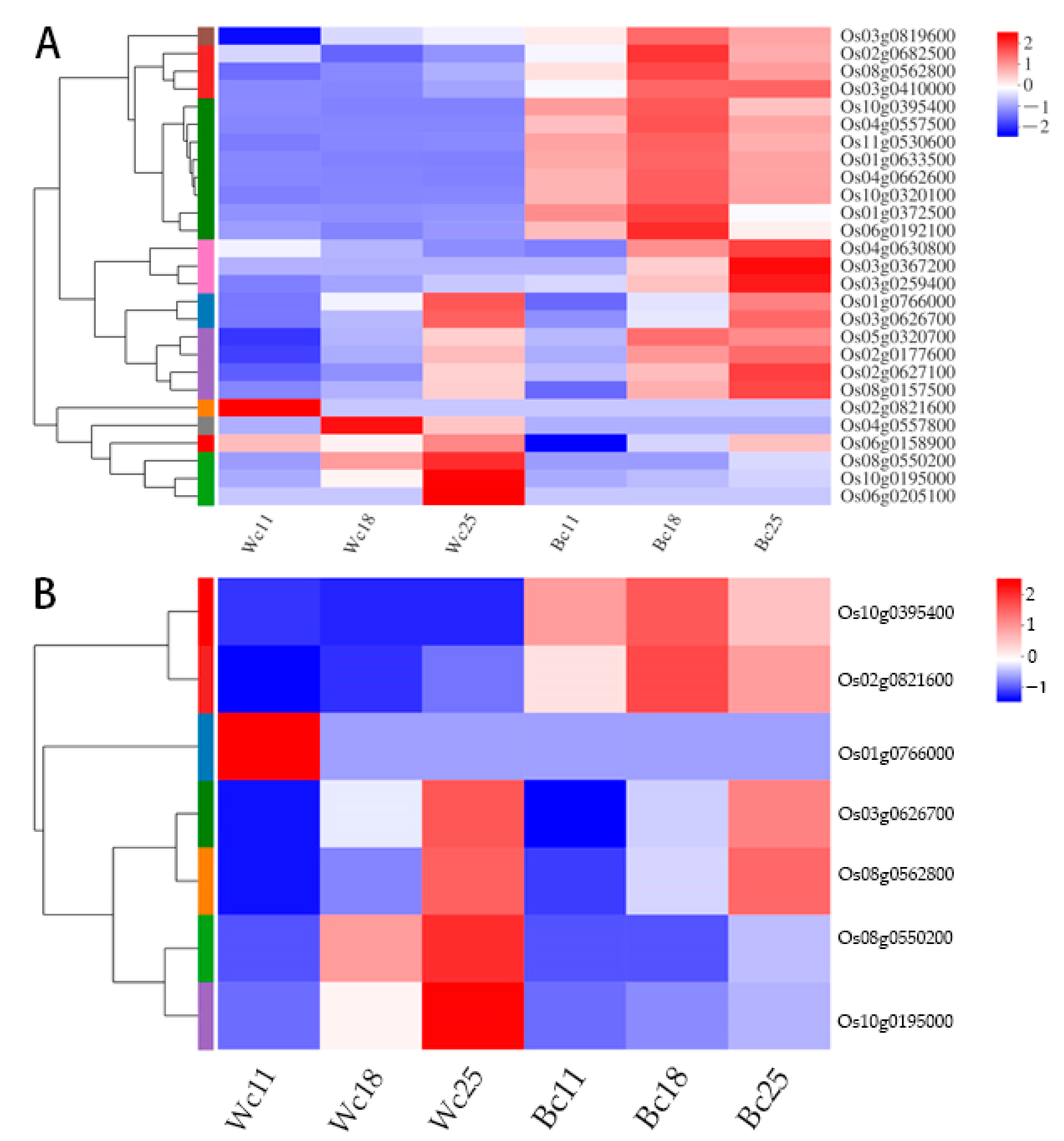
3.7. Expression of Anthocyanin Biosynthesis Genes and Transporters Genes in Rice Caryopsis
3.8. Validation of Transcriptome Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Aliphatic Index |
| ABC | ATP-Binding Cassette |
| AM | AnthoMATE |
| AVI | Anthocyanic Vacuolar Inclusion |
| bHLH | Basic helix-loop-helix |
| Bc | Black Caryopsis |
| C3G | Cyanidin-3-O-Glucoside |
| DEGs | Differentially Expressed Genes |
| DAF | Days After Flowering |
| ER | Endoplasmic Reticulum |
| FDR | False Discovery Rate |
| FW | Fresh Weight |
| GST | Glutathione-S-Transferase |
| GO | Gene Ontology |
| GRAVY | GRAND Average of Hydropathicity Index |
| HMMTOP | Hidden Markov Model Topology |
| HPLC | High-Performance Liquid Chromatography |
| I.I | Instability Index |
| JTT | Jones–Taylor–Thornton |
| KALA | Key Activator Loci for Anthocyanin |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LT | Ligandin Transportations |
| MATE | Multidrug and Toxic Compound Extrusion |
| MEME | Multiple Expectation Maximization for Motif Elicitation |
| ML | Maximum Likelihood |
| MRP | Multidrug Resistance-associated Proteins |
| MW | Molecular Weight |
| P3G | Peonidin-3-O-Glucoside |
| PVC | Pre-vacuolar Compartment |
| RSEM | RNA sequence by expectation maximization |
| ROS | Reactive Oxygen Species |
| SOPMA | Self-Optimized Prediction Method from Alignment |
| TAC | Total Anthocyanin Content |
| TMHMM | Transmembrane Helices Hidden Markov Model |
| TPM | Transcripts Per Million |
| VT | Vesicular Transport |
| Wc | White Caryopsis |
References
- Ali, I.; He, L.; Ullah, S.; Quan, Z.; Wei, S.; Iqbal, A.; Munsif, F.; Shah, T.; Xuan, Y.; Luo, Y.; et al. Biochar addition coupled with nitrogen fertilization impacts on soil quality, crop productivity, and nitrogen uptake under double-cropping system. Food Energy Secur. 2020, 9, e208. [Google Scholar] [CrossRef]
- Ranganathan, J.; Waite, R.; Searchinger, T.; Hanson, C. How to Sustainably Feed 10 Billion People by 2050, in 21 Charts. World Res. Ins. 2018. Available online: https://www.wri.org/blog/2018/12/how-sustainably-feed-10-billion-people-2050-21-.charts (accessed on 10 April 2020).
- Mbanjo, E.G.N.; Kretzschmar, T.; Jones, H.; Ereful, N.; Blanchard, C.; Boyd, L.A.; Sreenivasulu, N. The Genetic Basis and Nutritional Benefits of Pigmented Rice Grain. Front. Genet. 2020, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Terungwa, T.I.; Yuguda, M. The impact of rice production, consumption and importation in Nigeria: The political economy perspectives. Int. J. Sustain. Dev. World Policy 2014, 3, 90–99. [Google Scholar]
- Hao, J.; Zhu, H.; Zhang, Z.; Yang, S.; Li, H. Identification of anthocyanins in black rice (Oryza sativa L.) by UPLC/Q-TOF-MS and their in-vitro and in-vivo antioxidant activities. J. Cereal Sci. 2015, 64, 92–99. [Google Scholar] [CrossRef]
- Das, A.B.; Goud, V.; Das, C. Extraction of phenolic compounds and anthocyanin from black and purple rice bran (Oryza sativa L.) using ultrasound: A comparative analysis and phytochemical profiling. Ind. Crops Prod. 2017, 95, 332–341. [Google Scholar] [CrossRef]
- Jiamyangyuen, S.; Nuengchamnong, N.; Ngamdee, P. Bioactivity and chemical components of Thai rice in five stages of grain development. J. Cereal Sci. 2017, 74, 136–144. [Google Scholar] [CrossRef]
- Hemamalini, S.; Umamaheswari, D.S.; Lavanya, D.R.; Umamaheswara, R.D.C. Exploring the therapeutic potential and nutritional properties of’KaruppuKavuni’variety rice of Tamil Nadu. Int. J. Pharma Bio Sci. 2018, 9, 88–96. [Google Scholar] [CrossRef]
- Konczak, I.; Zhang, W. Anthocyanins—more than nature’s colours. J. Biomed. Biotechnol. 2004, 239–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazhen, S.; Wenju, W.; Panpan, Z.; Yuanyuan, Y.; Panpan, D.; Wusen, Z.; Yanling, W. Anthocyanins: Novel Antioxidants in Diseases Prevention and Human Health. In Flavonoids—A Coloring Model for Cheering up Life; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-N.; Chu, S.-C.; Chiou, H.-L.; Chiang, C.-L.; Yang, S.-F.; Hsieh, Y. Cancer, Cyanidin 3-glucoside and peonidin 3-glucoside inhibit tumor cell growth and induce apoptosis in vitro and suppress tumor growth in vivo. J. Nutr. 2005, 53, 232–243. [Google Scholar] [CrossRef]
- Luo, L.-P.; Han, B.; Yu, X.-P.; Chen, X.-Y.; Zhou, J.; Chen, W.; Zhu, Y.-F.; Peng, X.-L.; Zou, Q.; Li, S.-Y. Anti-metastasis activity of black rice anthocyanins against breast cancer: Analyses using an ErbB2 positive breast cancer cell line and tumoral xenograft model. Asian Pac. J. Cancer Prev. 2014, 15, 6219–6225. [Google Scholar] [CrossRef] [Green Version]
- Pojer, E.; Mattivi, F.; Johnson, D.; Stockley, C. The case for anthocyanin consumption to promote human health: A review. Compr. Rev. Food Sci. Food Saf. 2013, 12, 483–508. [Google Scholar] [CrossRef] [PubMed]
- Turturică, M.; Oancea, A.; Râpeanu, G.; Bahrim, G. Anthocyanins: Naturally occuring fruit pigments with functional properties. Ann. Univ. Dunarea Jos Galati Fascicle VI-Food Technol. 2015, 39, 100513018. [Google Scholar]
- Goufo, P.; Trindade, H. Rice antioxidants: Phenolic acids, flavonoids, anthocyanins, proanthocyanidins, tocopherols, tocotrienols, γ-oryzanol, and phytic acid. Food Sci. Nutr. 2014, 2, 75–104. [Google Scholar] [CrossRef] [PubMed]
- Gould, K. Nature’s Swiss army knife: The diverse protective roles of anthocyanins in leaves. J. Biomed. Biotechnol. 2004, 2004, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, L.; De Bie, E.; Vandeputte, G.E.; Veraverbeke, W.S.; Derycke, V.; De Man, W.; Delcour, J.A. Effect of milling on colour and nutritional properties of rice. Food Chem. 2007, 100, 1496–1503. [Google Scholar] [CrossRef]
- Reddy, C.K.; Kimi, L.; Haripriya, S.; Kang, N. Effects of polishing on proximate composition, physico-chemical characteristics, mineral composition and antioxidant properties of pigmented rice. Rice Sci. 2017, 24, 241–252. [Google Scholar] [CrossRef]
- Chen, M.-H.; McClung, A.M.; Bergman, C.J. Phenolic content, anthocyanins and antiradical capacity of diverse purple bran rice genotypes as compared to other bran colors. J. Cereal Sci. 2017, 77, 110–119. [Google Scholar] [CrossRef]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Mackon, E.; Mackon, G.J.D.E.; Ma, Y.; Kashif, M.H.; Ali, N.; Usman, B.; Liu, P. Recent insights into Anthocyanin Pigmentation, Synthesis, Trafficking, and Regulatory Mechanisms in Rice (Oryza sativa L.) Caryopsis. Biomolecules 2021, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. J. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Yu, S.; Zeng, D.; Liu, H.; Wang, H.; Yang, Z.; Xie, X.; Shen, R.; Tan, J.; Li, H. Development of “purple endosperm rice” by engineering anthocyanin biosynthesis in the endosperm with a high-efficiency transgene stacking system. Mol. Plant 2017, 10, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, S. Transport of flavonoids: From cytosolic synthesis to vacuolar accumulation. In The Science of Flavonoids; Springer: New York, NY, USA, 2006; pp. 123–146. [Google Scholar] [CrossRef]
- Grotewold, E. The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Grotewold, E.; Davies, K. Trafficking and sequestration of anthocyanins. Nat. Prod. Commun. 2008, 3. [Google Scholar] [CrossRef] [Green Version]
- Mueller, L.A.; Goodman, C.D.; Silady, R.A.; Walbot, V. AN9, a petunia glutathione S-transferase required for anthocyanin sequestration, is a flavonoid-binding protein. J. Plant Physiol. 2000, 123, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, H.; Huang, J.-R. Arabidopsis TT19 functions as a carrier to transport anthocyanin from the cytosol to tonoplasts. Mol. Plant 2012, 5, 387–400. [Google Scholar] [CrossRef]
- Jasinski, M.; Ducos, E.; Martinoia, E.; Boutry, M. The ATP-Binding Cassette Transporters: Structure, Function, and Gene Family Comparison between Rice and Arabidopsis. J. Plant Physiol. 2003, 131, 1169–1177. [Google Scholar] [CrossRef] [Green Version]
- Goodman, C.D.; Casati, P.; Walbot, V. A multidrug resistance–associated protein involved in anthocyanin transport in Zea mays. Plant Cell 2004, 16, 1812–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, C.; Conejero, G.; Torregrosa, L.; Cheynier, V.; Terrier, N.; Ageorges, A. In vivo grapevine anthocyanin transport involves vesicle-mediated trafficking and the contribution of anthoMATE transporters and GST. Plant J. 2011, 67, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. J. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-L.; Xie, X.-R.; Zhang, J.; Xiang, G.; Li, Y.; Wu, H.-B. In Silico Analysis of a MRP Transporter Gene Reveals Its Possible Role in Anthocyanins or Flavonoids Transport in Oryza sativa. Am. J. Plant Sci. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; An, W.; Wang, K.; Jiang, T.; Ren, Q.; Liang, W.; Wang, H. Expression profile analysis of MATE gene family in rice. Biol. Plant 2019, 63, 556–564. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef] [PubMed]
- The MEME Continued. Available online: http://meme-suite.org/ (accessed on 14 August 2020).
- HISAT: A Fast Spliced Aligner with Low Memory Requirements. Available online: http://0-ccb-jhu-edu.brum.beds.ac.uk/software/hisat2/index.shtml (accessed on 10 December 2020).
- StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Available online: https://0-ccb-jhu-edu.brum.beds.ac.uk/software/stringtie/index.shtml?t=example (accessed on 16 December 2020).
- RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. Available online: http://deweylab.biostat.wisc.edu/rsem/ (accessed on 20 December 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GOATOOLS: Tools for Gene Ontology (version v0.6.5). Available online: https://github.com/tanghaibao/GOatools (accessed on 5 January 2021).
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef] [PubMed]
- Rogers, E.E.; Guerinot, M.L. FRD3, a member of the multidrug and toxin efflux family, controls iron deficiency responses in Arabidopsis. Plant Cell 2002, 14, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, M.; Sharma, D.; Singh, M.; Tripathi, R.D.; Trivedi, P.K. Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci. Rep. 2002, 4, 3964. [Google Scholar] [CrossRef] [Green Version]
- M’mbone, M.E.; Cheng, W.; Xu, L.; Wang, Y.; Karanja, B.K.; Zhu, X.; Liu, L. Identification and transcript analysis of MATE genes involved in anthocyanin transport in radish (Raphanus sativus L). Sci. Hortic. 2018, 238, 195–203. [Google Scholar] [CrossRef]
- Gomez, C.; Terrier, N.; Torregrosa, L.; Vialet, S.; Fournier-Level, A.; Verries, C.; Souquet, J.-M.; Mazauric, J.-P.; Klein, M.; Cheynier, V. Grapevine MATE-type proteins act as vacuolar H+-dependent acylated anthocyanin transporters. J. Plant Physiol. 2009, 150, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Huhman, D.; Shadle, G.; He, X.-Z.; Sumner, L.W.; Tang, Y.; Dixon, R.A. MATE2 mediates vacuolar sequestration of flavonoid glycosides and glycoside malonates in Medicago truncatula. Plant Cell 2011, 23, 1536–1555. [Google Scholar] [CrossRef] [Green Version]
- Mathews, H.; Clendennen, S.K.; Caldwell, C.G.; Liu, X.L.; Connors, K.; Matheis, N.; Wagner, D.R. Activation tagging in tomato identifies a transcriptional regulator of anthocyanin biosynthesis, modification, and transport. Plant Cell 2003, 15, 1689–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.P.; Wilkins, C.; Demidchik, V.; Davies, J.M.; Glover, B.J. An Arabidopsis flavonoid transporter is required for anther dehiscence and pollen development. J. Exp. Bot. 2010, 61, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debeaujon, I.; Peeters, A.J.; Léon-Kloosterziel, K.M.; Koornneef, M. The TRANSPARENT TESTA12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell 2001, 13, 853–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, Y.; Lei, B.; Huang, H.L.; Li, J.N.; Yin, J.M.; Tang, Z.L.; Wang, R.; Chen, L. TRANSPARENT TESTA 12 genes from Brassica napus and parental species: Cloning, evolution, and differential involvement in yellow seed trait. Mol. Genet. Genom. 2009, 281, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diaz, R.; Ryngajllo, M.; Perez-Diaz, J.; Peña-Cortés, H.; Casaretto, J.A.; González-Villanueva, E.; Ruiz-Lara, S. VvMATE1 and VvMATE2 encode putative proanthocyanidin transporters expressed during berry development in Vitis vinifera L. Plant Cell Rep. 2014, 33, 1147–1159. [Google Scholar] [CrossRef]
- Zhao, J.; Dixon, R.A. MATE transporters facilitate vacuolar uptake of epicatechin 3′-O-glucoside for proanthocyanidin biosynthesis in Medicago truncatula and Arabidopsis. Plant Cell 2009, 21, 2323–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Keck, M.; Sagasser, M.; Niehaus, K.; Weisshaar, B.; Stracke, R. Two differentially expressed MATE factor genes from apple complement the Arabidopsis transparent testa12 mutant. Plant Biol. 2011, 13, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Tang, Y.-M.; Hu, Y.-Y.; Wang, Y.; Sun, B.; Wang, X.-R.; Tang, H.-R.; Chen, Q. FaTT12-1, a multidrug and toxin extrusion (MATE) member involved in proanthocyanidin transport in strawberry fruits. Sci. Hortic. 2018, 231, 158–165. [Google Scholar] [CrossRef]
- Min, X.; Jin, X.; Liu, W.; Wei, X.; Zhang, Z.; Ndayambaza, B.; Wang, Y. Transcriptome-wide characterization and functional analysis of MATE transporters in response to aluminum toxicity in Medicago sativa L. PeerJ 2019, 7, e6302. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, Y.; Liu, H.; Kang, L.; Geng, J.; Gai, Y.; Ding, Y.; Sun, H.; Li, Y. Identification andexpression analysis of MATE genes involved in flavonoid transport in blueberry plants. PLoS ONE 2015, 10, e0118578. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, Y.; Wang, W.; Gai, J.; Li, Y. Genome-wide analysis of MATE transporters and expression patterns of a subgroup of MATE genes in response to aluminum toxicity in soybean. BMC Genom. 2016, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Darbani, B.; Motawia, M.S.; Olsen, C.E.; Nour-Eldin, H.H.; Møller, B.L.; Rook, F. The biosynthetic gene cluster for the cyanogenic glucoside dhurrin in Sorghum bicolor contains its co-expressed vacuolar MATE transporter. Sci. Rep. 2016, 6, 37079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeriglio, A.; Barreca, D.; Bellocco, E.; Trombetta, D. Chemistry, pharmacology and health benefits of anthocyanins. Phytother. Res. 2016, 30, 1265–1286. [Google Scholar] [CrossRef] [PubMed]
- Khoo, H.E.; Azlan, A.; Tang, S.T.; Lim, S.M. Anthocyanidins and anthocyanins: Colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. Res. 2017, 61, 1361779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landi, M.; Tattini, M.; Gould, K.S. Multiple functional roles of anthocyanins in plant-environment interactions. Environ. Exp. Bot. 2015, 119, 4–17. [Google Scholar] [CrossRef]
- Chunthaburee, S.; Sakuanrungsirikul, S.; Wongwarat, T.; Sanitchon, J.; Pattanagul, W.; Theerakulpisut, P. Changes in anthocyanin content and expression of anthocyanin synthesis genes in seedlings of black glutinous rice in response to salt stress. Asian. J. Plant Sci. 2016, 15, 56–65. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, A.; Wu, X.; Zhu, Z.; Yang, Z.; Zhu, Y.; Zha, D. Transcriptome analysis revealed expression of genes related to anthocyanin biosynthesis in eggplant (Solanum melongena L.) under high-temperature stress. BMC Plant Biol. 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poustka, F.; Irani, N.G.; Feller, A.; Lu, Y.; Pourcel, L.; Frame, K.; Grotewold, E. A trafficking pathway for anthocyanins overlaps with the endoplasmic reticulum-to-vacuole protein-sorting route in Arabidopsis and contributes to the formation of vacuolar inclusions. Plant Physiol. 2007, 145, 1323–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.O.D.; Randall, R.; BlakeKalff, M.M.A. Detoxification of xenobiotics in plant cells by glutathione conjugation and vacuolar compartmentalization: A fluorescent assay using monochlorobimane. Plant Cell Environ. 1997, 20, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Das, N.; Bhattacharya, S.; Bhattacharyya, S.; Maiti, M.K. Expression of rice MATE family transporter OsMATE2 modulates arsenic accumulation in tobacco and rice. Plant Mol. Biol. 2018, 98, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bei, X.; Gao, J.; Li, Y.; Yan, Y.; Hu, Y. The similar and different evolutionary trends of MATE family occurred between rice and Arabidopsis thaliana. BMC Plant Biol. 2016, 16, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.G.; Kim, J.H.; Min, S.Y.; Shin, K.-H.; Kim, J.H.; Kim, H.Y.; Ryu, S.N. Anthocyanin content in rice is related to expression levels of anthocyanin biosynthetic genes. J. Plant Biol. 2007, 50, 156–160. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Jiang, H.; Sun, Y.; Tang, Y.; Yuan, Z.; Zhang, D. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006, 141, 1167–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, T.; Maeda, H.; Oguchi, T.; Yamaguchi, T.; Tanabe, N.; Ebana, K.; Yano, M.; Ebitani, T.; Izawa, T. The birth of a black rice gene and its local spread by introgression. Plant Cell 2015, 27, 2401–2414. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, Z.; Chen, C.; Wu, W.; Ren, N.; Jiang, C.; Yu, J.; Zhao, Y.; Zheng, X.; Yang, Q. The C–S–A gene system regulates hull pigmentation and reveals evolution of anthocyanin biosynthesis pathway in rice. J. Exp. Bot. 2018, 69, 1485–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Xu, F.; Sun, X.; Bao, J.; Beta, T. Identification and quantification of phenolic acids and anthocyanins as antioxidants in bran, embryo and endosperm of white, red and black rice kernels (Oryza sativa L.). J. Cereal Sci. 2014, 59, 211–218. [Google Scholar] [CrossRef]
- Druka, A.; Kudrna, D.; Rostoks, N.; Brueggeman, R.; Von Wettstein, D.; Kleinhofs, A. Chalcone isomerase gene from rice (Oryza sativa) and barley (Hordeum vulgare): Physical, genetic and mutation mapping. Gene 2003, 302, 171–178. [Google Scholar] [CrossRef]
- Reddy, A.R.; Scheffler, B.; Madhuri, G.; Srivastava, M.N.; Kumar, A.; Sathyanarayanan, P.V.; Mohan, M. Chalcone synthase in rice (Oryza sativa L.): Detection of the CHS protein in seedlings and molecular mapping of the chs locus. Plant Mol. Biol. 1996, 32, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Martinoia, E.; Grill, E.; Tommasini, R.; Kreuz, K.; Amrhein, N. ATP-dependent glutathione S-conjugate’export’pump in the vacuolar membrane of plants. Nature 1993, 364, 247–249. [Google Scholar] [CrossRef]
- Martinoia, E.; Maeshima, M.; Neuhaus, H.E. Vacuolar transporters and their essential role in plant metabolism. J. Exp. Bot. 2007, 58, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, K.; Sugiyama, A.; Morita, M.; Shitan, N. Secondary transport as an efficient membrane transport mechanism for plant secondary metabolites. Phytochem. Rev. 2008, 7, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Shitan, N.; Sawada, K.; Van Montagu, M.C.; Inzé, D.; Rischer, H.; Yazaki, K. Vacuolar transport of nicotine is mediated by a multidrug and toxic compound extrusion (MATE) transporter in Nicotiana tabacum. Proc. Nat. Acad. Sci. USA 2009, 106, 2447–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinova, K.; Pourcel, L.; Weder, B.; Schwarz, M.; Barron, D.; Routaboul, J.M.; Klein, M. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H+-antiporter active in proanthocyanidin-accumulating cells of the seed coat. Plant Cell 2007, 19, 2023–2038. [Google Scholar] [CrossRef] [Green Version]
- Roytrakul, S.; Verpoorte, R. Role of vacuolar transporter proteins in plant secondary metabolism: Catharanthus roseus cell culture. Phytochem. Rev. 2007, 6, 383–396. [Google Scholar] [CrossRef] [Green Version]
- Mackon, E.; Ma, Y.; Jeazet Dongho Epse Mackon, G.C.; Li, Q.; Zhou, Q.; Liu, P. Subcellular Localization and Vesicular Structures of Anthocyanin Pigmentation by Fluorescence Imaging of Black Rice (Oryza sativa L.) Stigma Protoplast. Plants 2021, 10, 685. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Anthocyanin | Rice Developmental Stages | |||||||
|---|---|---|---|---|---|---|---|---|
| Milk (11 DAF) | Dough (18 DAF) | Mature (25 DAF) | Fully Ripe (35 DAF) | |||||
| Wc11 | Bc11 | Wc18 | Bc18 | Wc25 | Bc25 | Wc35 | Bc35 | |
| Pt3G | ND | ND | ND | ND | ND | ND | ND | ND |
| P3G | ND | 163.75 ± 5 | ND | 295.33 ± 4 | ND | 97.52 ± 10 | ND | 35.98 ± 7 |
| C3G | ND | 1600.96 ± 15 | ND | 4206.91 ± 18 | ND | 1212.87 ± 10 | ND | 814.59 ± 5.5 |
| Sample ID | Raw Reads | Clean Reads | Total Mapped Reads | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|
| Wc11-rep1 | 46234788 | 45716764 | 42690828 (93.38%) | 98.81 | 96.31 | 51.99 |
| Wc11-rep2 | 47796190 | 47120470 | 43973125 (93.32%) | 98.74 | 96.12 | 51.79 |
| Wc18-rep1 | 53602144 | 53141526 | 48976170 (92.16%) | 98.69 | 95.89 | 52.4 |
| Wc18-rep2 | 51253398 | 50855026 | 47207849 (92.83%) | 98.83 | 96.28 | 52.22 |
| Wc25-rep1 | 53062226 | 52561118 | 48421904 (92.12%) | 98.74 | 96.16 | 54.88 |
| Wc25-rep2 | 56203098 | 55792292 | 51481571 (92.27%) | 98.87 | 96.4 | 53.63 |
| Bc11-rep1 | 44487444 | 43814814 | 41492963 (94.7%) | 98.79 | 96.27 | 51.4 |
| Bc11-rep2 | 47527712 | 46925570 | 44457493 (94.74%) | 98.78 | 96.17 | 51.67 |
| Bc18-rep1 | 54482624 | 54089936 | 51460136 (95.14%) | 98.8 | 96.21 | 52.01 |
| Bc18-rep2 | 54430818 | 53880872 | 51159662 (94.95%) | 98.71 | 95.99 | 52.25 |
| Bc25-rep1 | 57726156 | 57135956 | 54078567 (94.65%) | 98.78 | 96.23 | 54.57 |
| Bc25-rep2 | 49975758 | 49497564 | 46688293 (94.32%) | 98.78 | 96.21 | 54.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mackon, E.; Ma, Y.; Jeazet Dongho Epse Mackon, G.C.; Usman, B.; Zhao, Y.; Li, Q.; Liu, P. Computational and Transcriptomic Analysis Unraveled OsMATE34 as a Putative Anthocyanin Transporter in Black Rice (Oryza sativa L.) Caryopsis. Genes 2021, 12, 583. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040583
Mackon E, Ma Y, Jeazet Dongho Epse Mackon GC, Usman B, Zhao Y, Li Q, Liu P. Computational and Transcriptomic Analysis Unraveled OsMATE34 as a Putative Anthocyanin Transporter in Black Rice (Oryza sativa L.) Caryopsis. Genes. 2021; 12(4):583. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040583
Chicago/Turabian StyleMackon, Enerand, Yafei Ma, Guibeline Charlie Jeazet Dongho Epse Mackon, Babar Usman, Yitong Zhao, Qiufeng Li, and Piqing Liu. 2021. "Computational and Transcriptomic Analysis Unraveled OsMATE34 as a Putative Anthocyanin Transporter in Black Rice (Oryza sativa L.) Caryopsis" Genes 12, no. 4: 583. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040583