
Further Delineation of Phenotype and Genotype of Primary Microcephaly Syndrome with Cortical Malformations Associated with Mutations in the WDR62 Gene

 , , ,
, , ,  , , and
, , and 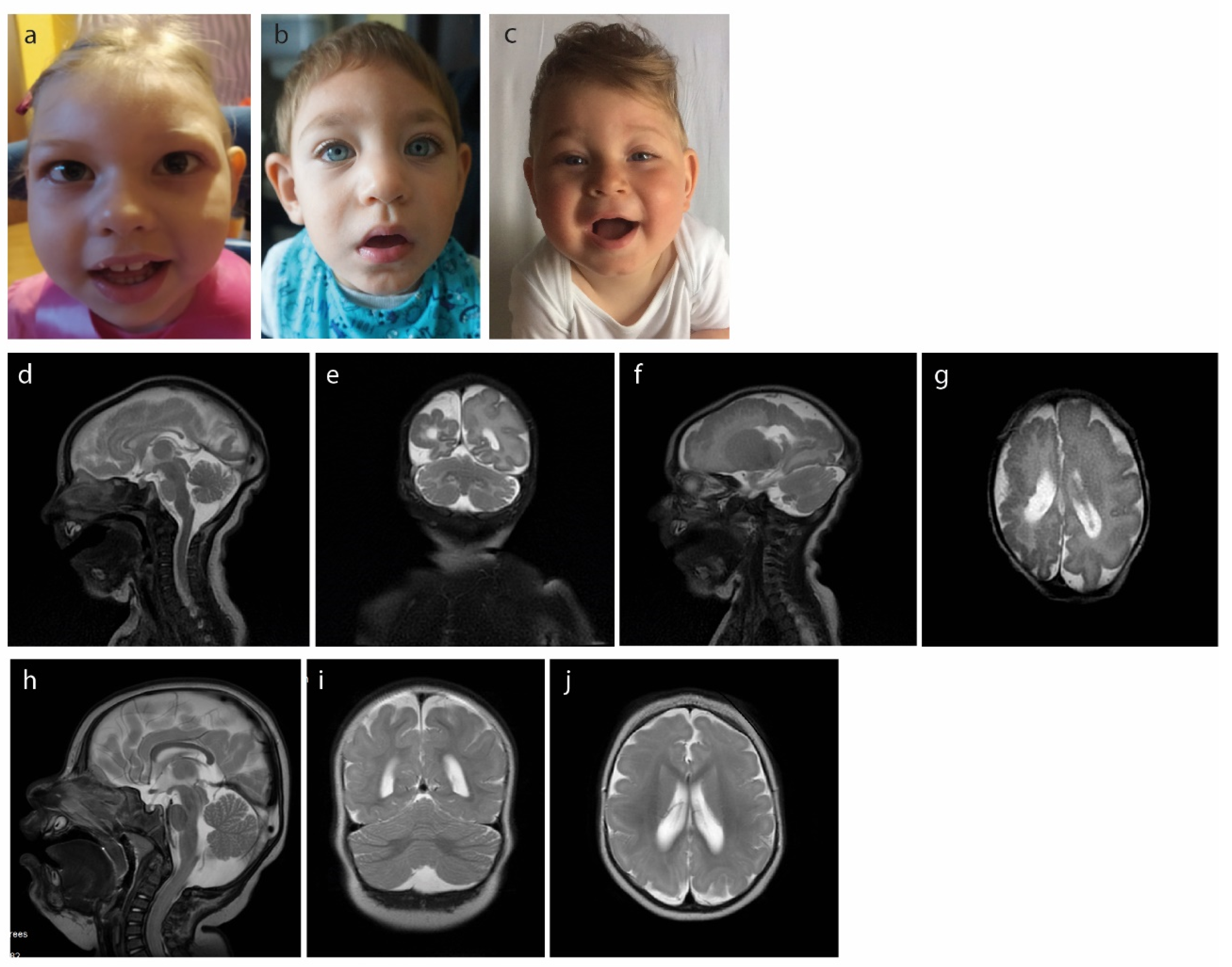{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Clinical Results
2.1. Case1
2.2. Case 2
2.3. Case 3
3. Methods and Genetic Results
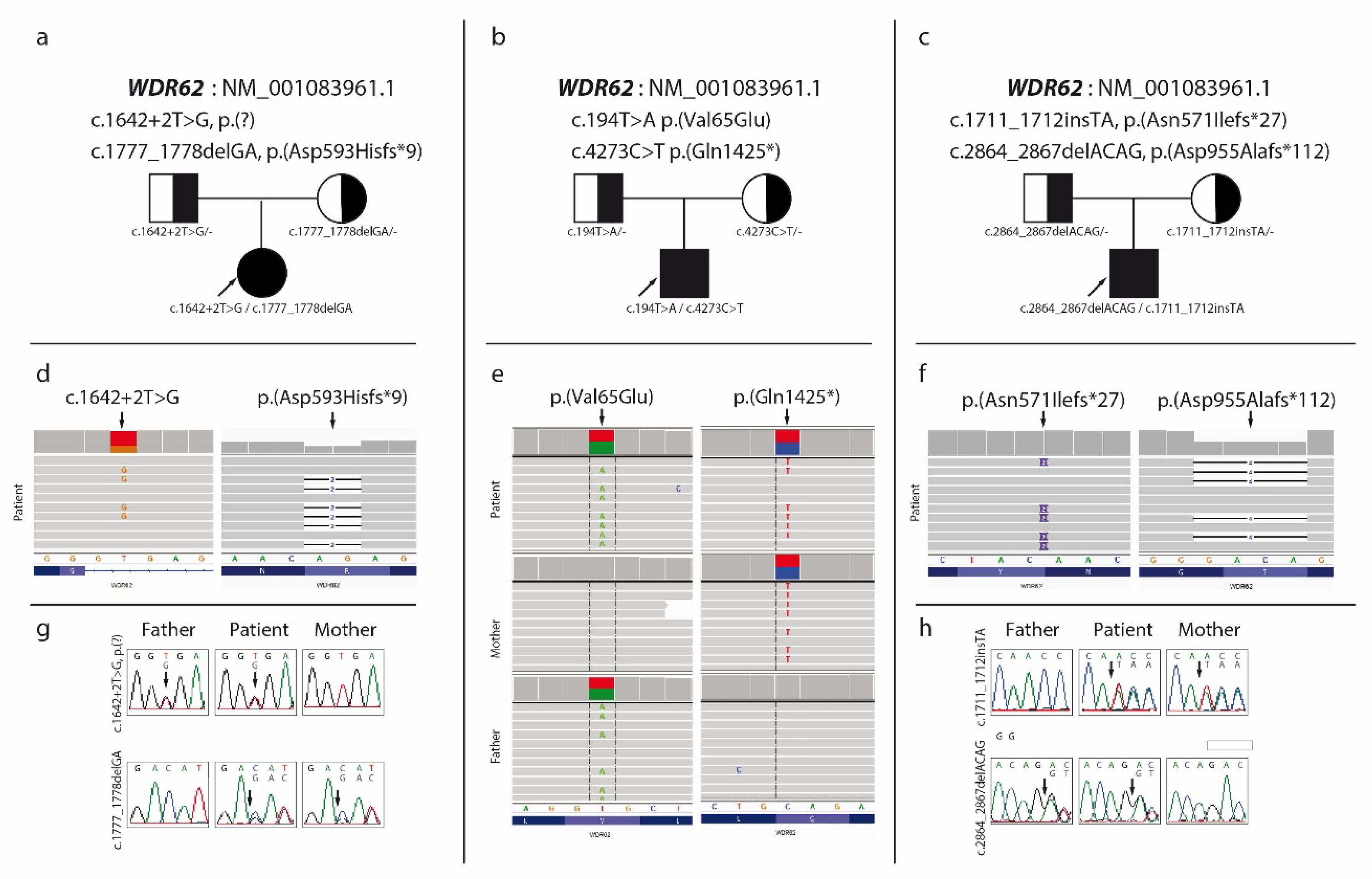
3.1. Case 1
3.2. Case 2
3.3. Case 3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duerinckx, S.; Abramowicz, M. The genetics of congenitally small brains. Semin. Cell Dev. Biol. 2018, 76, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Kazmi, S.K.; Amin, M.; Asif, Z.; Islam, U.; Shahid, K.; Tehreem, S. Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH). Genet. Res. 2018, 100, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Nicholas, A.K.; Khurshid, M.; Désir, J.; Carvalho, O.P.; Cox, J.J.; Thornton, G.; Kausar, R.; Ansar, M.; Ahmad, W.; Verloes, A.; et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat. Genet. 2010, 42, 1010–1014. [Google Scholar] [CrossRef]
- Le Goff, C.; Mahaut, C.; Abhyankar, A.; Le Goff, W.; Serre, V.; Afenjar, A.; Destrée, A.; Di Rocco, M.; Héron, D.; Jacquemont, S.; et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat. Genet. 2011, 44, 85–88. [Google Scholar] [CrossRef]
- Farag, H.G.; Froehler, S.; Oexle, K.; Ravindran, E.; Schindler, D.; Staab, T.; Huebner, A.; Kraemer, N.; Chen, W.; Kaindl, A.M. Abnormal centrosome and spindle morphology in a patient with autosomal recessive primary microcephaly type 2 due to compound heterozygous WDR62 gene mutation. Orphanet J. Rare Dis. 2013, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Salowsky, R.; Heiss, N.S.; Benner, A.; Wittig, R.; Poustka, A. Basal transcription activity of the dyskeratosis congenita gene is mediated by Sp1 and Sp3 and a patient mutation in a Sp1 binding site is associated with decreased promoter activity. Gene 2002, 293, 9–19. [Google Scholar] [CrossRef]
- Knight, S.; Vulliamy, T.; Morgan, B.; Devriendt, K.; Mason, P.; Dokal, I. Identification of novel DKC1 mutations in patients with dyskeratosis congenita: Implications for pathophysiology and diagnosis. Qual. Life Res. 2001, 108, 299–303. [Google Scholar] [CrossRef]
- Mahmood, S.; Ahmad, W.; Hassan, M.J. Autosomal recessive primary microcephaly (MCPH): Clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J. Rare Dis. 2011, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Peyre, E.; Morin, X. An oblique view on the role of spindle orientation in vertebrate neurogenesis. Dev. Growth Differ. 2012, 54, 287–305. [Google Scholar] [CrossRef]
- Yu, T.W.; Mochida, G.H.; Tischfield, D.J.; Sgaier, S.K.; Flores-Sarnat, L.; Sergi, C.M.; Topçu, M.; McDonald, M.T.; Barry, B.J.; Felie, J.M.; et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet. 2010, 42, 1015–1020. [Google Scholar] [CrossRef] [Green Version]
- Bacino, C.A.; Arriola, L.A.; Wiszniewska, J.; Bonnen, P.E. WDR62 missense mutation in a consanguineous family with primary microcephaly. Am. J. Med. Genet. Part A 2012, 158, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Chen, H.; Huang, H.; Wu, J.; Yang, Z.; Deng, W.; Chen, D.; Deng, J.; Su, Y.; Li, Y.; et al. Novel mutations c.28G>T (p.Ala10Ser) and c.189G>T (p.Glu63Asp) in WDR62 associated with early onset acanthosis and hyperkeratosis in a patient with autosomal recessive microcephaly type 2. Oncotarget 2016, 7, 78363–78371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastaki, F.; Mohamed, M.; Nair, P.; Saif, F.; Tawfiq, N.; Aithala, G.; El-Halik, M.; Al-Ali, M.; Hamzeh, A.R. Novel splice-site mutation inWDR62revealed by whole-exome sequencing in a Sudanese family with primary microcephaly. Congenit. Anom. 2015, 56, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Bhat, V.; Girimaji, S.; Mohan, G.; Arvinda, H.; Singhmar, P.; Duvvari, M.R.; Kumar, A. Mutations in WDR62, encoding a centrosomal and nuclear protein, in Indian primary microcephaly families with cortical malformations. Clin. Genet. 2011, 80, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Bilguvar, K.; Ozturk, A.K.; Louvi, A.; Kwan, K.Y.; Choi, M.; Tatli, B.; Yalnizoglu, D.; Tuysuz, B.; Caglayan, A.O.; Gokben, S.; et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 2010, 467, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Baig, J.M.; Moawia, A.; Ahmad, I.; Iqbal, M.; Waseem, S.S.; Asif, M.; Abdullah, U.; Makhdoom, E.U.H.; Kaygusuz, E.; et al. An update of pathogenic variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 underlying autosomal recessive primary microcephaly in 32 consanguineous families from Pakistan. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Bakhtiar, S.M.; Farooq, M.; Anjum, I.; Janzén, E.; Toliat, M.R.; Eiberg, H.; Kjaer, K.; Tommerup, N.; Noegel, A.; et al. Genetic heterogeneity in Pakistani microcephaly families. Clin. Genet. 2012, 83, 446–451. [Google Scholar] [CrossRef]
- Kousar, R.; Hassan, M.J.; Khan, B.; Basit, S.; Mahmood, S.; Mir, A.; Ahmad, W.; Ansar, M. Mutations in WDR62 gene in Pakistani families with autosomal recessive primary microcephaly. BMC Neurol. 2011, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- McDonell, L.M.; Chardon, J.W.; Schwartzentruber, J.; Foster, D.; Beaulieu, C.L.; Majewski, J.; Bulman, D.E.; Boycott, K.M. The utility of exome sequencing for genetic diagnosis in a familial microcephaly epilepsy syndrome. BMC Neurol. 2014, 14, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, M.M.; Raza, S.I.; Basit, S.; Kousar, R.; Ahmad, W.; Ansar, M. A novel WDR62 mutation causes primary microcephaly in a Pakistani family. Mol. Biol. Rep. 2013, 40, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Akutsu, S.N.; Fukumitsu, A.; Morino, H.; Masatsuna, Y.; Hosoba, K.; Kawakami, H.; Yamamoto, T.; Shimizu, K.; Ohashi, H.; et al. PLK1-mediated phosphorylation of WDR62/MCPH2 ensures proper mitotic spindle orientation. Hum. Mol. Genet. 2017, 26, 4429–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdock, D.R.; Clark, G.D.; Bainbridge, M.N.; Newsham, I.; Wu, Y.-Q.; Muzny, D.M.; Cheung, S.W.; Gibbs, R.A.; Ramocki, M.B. Whole-exome sequencing identifies compound heterozygous mutations in WDR62 in siblings with recurrent polymicrogyria. Am. J. Med. Genet. Part A 2011, 155, 2071–2077. [Google Scholar] [CrossRef] [Green Version]
- Nardello, R.; Fontana, A.; Antona, V.; Beninati, A.; Mangano, G.D.; Stallone, M.C.; Mangano, S. A novel mutation of WDR62 gene associated with severe phenotype including infantile spasm, microcephaly, and intellectual disability. Brain Dev. 2018, 40, 58–64. [Google Scholar] [CrossRef]
- Naseer, M.I.; Rasool, M.; Abdulkareem, A.A.; Chaudhary, A.G.; Zaidi, S.K.; Al-Qahtani, M.H. Novel compound heterozygous mutations in WDR62 gene leading to developmental delay and Primary Microcephaly in Saudi Family. Pak. J. Med. Sci. 2019, 35, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Naseer, M.I.; Rasool, M.; Sogaty, S.; Chaudhary, R.A.; Mansour, H.M.; Chaudhary, A.G.; Abuzenadah, A.M.; Al-Qahtani, M.H. A novel WDR62 mutation causes primary microcephaly in a large consanguineous Saudi family. Ann. Saudi Med. 2017, 37, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Poulton, C.J.; Schot, R.; Seufert, K.; Lequin, M.H.; Accogli, A.; Annunzio, G.D.; Villard, L.; Philip, N.; De Coo, R.; Catsman-Berrevoets, C.; et al. Severe presentation ofWDR62mutation: Is there a role for modifying genetic factors? Am. J. Med. Genet. Part A 2014, 164, 2161–2171. [Google Scholar] [CrossRef]
- Rupp, V.; Rauf, S.; Naveed, I.; Windpassinger, C.; Mir, A.; Christian, W. A novel single base pair duplication in WDR62 causes primary microcephaly. BMC Med. Genet. 2014, 15, 107. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Khan, A.; Han, S.; Zhang, X. Molecular analysis of 23 Pakistani families with autosomal recessive primary microcephaly using targeted next-generation sequencing. J. Hum. Genet. 2016, 62, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Ding, X.; Zeng, Q.; Zou, H.; Liao, J.; Cao, D. A case report of microcephaly and refractory West syndrome associated with WDR62 mutation. Acta Epileptol. 2020, 2, 1–6. [Google Scholar] [CrossRef]
- Zombor, M.; Kalmár, T.; Nagy, N.; Berényi, M.; Telcs, B.; Maróti, Z.; Brandau, O.; Sztriha, L. A novel WDR62 missense mutation in microcephaly with abnormal cortical architecture and review of the literature. J. Appl. Genet. 2019, 60, 151–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.G.; Lee, D.-W.; Kim, J.; Jang, J.-H.; Lee, S.-M.; Jang, D.-H. Two Novel Mutations (c.883-4_890del and c.1684C>G) of WDR62 Gene Associated with Autosomal Recessive Primary Microcephaly: A Case Report. Front. Pediatr. 2019, 7, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaouad, I.C.; Zrhidri, A.; Jdioui, W.; Lyahyai, J.; Raymond, L.; Egéa, G.; Taoudi, M.; El Mouatassim, S.; Sefiani, A. A novel non sense mutation in WDR62 causes autosomal recessive primary microcephaly: A case report. BMC Med. Genet. 2018, 19, 118. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slezak, R.; Smigiel, R.; Obersztyn, E.; Pollak, A.; Dawidziuk, M.; Wiszniewski, W.; Bekiesinska-Figatowska, M.; Rydzanicz, M.; Ploski, R.; Gawlinski, P. Further Delineation of Phenotype and Genotype of Primary Microcephaly Syndrome with Cortical Malformations Associated with Mutations in the WDR62 Gene. Genes 2021, 12, 594. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040594
Slezak R, Smigiel R, Obersztyn E, Pollak A, Dawidziuk M, Wiszniewski W, Bekiesinska-Figatowska M, Rydzanicz M, Ploski R, Gawlinski P. Further Delineation of Phenotype and Genotype of Primary Microcephaly Syndrome with Cortical Malformations Associated with Mutations in the WDR62 Gene. Genes. 2021; 12(4):594. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040594
Chicago/Turabian StyleSlezak, Ryszard, Robert Smigiel, Ewa Obersztyn, Agnieszka Pollak, Mateusz Dawidziuk, Wojciech Wiszniewski, Monika Bekiesinska-Figatowska, Malgorzata Rydzanicz, Rafal Ploski, and Pawel Gawlinski. 2021. "Further Delineation of Phenotype and Genotype of Primary Microcephaly Syndrome with Cortical Malformations Associated with Mutations in the WDR62 Gene" Genes 12, no. 4: 594. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12040594