
Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes



1
Department of Experimental Medicine, Sapienza University of Rome, 00161 Rome, Italy
2
Immunovirology Unit, Department of Clinical Sciences, Skåne University Hospital, Lund University, 21428 Malmo, Sweden
*
Author to whom correspondence should be addressed.
Genes 2021, 12(6), 887; https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060887
Submission received: 24 May 2021
/
Revised: 6 June 2021
/
Accepted: 7 June 2021
/
Published: 8 June 2021
(This article belongs to the Special Issue Deciphering Epigenetic Signature in Human Health and Disease)
Abstract
:Organ-specific autoimmune diseases, such as type 1 diabetes, are believed to result from T-cell-mediated damage of the target tissue. The immune-mediated tissue injury, in turn, is known to depend on complex interactions between genetic and environmental factors. Nevertheless, the mechanisms whereby environmental factors contribute to the pathogenesis of autoimmune diseases remain elusive and represent a major untapped target to develop novel strategies for disease prevention. Given the impact of the early environment on the developing immune system, epigenetic changes induced by maternal factors during fetal life have been linked to a likelihood of developing an autoimmune disease later in life. In humans, DNA methylation is the epigenetic mechanism most extensively investigated. This review provides an overview of the critical role of DNA methylation changes induced by prenatal maternal conditions contributing to the increased risk of immune-mediated diseases on the offspring, with a particular focus on T1D. A deeper understanding of epigenetic alterations induced by environmental stressors during fetal life may be pivotal for developing targeted prevention strategies of type 1 diabetes by modifying the maternal environment.
1. Introduction
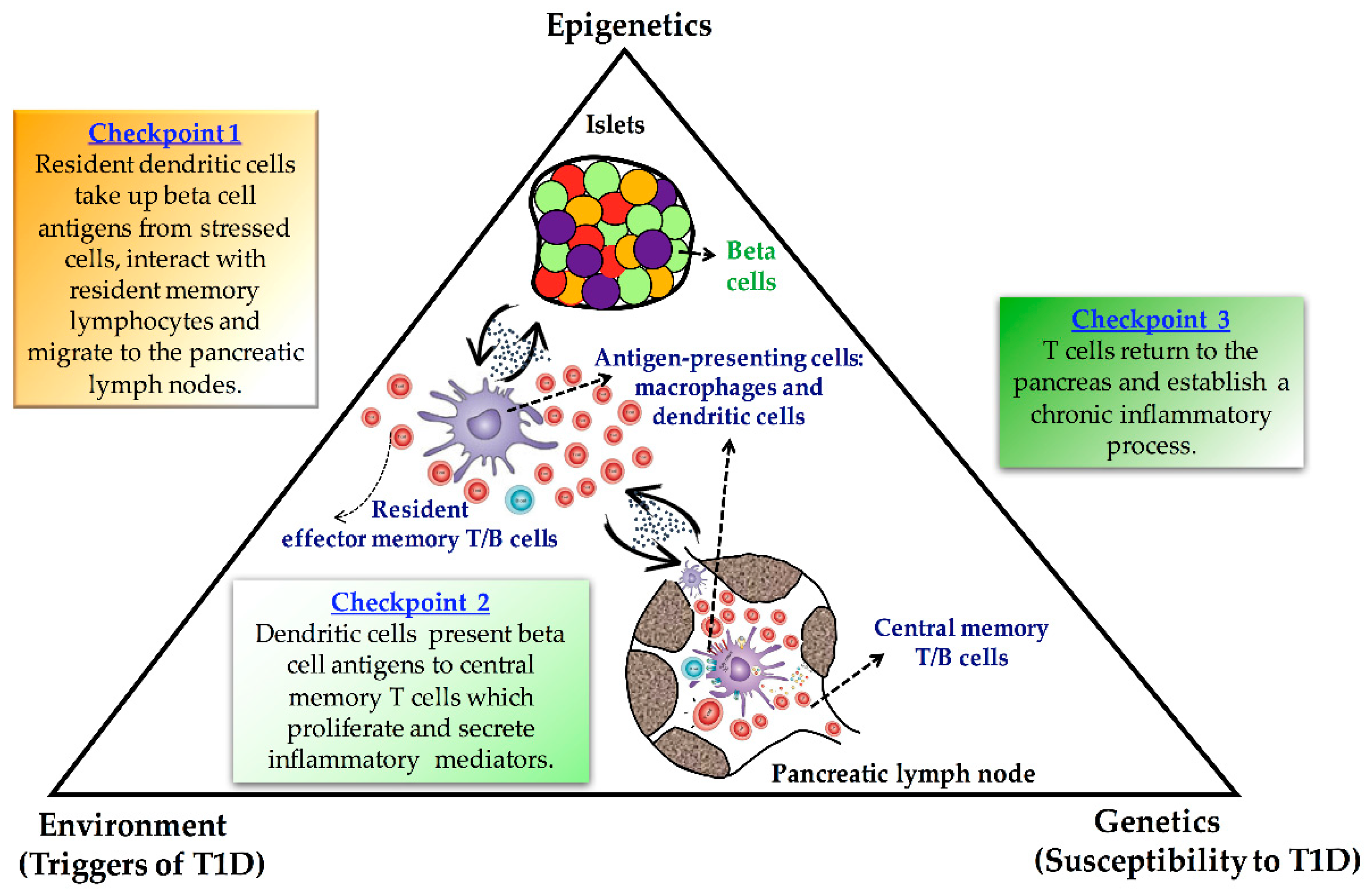
Type 1 diabetes (T1D) is considered a cell-mediated autoimmune disease characterized by insulin deficiency resulting from pancreatic beta cell dysfunction [1,2]. Although the discovery of islet cell autoantibodies in 1974 shaped thinking on the pathogenesis of T1D, leading to its classification as autoimmune in nature, the etiology of the disease remains unknown. Disease-associated genes are clearly important, but numerous studies, especially those on monozygotic twins, show that heritable factors account for only 30–50% of disease susceptibility [3,4]. These findings suggest that, besides genetic contribution, environmental influences largely determine the penetrance of T1D in a genetically susceptible population. Attention in the research community has therefore focused on two major questions: (i) what are the immune mechanisms that lead to T1D, and (ii) how does interaction with environmental factors contribute to these? Addressing question (i), it is known that the immune mechanisms that lead to disease involve the generation of islet autoreactive, pro-inflammatory T cells. This process, in turn, is known to depend upon activated dendritic cells. Addressing question (ii), increasing epidemiological evidence has linked environmental agents such as diet, microbial burden, drugs, exposure to chemicals and pollutants, or country latitude with the widespread prevalence of T1D over the last decades [5,6,7,8]. Many of these environmental factors display their role in influencing disease susceptibility through changes in gene expression without altering the DNA sequence, which has been termed epigenetics [9]. Thus, epigenetic processes most probably constitute a key mechanism that bridges the gap between environmental and genetic factors in the autoimmune destruction of the pancreatic beta cells (Figure 1).
The major epigenetic mechanisms include DNA methylation, histone protein posttranslational modifications, noncoding RNA regulation, and RNA editing [10]. In mammalian species, including humans, DNA methylation is the epigenetic mechanism most extensively investigated and has a critical role in controlling gene expression [11]. Since cell type-specific DNA methylation patterns are established during embryogenesis and fetal development through a programmed process, the prenatal stages represent windows of potential vulnerability to environmental exposure-related epigenetic alterations [12]. This review briefly discusses evidence on DNA methylation alterations induced by in utero environment that may affect the risk of immune-mediated diseases on the offspring, focusing on T1D.
2. Fetal Epigenetic Imprinting and Maternal Factors
Genomic imprinting refers to a parent-to-offspring transmission, where epigenetic mechanisms restrict gene expression to a single allele determined by parental origin. Thus, the control of gene expression by epigenetic inheritance confers a parent-of-origin-specific mark [13,14,15]. It has long been recognized that DNA methylation is the main mechanism responsible for establishing the imprint on one of the parental chromosomes. In humans, DNA methylation primarily occurs at cytosines in CG dinucleotides (commonly annotated as CpG, where ‘p’ represents the phosphodiester bond linking cytosine- and guanosine-containing nucleotides). Most gene promoter regions contain these CpG-rich stretches of DNA (≈500 bp), called CpG-island, and almost half of the human genes initiate transcription from CpG-islands [16]. As a consequence, methylation of promoter CpG islands is associated with transcriptional repression [17].
Although the diploidic state confers protection towards the consequences of genetic aberration in one gene copy during embryogenesis and fetal development, approximately 1% of the human protein-coding genome is imprinted [18]. Most of these genes are organized in clusters and expressed in the placenta [19,20]. As a direct consequence of fetal genomic imprinting, the offspring’s final phenotype is a result not only of gene sequence variation per se, but also of structural epigenetic modifications, partially obscuring any genotype–phenotype association. Hence, along with mitochondrial heritability and changes induced by in utero environment, imprinting may help explain how parent-of-origin transmission influences offspring phenotype [21]. Importantly, since this epigenetic gene-marking phenomenon occurs in germline cells, genomic imprinting modifications can be stably transmitted to several generations of cells until they are reset or lost under specific conditions [22,23].
3. Non-Imprinting Epigenetic Changes in Prenatal Life
Besides genomic imprinting, epigenetic modifications also occur in non-imprinted genes due to exposure to environmental factors, which exert their action predominantly by inducing different methylation profiles in CpG islets of the gene [24,25,26,27]. Like genomic imprinting, non-imprinting-related epigenetic changes are stable and heritable across generations of cells and organisms [28,29,30]. Therefore, non-imprinting epigenetic changes can be viewed as a functionally adaptive rearrangement of gene expression under environmental pressure. Fetal exposures to environmental and maternal factors may induce permanent physiological changes, termed “programming”, potentially leading to a variety of diseases later in life [31,32,33,34,35]. Indeed, both animal [36,37,38] and human [39,40,41] studies have indicated that environmental exposure experienced in utero may determine offspring phenotypic outcomes through epigenetic modulation of gene transcription. For example, it has been shown that maternal diet and nutrition patterns early in life predispose to increased cardiovascular risk, metabolic disorders, and immune impairment [42,43,44]. Moreover, insufficient intake of fruits and vegetables and high consumption of modern processed foods during pregnancy have been associated with systemic low-grade systematic inflammation [45,46]. Such maternal inflammation is believed to pass an inflammatory ‘code’ through epigenetic modifications to the offspring and influence the programming of the offspring’s immune system [47].
Although the existence of a “legacy” leading to permanent effects of in utero and early-life environmental exposures on unfavorable outcomes later in life has been demonstrated in prospective studies, only recently has it been recognized that these effects are mediated through epigenetic mechanisms. Thus, a genotype–phenotype mismatch could be partially attributable to external pressures that can reprogram the expression of genes related to immunity and metabolism, thereby leading to a pathological phenotype. This fits with data showing that, in mice, a maternal diet supplemented with methyl donors enhanced the severity of allergic airway disease inherited transgenerationally [48]. Therefore, changes in the DNA methylation pattern of target genes during the embryonic period could modify allergic airway disease’s heritable risk. Additionally, there is evidence in humans that site-specific changes in epigenetic marking at the Retinoid-X Receptor alpha (RXRA) promoter region in umbilical cord blood cells are negatively associated with maternal carbohydrate intake during early pregnancy. Remarkably, these epigenetic modifications correlated with childhood adiposity later in life [49]. Together, these studies support a link between non-imprinted epigenetics in fetal development and phenotypic changes in offspring.
4. Epigenetic Changes of Immune-Related Genes
A growing number of studies highlight the importance of epigenetic mechanisms in hematopoietic lineage choice [50], antigen-receptor rearrangement [51], allelic exclusion [52], and immune responses to pathogens [53,54]. Along with controlling T cell central tolerance in the thymus by processes related to methylated histones and miRNAs, epigenetic mechanisms also regulate peripheral tolerance. For example, it has been shown that the activity of Foxp3 protein, the master regulator for Treg cell development and immunosuppressive function, is regulated post-translationally by acetylation and deacetylation [55,56,57]. Indeed, alterations in this process lead to insufficiency in natural Tregs and impaired development and function of inducible Tregs [58,59]. In support of this notion, a study in mice demonstrated that treatment with the DNA methylation inhibitor 5-azacytidine causes experimentally induced autoimmune arthritis [60]. Studies in monozygotic twins discordant for psoriasis have also shown that changes in DNA methylation between unaffected and affected twins correlated with changes in the expression of genes involved in the immune response [61]. Finally, one study found that monozygotic twins discordant for T1D exhibit significant differences in methylation patterns in CD14+ monocytes [62].
Epidemiological studies investigating the effects of maternal stress on offspring have shown that prenatal exposure to maternal adverse life events results in lasting and broad functional DNA methylation changes in innate and adaptive immune genes and genes involved in glucose metabolism. In particular, the objective prenatal maternal stress experienced during the 1998 Quebec Ice Storm directly correlated with a specific DNA methylation pattern in CD3+ T cells, saliva, and whole peripheral blood of offspring, almost thirteen years after birth [63]. Interestingly, the long-lasting impact of traumatic stress on the methylation pattern of CpG sites was even detected in several genes involved in both T1D [63] and type 2 diabetes (T2D) pathways [63,64].
Given these findings, it appears plausible that fetal epigenetic changes triggered during the prenatal environment may induce long-lasting effects on offspring outcomes in later life. However, the lack of fetal cord blood cells did not allow these authors to demonstrate whether a stress-induced DNA methylation profile may already occur in the prenatal period or early in life.
5. Fetal Epigenetic Changes: Studies on Cord Blood Cells
Different conditions may influence the fetal immune system’s development during pregnancy and, consequently, the risk of immune-related diseases. For example, maternal obesity (body mass index (BMI) ≥ 30 kg/m2) has been associated with several alterations in the perinatal immune system. In particular, maternal BMI during pre-pregnancy or early gestation affects DNA methylation in the offspring’s peripheral blood cells [65,66]. In accordance, Sureshchandra et al. [67] revealed that maternal pre-pregnancy BMI correlates inversely with overall methylation levels in cord blood samples. Interestingly, the most significant methylation changes occurred within genes associated with cancer (WNT16) and diabetes (BTN3AI). An important study by Wilson et al. [68] showed a reduction of eosinophils and CD4+ T helper cells, reduced monocytes and dendritic cell responses to Toll-like receptor ligands, as wells as increased plasma IFN-γ and IL-6 levels in cord blood cells of newborns from obese in comparison with those from lean mothers.
Other evidence supporting that maternal lifestyle and environmental exposures can influence the epigenetic programming of the offspring’s immune system is provided by Nemoda et al. [69], who found that maternal depression affects T cell DNA methylation profiles in the offspring. However, the authors did not see significant DNA methylation changes associated with depression in T lymphocytes from the antepartum maternal samples. Therefore, changes in the prenatal environment induced by maternal depression may exert long-lasting effects on immune functions in the periphery and the central nervous system of the offspring.
Other researchers have found that in-utero exposures to environmental factors, such as cigarette smoking during pregnancy [70,71,72], maternal diet [73,74,75,76], and microbial exposures [77,78], also have a dramatic influence on the risk of allergic disease in the offspring by altering fetal lung development and immune function [79]. Results from “The Managing Asthma in Pregnancy (MAP) Study” provided the first demonstration that exposure to maternal asthma during pregnancy is associated with alterations in the DNA methylation profile of infants’ peripheral blood. Among the sixty-eight genes differentially methylated, key regulatory pathways concerning developmental, metabolic, and inflammatory processes were most involved [80]. Interestingly, prenatal exposure to maternal cigarette smoke has been linked to the abnormal DNA methylation status of the 5′-CpG-island in the thymic stromal lymphopoietin (TSLP [81]), a key immune cytokine gene involved in the pathogenesis of asthma [82,83], atopic dermatitis [84], and pediatric eosinophilic esophagitis [85].
Taken together, these studies illustrate that epigenetic changes induced by prenatal maternal conditions such as maternal obesity, maternal depression, or cigarette smoking during pregnancy confer an increased risk of immune-mediated diseases in the offspring. In this regard, a particular focus should be given to the study of maternal lifestyle factors in the development of autoimmune diseases, which are largely prevalent among women of reproductive age.
6. Epigenetics in T1D: The Missing Piece of the Puzzle
In T1D, insulin-producing beta cells are destroyed by autoimmune mechanisms, resulting in insulin-deficiency and hyperglycemia [1,2]. Although it is believed that genetic and environmental factors play critical roles in T1D development, a long-term puzzle in the diabetes field has been how autoreactive T cells mistakenly destroy beta cells. Thus, dissecting the epigenetic architecture at the crossroads between genes and the environment could reveal the missing piece of the T1D puzzle (Figure 1).
6.1. Genetics
Over the past thirty years, extensive population studies have provided an explanation for nearly 80% of the heritability of T1D [86,87]. The strongest genetic risk factor for T1D is attributable to the Human Leukocyte Antigen (HLA) class II alleles, which account for up to 50% of genetic T1D risk [88,89,90,91]. Outside of the class II region, the strongest susceptibility is conferred by HLA class I allele B*39 [92]. Among non-HLA genes, some loci weakly contribute to disease onset, such as the insulin gene (INS), tyrosine phosphatase non-receptor type 22 (PTPN22), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), interleukin 2 receptor α (IL2RA), C-type lectin domain containing 16A (CLEC16A), cathepsin H (CTSH), interferon-induced with helicase C domain 1 (IFIH1), CAPSL-IL7R, Th1 transcription factor STAT4, tyrosine phosphatase non-receptor type 2 (PTPN2), and others [93,94,95,96].
6.2. Genome Imprinting
In addition to the predisposing genes identified, the effect of a small number of T1D-associated genes may be mediated through imprinting. It is thus conceivable that impaired fetal imprinting can lead to T1D development in several conditions. Indeed, genetic imprinting on chromosome region 6q24 PLAGL1-HYMAI is associated with transient neonatal diabetes, a rare form of diabetes whereby an increased dosage at the chromosome 6q24 region leads to impaired glucose regulation and diabetes. Notably, near half of the cases of neonatal diabetes have the condition for life [97,98,99,100,101,102]. Moreover, impaired genomic imprinting seems to influence the development of polygenic T1D [103] and T2D [104].
6.3. Non-Imprinting Epigenetic Changes
Despite evidence linking genetics with disease T1D susceptibility, they are not likely the primary driver. It should be noted that T1D incidence has increased worldwide over the last few decades at an average of ~3% to 5% per year [105], which is too rapid to be explained only by enhanced genetic disease susceptibility in the background population. If this trend continues, T1D incidence will double in the next 20 years. Interest has therefore focused on environmental factors that might trigger and/or accelerate the disease. The role of environmental factors in T1D development is also supported by a plethora of findings demonstrating that the concordance rate in monozygotic twins for T1D ranges from 13% to 60% according to the age at disease onset, insulin genotype, and latitude [106,107,108,109,110,111]. Given the evident importance of the overriding environmental influence on T1D development, it appears plausible that environmental epigenetic modifications during prenatal development may be one of the factors that are associated with an increased risk for developing T1D [112]. In particular, exposure to an adverse in utero milieu may induce epigenetic effects on DNA, permanently modifying the expression of immune genes and islet cell function-related genes. Perhaps the most compelling evidence to date on the influence of the intrauterine environment on T1D risk comes from a “migration” study performed in Sweden, a country with the second-highest level of T1D in the world. This study demonstrated that being born in Sweden increases the risk for T1D even in children with an origin in low-incidence countries, whereas T1D risk did not vary in children immigrating to Sweden at an early age for adoption and immediately introduced into Swedish families [113]. In line with this observation, data from the Skåne area in the southern part of Sweden suggested that high exposure to air pollution (i.e., nitrogen oxides and ozone) during pregnancy represents a risk factor of developing T1D in offspring [114]. Indeed, evidence exists that nitrogen oxides act as an epigenetic regulator of gene expression by controlling histone posttranslational modifications [115]. Moreover, a study demonstrated that disruption of miRNA expression profiles by ozone inhalation is associated with inflammatory and immune response signaling [116]. Consistent with this, epidemiological studies have shown that children exposed to smoking during fetal life are at higher risk of developing T1D in childhood [117].
Decades of research have provided evidence suggesting that certain viruses, especially human enterovirus, are putative environment-derived disease modifiers in T1D [118,119,120]. Remarkably, maternal enteroviral infection during pregnancy has been considered a risk factor for T1D onset during childhood and adolescence in several studies [121,122,123,124,125,126,127,128,129,130,131,132,133,134]. In keeping with the crucial role of epigenetic modification in early development, it is tempting to speculate that maternal viral infection during pregnancy can give rise to stable changes in immune-related genes by epigenetic mechanisms. This is an attractive idea because, if confirmed, the infection-induced epigenetic modification could contribute significantly to the offspring’s risk of T1D later in life. In support of this notion, recent studies have shown that enterovirus can alter miRNA-directed suppression of pro-inflammatory factors within pancreatic beta cells [135] and pancreatic ductal-like cells [136,137]. Likewise, Rhinovirus (another important member of the Picornaviridae family, as human enteroviruses) affects both the methylation status and the expression of pro-inflammatory cytokines in epithelial cells [138]. Hence, non-imprinting epigenetic modifications induced by maternal viral infections may represent one mechanism through which viruses contribute to T1D.
6.4. DNA Methylation Signature in T1D
Although the non-structural genetic component of T1D susceptibility remains to be determined, remarkable progress has been made in elucidating the epigenetics of T1D. As with other autoimmune diseases, DNA methylation has been the most extensively studied epigenetic signature in T1D. The major studies are shown in Table 1.
Studies in monozygotic twins have been critical to strengthening the hypothesis that DNA methylation is involved in T1D etiology. A genome-wide DNA methylation analysis of monocytes from monozygotic twins discordant for T1D conducted by Rakyan and colleagues [62] revealed the presence of T1D-specific methylation variable positions (T1D-MVP) in the diabetic co-twins. They found that the epigenetic changes in autoantibodies-positive individuals occurred before the diagnosis of T1D, which excludes the possibility of an association between methylation profile and post-disease dysmetabolic environment. Remarkably, T1D-MVP-associated genes included several genes known to be associated with T1D or immune responses, such as HLA class II, HLA-DQB1, Regulatory Factor X-Associated Protein (RFXAP), Nuclear Factor Kappa B Subunit 1 (NFKB1A), Tumor Necrosis Factor (TNF), and Glutamate Decarboxylase 2 (GAD2). Of note, the GAD2 gene encodes the islet cell-specific (65 kDa) form of glutamic acid decarboxylase (GAD65), which is one of the major autoantigens in T1D [150]. Moreover, an epigenome-wide association study in 52 monozygotic twin pairs discordant for T1D in three immune effector cell types (that is, CD4+ T cells, CD19+ B cells, and CD14+CD16− monocytes) showed significant enrichment of differentially variable CpG positions in T1D twins when compared with their healthy co-twins and healthy controls [144]. It is also important to note that non-twin studies using T1D patients and healthy individuals have demonstrated differences in methylation profiles between T1D patients and controls [141,151]. Indeed, recent research has shown that DNA methylation is involved in regulating the genetic and environmental influence of T1D at the CTSH locus [149].
6.5. Maternal Autoantibodies and Their Role in T1D
Although the relationship between maternal antibody transmission and antibody-mediated diseases such as systemic lupus erythematosus [152,153] and thyroiditis [154,155] is widely recognized, the pathogenic role for maternal autoantibodies in T cell-mediated autoimmune diseases remains controversial. Part of this uncertainty is due to studies in nonobese diabetic (NOD) mice, a well-known animal model for T1D, showing that maternal transmission of beta cell-specific autoantibodies is necessary for inducing [156] or accelerating [157] the disease development. In contrast, more recent studies provide evidence that fetal exposure to insulin autoantibodies (IAA) did not increase the risk of diabetes development in NOD mice [158]. In humans, epidemiological data are also contradictory. Some studies have reported an increased frequency of beta cell-specific autoantibodies in cord blood of children who developed T1D, suggesting that this might represent a possible risk factor [159,160]. In contrast, the German BABYDIAB Study has demonstrated that offspring born to mothers with T1D who were positive for autoantibodies against islet-specific autoantigens linked to T1D (namely GAD65 and/or tyrosine phosphatase-related islet antigen 2 tyrosine phosphatase-related islet antigen 2, IA-2) at birth were at lower risk of T1D than offspring who were autoantibody-negative. Notably, the risk remained reduced after adjustment for potential independent confounders, such as maternal diabetes duration, birth weight, and gestational age [161], suggesting a protective role of fetal exposure to islet autoantibodies against T1D in offspring. In support of this hypothesis, accumulating data from epidemiological studies have revealed that the risk of developing T1D is low in infants born to mothers with T1D [162,163,164,165,166,167].
In the context of the Diabetes Prediction Study in Skåne (DiPiS), we studied the inflammatory, autoantibodies, and lymphocyte profiles in cord blood cells of children born to mothers either with T1D, gestational diabetes, or healthy mothers [168]. Interestingly, cord blood from children born to mothers with T1D showed increased IL-1β, IL-8, and TNFα levels and a higher frequency of CD4+ CD25+ T cells. Particularly, the CD4+ CD25+ T cells’ increase correlated with the anti-GAD65 antibodies’ titer. Remarkably, early modifications of inflammatory and immune patterns were absent in children born to mothers with gestational diabetes and without the islets’ autoantibody [168]. These results rule out the possibility that early changes in the immune system may have been induced by other factors linked to maternal diabetes, such as hyperglycemia. Overall, these data suggest that fetal/early-in-life epigenetic mechanisms might be involved in the susceptibility to islets’ autoimmunity and T1D.
7. Conclusions
The studies outlined here provide converging evidence to suggest that maternal factors are associated with increased risk for developing autoimmune diseases, such as T1D, through epigenetic changes in fetal life. However, there remains skepticism about whether in utero exposure to environmental factors may modify the immune profile and, subsequently, the risk of T1D later in life through epigenetic modifications. Therefore, additional birth cohort studies with long-term follow-up are needed to gain a more comprehensive understanding of how environmental cues during intrauterine life modulate the developing immune system. The use of Guthrie cards, state-of-the-art automated platforms for high-throughput epigenomics, and single-cell genomics in cord blood samples in established prospective cohorts hold promise to facilitate our understanding of gene–environment interaction in early life [169]. Identification of epigenetic modifications induced by prenatal environmental exposures associated with a higher risk of autoimmune diseases and T1D later in life will be of utmost importance, as this may provide better for disease prevention strategies already in utero.
Author Contributions
Conceptualization: I.B., J.A., L.S. and C.M.C.; writing—original draft preparation: I.B. and L.S.; writing—review and editing: I.B., J.A., L.S. and C.M.C.; project administration: J.A.; funding acquisition: C.M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Swedish Research Council (Dnr 2018-03196), Barndiabetesfonden (The Swedish Child Diabetes Foundation), Diabetesfonden, Strategic Research Area Exodiab, Dnr 2009-1039, and by the Swedish Foundation for Strategic Research Dnr IRC15-0067 (LUDC-IRC). The article processing charges (APC) were partially funded by the Lund University’s APC-fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. All data included in this review have been previously published. If further specific data is needed, it may be provided by the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no competing financial interest. The funders had no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
References
- Roep, B.O. The role of T-cells in the pathogenesis of type 1 diabetes: From cause to cure. Diabetologia 2003, 46, 305–321. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Gauderman, W.J.; Cozen, W.; Hamilton, A.S.; Burnett, M.E.; Mack, T.M. Differential twin concordance for multiple sclerosis by latitude of birthplace. Ann. Neurol. 2006, 60, 56–64. [Google Scholar] [CrossRef]
- Vojdani, A. A Potential link between environmental triggers and autoimmunity. Autoimmune Dis. 2014, 437231. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.F.; Wang, G. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol. 2018, 7, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Wang, H. Environmental exposures and autoimmune diseases: Contribution of gut microbiome. Front. Immunol. 2020, 10, 3094. [Google Scholar] [CrossRef] [PubMed]
- Predieri, B.; Bruzzi, P.; Bigi, E.; Ciancia, S.; Madeo, S.F.; Lucaccioni, L.; Lughetti, L. Endocrine disrupting chemicals and type 1 diabetes. Int. J. Mol. Sci. 2020, 21, 2937. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Gibney, E.; Nolan, C. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y. Modern epigenetics methods in biological research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C.; Bourc’his, D. The discovery and importance of genomic imprinting. eLife 2018, 7, e42368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting Group. Genomic imprinting and physiological processes in mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock, T.J.; Paracchini, S.; Gardner, A. Genomic imprinting as a window into human language evolution. Bioessays 2019, 41, e1800212. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Schübeler, D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr. Opin. Cell. Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [Green Version]
- Kaneko-Ishino, T.; Ishino, F. Evolution of viviparity in mammals: What genomic imprinting tells us about mammalian placental evolution. Reprod. Fertil. Dev. 2019, 31, 1219–1227. [Google Scholar] [CrossRef]
- Hanna, C.W. Placental imprinting: Emerging mechanisms and functions. PLoS Genet. 2020, 16, e1008709. [Google Scholar] [CrossRef]
- Millership, S.J.; Van de Pette, M.; Withers, D.J. Genomic imprinting and its effects on postnatal growth and adult metabolism. Cell. Mol. Life. Sci. 2019, 76, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Trasler, J.M. Gamete imprinting: Setting epigenetic patterns for the next generation. Reprod Fertil. Dev. 2006, 18, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thamban, T.; Agarwaal, V.; Khosla, S. Role of genomic imprinting in mammalian development. J. Biosci. 2020, 45, 20. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Dolinoy, D.C.; Mancuso, P. Perinatal bisphenol A exposures increase production of pro-inflammatory mediators in bone marrow-derived mast cells of adult mice. Immunotoxicology 2014, 11, 205–212. [Google Scholar] [CrossRef]
- Jahreis, S.; Trump, S.; Bauer, M.; Bauer, T.; Thürmann, L.; Feltens, R.; Wang, Q.; Gu, L.; Grützmann, K.; Röder, S.; et al. Maternal phthalate exposure promotes allergic airway inflammation over 2 generations through epigenetic modifications. J. Allergy Clin. Immunol. 2018, 141, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Ringh, M.V.; Hagemann-Jensen, M.; Needhamsen, M.; Kular, L.; Breeze, C.E.; Sjöholm, L.K.; Slavec, L.; Kullber, S.; Wahlström, J.; Grunewald, J.; et al. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine 2019, 46, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Puttabyatappa, M.; Zeng, L.; Vazquez, D.; Pennathur, S.; Padmanabhan, V. Developmental programming: Prenatal bisphenol A treatment disrupts mediators of placental function in sheep. Chemosphere 2020, 243, 125301. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Non-genomic transgenerational inheritance of disease risk. Bioessays 2007, 29, 145–154. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr. Res. 2007, 61, 5R–10R. [Google Scholar] [CrossRef]
- Miska, E.A.; Ferguson-Smith, A.C. Transgenerational inheritance: Models and mechanisms of non-DNA sequence-based inheritance. Science 2016, 354, 59–63. [Google Scholar] [CrossRef]
- Barker, D.J. In utero programming of chronic disease. Clin. Sci. 1998, 95, 115–128. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, L. Epigenetics and fetal metabolic programming: A call for integrated research on larger cohorts. Diabetes 2013, 62, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Zierath, J.R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat. Rev. Endocrinol. 2016, 12, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Dong, H.; Becker, A.S.; Dapito, D.H.; Modica, S.; Grandl, G.; Opitz, L.; Efthymiou, V.; Straub, L.G.; Sarker, G.; et al. Cold-induced epigenetic programming of the sperm enhances brown adipose tissue activity in the offspring. Nat. Med. 2018, 24, 1776. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Hiramatsu, Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef] [Green Version]
- Masuyama, H.; Mitsui, T.; Nobumoto, E.; Hiramatsu, Y. The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology 2015, 156, 2482–2491. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Xiong, J.W.; Zhang, Y.J.; Cao, X.Y.; Sun, P.; Wu, J.; Chen, L. Exposure of pregnant mice to triclosan causes hyperphagic obesity of offspring via the hypermethylation of proopiomelanocortin promoter. Arch. Toxicol. 2019, 93, 547–558. [Google Scholar] [CrossRef]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [Green Version]
- Hjort, L.; Martino, D.; Grunnet, L.G.; Naeem, H.; Maksimovic, J.; Olsson, A.H.; Zhang, C.; Ling, C.; Olsen, S.F.; Saffery, R.; et al. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI Insight 2018, 3, e122572. [Google Scholar] [CrossRef]
- Caffrey, A.; Irwin, R.E.; McNulty, H.; Strain, J.J.; Lees-Murdock, D.J.; McNulty, B.A.; Ward, M.; Walsh, C.P.; Pentieva, K. Gene-specific DNA methylation in newborns in response to folic acid supplementation during the second and third trimesters of pregnancy: Epigenetic analysis from a randomized controlled trial. Am. J. Clin. Nutr. 2018, 107, 566–575. [Google Scholar] [CrossRef]
- Block, T.; El-Osta, A. Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis 2017, 266, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Dolk, H.; McCullough, N.; Callaghan, S.; Casey, F.; Craig, B.; Given, J.; Loane, M.; Lagan, B.M.; Bunting, B.; Boyle, B.; et al. Risk factors for congenital heart disease: The Baby Hearts Study, a population-based case-control study. PLoS ONE 2020, 15, e0227908. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2021, 70, 29–49. [Google Scholar] [CrossRef]
- Hosseini, B.; Berthon, B.S.; Saedisomeolia, A.; Starkey, M.R.; Collison, A.; Wark, P.A.B.; Wood, L.G. Effects of fruit and vegetable consumption on inflammatory biomarkers and immune cell populations: A systematic literature review and meta-analysis. Am. J. Clin. Nutr. 2018, 108, 136–155. [Google Scholar] [CrossRef] [Green Version]
- Palatnik, A.; Moosreiner, A.; Olivier-Van Stichelen, S. Consumption of non-nutritive sweeteners during pregnancy. Am. J. Obstet. Gynecol. 2020, 223, 211–218. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Hollingsworth, J.W.; Maruoka, S.; Boon, K.; Garantziotis, S.; Li, Z.; Tomfohr, J.; Bailey, N.; Potts, E.N.; Whitehead, G.; Brass, D.M.; et al. In utero supplementation with methyl donors enhances allergic airway disease in mice. J. Clin. Investig. 2008, 118, 3462–3469. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Kurotaki, D.; Kawase, W.; Sasaki, H.; Nakabayashi, J.; Nishiyama, A.; Morse, H.C., 3rd; Ozato, K.; Suzuki, Y.; Tamura, T. Epigenetic control of early dendritic cell lineage specification by the transcription factor IRF8 in mice. Blood 2019, 133, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Spicuglia, S.; Zacarias-Cabeza, J.; Pekowska, A.; Ferrier, P. Epigenetic regulation of antigen receptor gene rearrangement. F1000 Biol. Rep. 2010, 2, 23. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat. Rev. Immunol. 2004, 4, 753–761. [Google Scholar] [CrossRef]
- Gutierrez, M.J.; Nino, G.; Hong, X.; Wang, X. Epigenomics and early life human humoral immunity: Novel paradigms and research pportunities. Front. Immunol. 2020, 11, 1766. [Google Scholar] [CrossRef] [PubMed]
- Crimi, E.; Benincasa, G.; Figueroa-Marrero, N.; Galdiero, M.; Napoli, C. Epigenetic susceptibility to severe respiratory viral infections and its therapeutic implications: A narrative review. Br. J. Anaesth. 2020, 125, 1002–1017. [Google Scholar] [CrossRef]
- Kwon, H.S.; Lim, H.W.; Wu, J.; Schnölzer, M.; Verdin, E.; Ott, M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J. Immunol. 2012, 188, 2712–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, G.; Song, X.; Fujimoto, S.; Piccirillo, C.A.; Nagai, Y.; Greene, M.I. Foxp3 Posttranslational modifications and Treg suppressive activity. Front. Immunol. 2019, 10, 2486. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yang, C.; Pan, F. Posttranslational regulations of Foxp3 in Treg cells and their therapeutic applications. Front. Immunol. 2021, 12, 626172. [Google Scholar] [CrossRef]
- Beyer, M.; Thabet, Y.; Müller, R.U.; Sadlon, T.; Classen, S.; Lahl, K.; Basu, S.; Zhou, X.; Bailey-Bucktrout, S.L.; Krebs, W.; et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat. Immunol. 2011, 12, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Bettini, M.L.; Pan, F.; Bettini, M.; Finkelstein, D.; Rehg, J.E.; Floess, S.; Bell, B.D.; Ziegler, S.F.; Huehn, J.; Pardoll, D.M.; et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity 2012, 36, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Tóth, D.M.; Ocskó, T.; Balog, A.; Markovics, A.; Mikecz, K.; Kovács, L.; Jolly, M.; Bukiej, A.A.; Ruthberg, A.D.; Vida, A.; et al. Amelioration of autoimmune arthritis in mice treated with the DNA methyltransferase inhibitor 5′-Azacytidine. Arthritis Rheumatol. 2019, 71, 1265–1275. [Google Scholar] [CrossRef] [Green Version]
- Gervin, K.; Vigeland, M.D.; Mattingsdal, M.; Hammerø, M.; Nygård, H.; Olsen, A.O.; Brandt, I.; Harris, J.R.; Undlien, D.E.; Lyle, R. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: Identification of epigenetically dysregulated genes. PLoS Genet. 2012, 8, e1002454. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.A.; Manfras, B.; et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lei, L.; Massart, R.; Suderman, M.J.; Machnes, Z.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS ONE 2014, 9, e107653. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lei, L.; Dancause, K.N.; Elgbeili, G.; Massart, R.; Szyf, M.; Liu, A.; Laplante, D.P.; King, S. DNA methylation mediates the impact of exposure to prenatal maternal stress on BMI and central adiposity in children at age 13½ years: Project Ice Storm. Epigenetics 2015, 10, 749–761. [Google Scholar] [CrossRef]
- Sen, S.; Iyer, C.; Klebenov, D.; Histed, A.; Aviles, J.A.; Meydani, S.N. Obesity impairs cell-mediated immunity during the second trimester of pregnancy. Am. J. Obstet. Gynecol. 2013, 208, 139.e1–139.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, H.; Xu, Y.; An, C.; Zhong, L.; Wang, X.; Zhang, Y.; Chen, H.; Zhang, J.; Wang, Z. Fetal growth is associated with maternal fasting plasma glucose at first prenatal visit. PLoS ONE 2014, 9, e116352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureshchandra, S.; Wilson, R.M.; Rais, M.; Marshall, N.E.; Purnell, J.Q.; Thornburg, K.L.; Messaoudi, I. Maternal Pregravid Obesity Remodels the DNA Methylation Landscape of Cord Blood Monocytes Disrupting Their Inflammatory Program. J. Immunol. 2017, 199, 2729–2744. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.M.; Marshall, N.E.; Jeske, D.R.; Purnell, J.Q.; Thornburg, K.; Messaoudi, I. Maternal obesity alters immune cell frequencies and responses in umbilical cord blood samples. Pediatr. Allergy Immunol. 2015, 26, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Nemoda, Z.; Massart, R.; Suderman, M.; Hallett, M.; Li, T.; Coote, M.; Cody, N.; Sun, Z.S.; Soares, C.N.; Turecki, G.; et al. Maternal depression is associated with DNA methylation changes in cord blood T lymphocytes and adult hippocampi. Transl. Psychiatry 2015, 5, e545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Gundavarapu, S.; Peña-Philippides, J.; Rir-Sima-ah, J.; Mishra, N.; Wilder, J.; Langley, R.J.; Smith, K.R.; Sopori, M.L. Prenatal secondhand cigarette smoke promotes Th2 polarization and impairs goblet cell differentiation and airway mucus formation. J. Immunol. 2011, 187, 4542–4552. [Google Scholar] [CrossRef]
- Savran, O.; Ulrik, C.S. Early life insults as determinants of chronic obstructive pulmonary disease in adult life. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drago, G.; Ruggieri, S.; Cuttitta, G.; La Grutta, S.; Ferrante, G.; Cibella, F. Determinants of Allergic Sensitization, Asthma and Lung Function: Results from a Cross-Sectional Study in Italian Schoolchildren. Int. J. Environ. Res. Public. Health 2020, 17, 5087. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Chiang, B.L.; Wang, L.C. Maternal Nutritional Status and Development of Atopic Dermatitis in Their Offspring. Clin. Rev. Allergy Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Venter, C.; Agostoni, C.; Arshad, S.H.; Ben-Abdallah, M.; Du Toit, G.; Fleischer, D.M.; Greenhawt, M.; Glueck, D.H.; Groetch, M.; Lunjani, N.; et al. Dietary factors during pregnancy and atopic outcomes in childhood: A systematic review from the European Academy of Allergy and Clinical Immunology. Pediatr. Allergy Immunol. 2020, 31, 889–912. [Google Scholar] [CrossRef]
- Ogawa, K.; Pak, K.; Yamamoto-Hanada, K.; Ishitsuka, K.; Sasaki, H.; Mezawa, H.; Saito-Abe, M.; Sato, M.; Yang, L.; Nishizato, M.; et al. Association between maternal vegetable intake during pregnancy and allergy in offspring: Japan Environment and Children’s Study. PLoS ONE 2021, 16, e0245782. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Alashkar Alhamwe, B.; Caraballo, L.; Ding, M.; Ferrante, A.; Garn, H.; Garssen, J.; Hii, C.S.; Irvine, J.; Llinás-Caballero, K.; et al. Perinatal and Early-Life Nutrition, Epigenetics, and Allergy. Nutrients 2021, 13, 724. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tu, C.; Yu, J.; Chen, M.; Tan, C.; Zheng, X.; Wang, Z.; Liang, Y.; Wang, K.; Wu, J.; et al. Maternal microbiome regulation prevents early allergic airway diseases in mouse offspring. Pediatr. Allergy Immunol. 2020, 31, 962–973. [Google Scholar] [CrossRef]
- Forsberg, A.; Abrahamsson, T.R.; Nilsson, L.; Ernerudh, J.; Duchén, K.; Jenmalm, M.C. Changes in peripheral immune populations during pregnancy and modulation by probiotics and ω-3 fatty acids. Sci. Rep. 2020, 10, 18723. [Google Scholar] [CrossRef]
- Fiuza, B.S.D.; Fonseca, H.F.; Meirelles, P.M.; Marques, C.R.; da Silva, T.M.; Figueiredo, C.A. Understanding Asthma and Allergies by the Lens of Biodiversity and Epigenetic Changes. Front. Immunol. 2021, 12, 623737. [Google Scholar] [CrossRef]
- Gunawardhana, L.P.; Baines, K.J.; Mattes, J.; Murphy, V.E.; Simpson, J.L.; Gibson, P.G. Differential DNA methylation profiles of infants exposed to maternal asthma during pregnancy. Pediatr. Pulmonol. 2014, 49, 852–862. [Google Scholar] [CrossRef]
- Wang, I.J.; Chen, S.L.; Lu, T.P.; Chuang, E.Y.; Chen, P.C. Prenatal smoke exposure, DNA methylation, and childhood atopic dermatitis. Clin. Exp. Allergy 2013, 43, 535–543. [Google Scholar] [CrossRef]
- Paplinska-Goryca, M.; Misiukiewicz-Stepien, P.; Proboszcz, M.; Nejman-Gryz, P.; Gorska, K.; Krenke, R. The Expressions of TSLP, IL-33, and IL-17A in Monocyte Derived Dendritic Cells from Asthma and COPD Patients are Related to Epithelial-Macrophage Interactions. Cells 2020, 9, 1944. [Google Scholar] [CrossRef] [PubMed]
- Paplińska-Goryca, M.; Nejman-Gryz, P.; Proboszcz, M.; Kwiecień, I.; Hermanowicz-Salamon, J.; Grabczak, E.M.; Krenke, R. Expression of TSLP and IL-33 receptors on sputum macrophages of asthma patients and healthy subjects. J. Asthma 2020, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berna, R.; Mitra, N.; Lou, C.; Wan, J.; Hoffstad, O.; Wubbenhorst, B.; Nathanson, K.L.; Margolis, D.J. TSLP and IL-7R Variants Are Associated with Persistent Atopic Dermatitis. J. Investig. Dermatol. 2021, 141, 446–450.e2. [Google Scholar] [CrossRef]
- O’Shea, K.M.; Aceves, S.S.; Dellon, E.S.; Gupta, S.K.; Spergel, J.M.; Furuta, G.T.; Rothenberg, M.E. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018, 154, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Jerram, S.T.; Leslie, R.D. The genetic architecture of type 1 diabetes. Genes 2017, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Hwang, J.S. Genetic aspects of type 1 diabetes. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Deutsch, A.J.; Lenz, T.L.; Onengut-Gumuscu, S.; Han, B.; Chen, W.-M.; Howson, J.M.M.; Todd, J.A.; de Bakker, P.I.W.; Rich, S.S.; et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet. 2015, 47, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Van Lummel, M.; Van Veelen, P.A.; De Ru, A.H.; Janssen, G.M.; Pool, J.; Laban, S.; Joosten, A.M.; Nikolic, T.; Drijfhout, J.W.; Mearin, M.L. Dendritic Cells Guide Islet Autoimmunity through a Restricted and Uniquely Processed Peptidome Presented by High-Risk HLA-DR. J. Immunol. 2016, 196, 3253–3263. [Google Scholar] [CrossRef]
- Van Lummel, M.; van Veelen, P.A.; de Ru, A.H.; Pool, J.; Nikolic, T.; Laban, S.; Joosten, A.; Drijfhout, J.W.; Gómez-Touriño, I.; Arif, S.; et al. Discovery of a Selective Islet Peptidome Presented by the Highest-Risk HLA-DQ8trans Molecule. Diabetes 2016, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Nejentsev, S.; Howson, J.; Walker, N.M.; Szeszko, J.; Field, S.F.; Stevens, H.E.; Reynolds, P.; Hardy, M.; King, E.; Masters, J.; et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007, 450, 887–892. [Google Scholar] [CrossRef]
- Pugliese, A.; Zeller, M.; Fernandez, A., Jr.; Zalcberg, L.J.; Bartlett, R.J.; Ricordi, C.; Pietropaolo, M.; Eisenbarth, G.S.; Bennett, S.T.; Patel, D.D. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 1997, 15, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Vafiadis, P.; Bennett, S.T.; Todd, J.A.; Nadeau, J.; Grabs, R.; Goodyer, C.G.; Wickramasinghe, S.; Colle, E.; Polychronakos, C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 1997, 15, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Muhlendahl, K.E.; Herkenhoff, H. Long-term course of neonatal diabetes. N. Engl. J. Med. 1995, 333, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Shield, J.P.; Gardner, R.J.; Wadsworth, E.J.; Whiteford, M.L.; James, R.S.; Robinson, D.O.; Baum, J.D.; Temple, I.K. Aetiopathology and genetic basis of neonatal diabetes. Arch. Dis. Child. Fetal Neonatal 1997, 76, F39–F42. [Google Scholar] [CrossRef] [Green Version]
- Arima, T.; Kamikihara, T.; Hayashida, T.; Kato, K.; Inoue, T.; Shirayoshi, Y.; Oshimura, M.; Soejima, H.; Mukai, T.; Wake, N. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005, 33, 2650–2660. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.M.; Mackay, D.J.; Edghill, E.L.; Gloyn, A.L.; Robinson, D.; Shield, J.P.; Temple, K.; Ellard, S.; Hattersley, A.T. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007, 56, 1930–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slingerland, A.S.; Shields, B.M.; Flanagan, S.E.; Bruining, G.J.; Noordam, K.; Gach, A.; Mlynarski, W.; Malecki, M.T.; Hattersley, A.T.; Ellard, S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 2009, 52, 1683–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, D.J.; Temple, I.K. Transient neonatal diabetes mellitus type 1. Am. J. Med. Genet. C. Semin. Med. Genet. 2010, 154C, 335–342. [Google Scholar] [CrossRef]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Jonasdottir, A.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Tuomilehto, J. The emerging global epidemic of type 1 diabetes. Curr. Diabetes Rep. 2013, 13, 795–804. [Google Scholar] [CrossRef]
- Kyvik, K.O.; Green, A.; Beck-Nielsen, H. Concordance rates of insulin dependent diabetes mellitus: A population based study of young Danish twins. BMJ 1995, 311, 913–917. [Google Scholar] [CrossRef] [Green Version]
- Fava, D.; Gardner, S.; Pyke, D.; Leslie, R.D. Evidence that the age at diagnosis of IDDM is genetically determined. Diabetes Care 1998, 21, 925–929. [Google Scholar] [CrossRef]
- Metcalfe, K.A.; Hitman, G.A.; Rowe, R.E.; Hawa, M.; Huang, X.; Stewart, T.; Leslie, R.D. Concordance for type 1 diabetes in identical twins is affected by insulin genotype. Diabetes Care 2001, 24, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Yu, L.; Hawa, M.; Mackenzie, T.; Pyke, D.A.; Eisenbarth, G.S.; Leslie, R.D. Heterogeneity of type I diabetes: Analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001, 44, 354–362. [Google Scholar] [CrossRef]
- Hyttinen, V.; Kaprio, J.; Kinnunen, L.; Koskenvuo, M.; Tuomilehto, J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: A nationwide follow-up study. Diabetes 2003, 52, 1052–1055. [Google Scholar] [CrossRef] [Green Version]
- Triolo, T.M.; Fouts, A.; Pyle, L.; Yu, L.; Gottlieb, P.A.; Steck, A.K.; Type 1 Diabetes TrialNet Study Group. Identical and Nonidentical Twins: Risk and factors involved in development of islet autoimmunity and type 1 diabetes. Diabetes Care 2019, 42, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Blunk, I.; Thomsen, H.; Reinsch, N.; Mayer, M.; Försti, A.; Sundquist, J.; Sundquist, K.; Hemminki, K. Genomic imprinting analyses identify maternal effects as a cause of phenotypic variability in type 1 diabetes and rheumatoid arthritis. Sci. Rep. 2020, 10, 11562. [Google Scholar] [CrossRef]
- Söderström, U.; Aman, J.; Hjern, A. Being born in Sweden increases the risk for type 1 diabetes-a study of migration of children to Sweden as a natural experiment. Acta Paediatr. 2012, 101, 73–77. [Google Scholar] [CrossRef]
- Malmqvist, E.; Larsson, H.E.; Jönsson, I.; Rignell-Hydbom, A.; Ivarsson, S.A.; Tinnerberg, H.; Stroh, E.; Rittner, R.; Jakobsson, K.; Swietlicki, E.; et al. Maternal exposure to air pollution and type 1 diabetes-Accounting for genetic factors. Environ. Res. 2015, 140, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, D.; Hickok, J.R.; Bovee, R.C.; Pham, V.; Mantell, L.L.; Bahroos, N.; Kanabar, P.; Cao, X.J.; Maienschein-Cline, M.; Garcia, B.; et al. Nitric oxide regulates gene expression in cancers by controlling histone posttranslational modifications. Cancer Res. 2015, 5, 1582. [Google Scholar] [CrossRef] [Green Version]
- Fry, R.C.; Rager, J.E.; Bauer, R.; Sebastian, E.; Peden, D.B.; Jaspers, I.; Alexis, N.E. Air toxics and epigenetic effects: Ozone altered microRNAs in the sputum of human subjects. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2014, 306, L1129–L1137. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, K.; Jönsson, I.; Malmqvist, E.; Larsson, H.E.; Rylander, L. Maternal smoking during pregnancy and offspring type 1 diabetes mellitus risk: Accounting for HLA haplotype. Eur J. Epidemiol. 2015, 30, 231–238. [Google Scholar] [CrossRef]
- Gundersen, E. Is diabetes of infectious origin? J. Infect. Dis. 1927, 41, 197–202. [Google Scholar] [CrossRef]
- Sarmiento, L.; Cubas-Dueñas, I.; Cabrera-Rode, E. Evidence of association between type 1 diabetes and exposure to enterovirus in Cuban children and adolescents. MEDICC Rev. 2013, 15, 29–32. [Google Scholar] [CrossRef]
- Geravandi, S.; Liu, H.; Maedler, K. Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D. Microorganisms 2020, 8, 1017. [Google Scholar] [CrossRef]
- Dahlquist, G.; Frisk, G.; Ivarsson, S.A.; Svanberg, L.; Forsgren, M.; Diderholm, H. Indications that maternal coxsackie B virus infection during pregnancy is a risk factor for childhood- onset IDDM. Diabetologia 1995, 38, 1371–1373. [Google Scholar] [CrossRef]
- Dahlquist, G.G.; Ivarsson, S.; Lingberg, B.; Forsgren, M. Maternal enteroviral infection in pregnancy is a risk factor for childhood IDDM-A population based case control study. Diabetes 1995, 44, 408–413. [Google Scholar] [CrossRef]
- Dalhquist, G.; Kalen, B. Time-space clustering of time at birth in childhood onset diabetes. Diabetes Care 1996, 19, 328–332. [Google Scholar] [CrossRef]
- Dahlquist, G. Viruses and other perinatal exposure as initiating events for β-cell destruction. Ann. Med. 1997, 29, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Dahlquist, G.G.; Bowman, J.E.; Juto, P. Enteroviral RNA and IGM antibodies in early pregnancy is a risk factor for childhood onset IDDM. Diabetes Care 1999, 22, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Viskari, H.R.; Roivainen, M.; Reunanen, A.; Pitkäniemi, J.; Sadeharju, K.; Koskela, P.; Hovi, T.; Leinikki, P.; Vilja, P.; Tuomilehto, J.; et al. Maternal first-trimester enterovirus infection and future risk of type 1 diabetes in the exposed fetus. Diabetes 2002, 51, 2568–2571. [Google Scholar] [CrossRef] [Green Version]
- Dahlquist, G.G.; Forsberg, J.; Hagenfeldt, L.; Boman, J.; Juto, P. Increased prevalence of enteroviral RNA in blood spots from newborn children who later developed type 1 diabetes: A population-based case-control study. Diabetes Care 2004, 27, 285–286. [Google Scholar] [CrossRef] [Green Version]
- Elfving, M.; Svensson, J.; Oikarinen, S.; Jonsson, B.; Olofsson, P.; Sundkvist, G.; Lindberg, B.; Lernmark, A.; Hyöty, H.; Ivarsson, S.A. Maternal enterovirus infection during pregnancy as a risk factor in offspring diagnosed with type 1diabetes between 15 and 30 years of age. Exp. Diabetes Res. 2008, 2008, 271958. [Google Scholar] [CrossRef] [Green Version]
- Rešić Lindehammer, S.; Honkanen, H.; Nix, W.A.; Oikarinen, M.; Lynch, K.F.; Jönsson, I.; Marsal, K.; Oberste, S.; Hyöty, H.; Lernmark, Å. Seroconversion to islet autoantibodies after enterovirus infection in early pregnancy. Viral Immunol. 2012, 25, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viskari, H.; Knip, M.; Tauriainen, S.; Huhtala, H.; Veijola, R.; Ilonen, J.; Simell, O.; Surcel, H.M.; Hyöty, H. Maternal enterovirus infection as a risk factor for type 1 diabetes in the exposed offspring. Diabetes Care 2012, 35, 1328–1332. [Google Scholar] [CrossRef] [Green Version]
- Penno, M.; Couper, J.J.; Craig, M.E.; Colman, P.G.; Rawlinson, W.D.; Cotterill, A.M.; Jones, T.W.; Harrison, L.C.; ENDIA Study Group. Environmental determinants of islet autoimmunity (ENDIA): A pregnancy to early life cohort study in children at-risk of type 1 diabetes. BMC Pediatr. 2013, 13, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.W.; Kim, K.; Rawlinson, W.D.; Craig, M.E. Maternal virus infections in pregnancy and type 1 diabetes in their offspring: Systematic review and meta-analysis of observational studies. Rev. Med. Virol. 2018, 28, e1974. [Google Scholar] [CrossRef] [PubMed]
- Wook Kim, K.; Allen, D.W.; Briese, T.; Couper, J.J.; Barry, S.C.; Colman, P.G.; Cotterill, A.M.; Davis, E.A.; Giles, L.C.; Harrison, L.C.; et al. Distinct gut virome profile of pregnant women with type 1 diabetes in the ENDIA study. Open Forum Infect. Dis. 2019, 6, ofz025. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.E.; Kim, K.W.; Isaacs, S.R.; Penno, M.A.; Hamilton-Williams, E.E.; Couper, J.J.; Rawlinson, W.D. Early-life factors contributing to type 1 diabetes. Diabetologia 2019, 62, 1823–1834. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Ho, A.; Alshabee-Akil, A.; Hardikar, A.A.; Kay, T.W.; Rawlinson, W.D.; Craig, M.E. Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 2016, 65, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alidjinou, E.K.; Engelmann, I.; Bossu, J.; Villenet, C.; Figeac, M.; Romond, M.B.; Sané, F.; Hober, D. Persistence of Coxsackievirus B4 in pancreatic ductal-like cells results in cellular and viral changes. Virulence 2017, 8, 1229–1244. [Google Scholar] [CrossRef]
- Engelmann, I.; Alidjinou, E.K.; Bertin, A.; Bossu, J.; Villenet, C.; Figeac, M.; Sane, F.; Hober, D. Persistent coxsackievirus B4 infection induces microRNA dysregulation in human pancreatic cells. Cell. Mol. Life Sci. 2017, 74, 3851–3861. [Google Scholar] [CrossRef] [PubMed]
- McErlean, P.; Favoreto, S., Jr.; Costa, F.F.; Shen, J.; Quraishi, J.; Biyasheva, A.; Cooper, J.J.; Scholtens, D.M.; Vanin, E.F.; De Bonaldo, M.F.; et al. Human rhinovirus infection causes different DNA methylation changes in nasal epithelial cells from healthy and asthmatic subjects. BMC Med. Genom. 2014, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Teschendorff, A.E.; Rakyan, V.K.; Maxwell, A.P.; Beck, S.; Savage, D.A. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genom. 2010, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Fradin, D.; Le, F.S.; Mille, C.; Naoui, N.; Groves, C.; Zelenika, D.; McCarthy, M.I.; Lathrop, M.; Bougnères, P. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS ONE 2012, 7, e36278. [Google Scholar] [CrossRef]
- Belot, M.P.; Fradin, D.; Mai, N.; Le Fur, S.; Zélénika, D.; Kerr-Conte, J.; Pattou, F.; Lucas, B.; Bougnères, P. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PLoS ONE 2013, 8, e68093. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Falhammar, H.; Gu, H.F.; Brismar, K. Epigenetic analyses of the insulin-like growth factor binding protein 1 gene in type 1 diabetes and diabetic nephropathy. Clin. Epigenet. 2014, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Agardh, E.; Lundstig, A.; Perfilyev, A.; Volkov, P.; Freiburghaus, T.; Lindholm, E.; Rönn, T.; Agardh, C.D.; Ling, C. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015, 13, 182. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef]
- Sklenarova, J.; Petruzelkova, L.; Kolouskova, S.; Lebl, J.; Sumnik, Z.; Cinek, O. Glucokinase gene may be a more suitable target than the insulin gene for detection of β cell death. Endocrinology 2017, 158, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Richardson, T.G.; McArdle, W.L.; Relton, C.L.; Gillespie, K.M.; Suderman, M.; Hemani, G. Identification of loci where DNA methylation potentially mediates genetic risk of type 1 diabetes. J. Autoimmun. 2018, 93, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Simmons, K.M.; Fouts, A.; Pyle, L.; Clark, P.; Dong, F.; Yu, L.; Usmani-Brown, S.; Gottlieb, P.; Herold, K.C.; Steck, A.K. Type 1 diabetes TrialNet Study Group. Unmethylated insulin as an adjunctive marker of β cell death and progression to type 1 diabetes in participants at risk for diabetes. Int. J. Mol. Sci. 2019, 20, 3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carry, P.M.; Vanderlinden, L.A.; Johnson, R.K.; Dong, F.; Steck, A.K.; Frohnert, B.I.; Rewers, M.; Yang, I.V.; Kechris, K.; Norris, J.M. DNA methylation near the INS gene is associated with INS genetic variation (rs689) and type 1 diabetes in the Diabetes Autoimmunity Study in the Young. Pediatr. Diabetes 2020, 21, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Stefan-Lifshitz, M.; Tomer, Y. Genetic and environmental factors regulate the type 1 diabetes gene CTSH via differential DNA methylation. J. Biol. Chem. 2021, 13, 100774. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.; Faresjö, M.; Hjorth, M.; Axelsson, S.; Chéramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Cepek, P.; Zajacova, M.; Kotrbova-Kozak, A.; Silhova, E.; Cerna, M. DNA methylation and mRNA expression of HLA-DQA1 alleles in type 1 diabetes mellitus. Immunology 2016, 148, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Van Kerckhove, C. Lupus erythematosus in childhood: Effect of maternal factors beyond neonatal disease? Clin. Rheumatol. 1999, 9, 168–170. [Google Scholar] [CrossRef]
- Erden, A.; Fanouriakis, A.; Kiliç, L.; Sari, A.; Armağan, B.; Bilgin, E.; Şener, Y.Z.; Hymabaccus, B.; Gürler, F.; Ceylan, S.; et al. Geoepidemiology and clinical characteristics of neonatal lupus erythematosus: A systematic literature review of individual patients’ data. Turk. J. Med. Sci. 2020, 50, 281–290. [Google Scholar] [CrossRef]
- Joshi, K.; Zacharin, M. Hyperthyroidism in an infant of a mother with autoimmune hypothyroidism with positive TSH receptor antibodies. J. Pediatr. Endocrinol. Metab. 2018, 31, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Delay, F.; Dochez, V.; Biquard, F.; Cheve, M.T.; Gillard, P.; Arthuis, C.J.; Winer, N. Management of fetal goiters: 6-year retrospective observational study in three prenatal diagnosis and treatment centers of the Pays De Loire Perinatal Network. J. Matern. Fetal Neonatal Med. 2020, 33, 2561–2569. [Google Scholar] [CrossRef]
- Greeley, S.A.; Katsumata, M.; Yu, L.; Eisenbarth, G.S.; Moore, D.J.; Goodarzi, H.; Barker, C.F.; Naji, A.; Noorchashm, H. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat. Med. 2002, 8, 399–402. [Google Scholar] [CrossRef]
- Silva, D.G.; Daley, S.R.; Hogan, J.; Lee, S.K.; The, C.E.; Hu, D.Y.; Lam, K.P.; Goodnow, C.C.; Vinuesa, C.G. Anti-islet autoantibodies trigger autoimmune diabetes in the presence of an increased frequency of islet-reactive CD4 T cells. Diabetes 2011, 60, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- Koczwara, K.; Ziegler, A.G.; Bonifacio, E. Maternal immunity to insulin does not affect diabetes risk in progeny of non-obese diabetic mice. Clin. Exp. Immunol. 2004, 136, 56–59. [Google Scholar] [CrossRef]
- Lindberg, B.; Ivarsson, S.A.; Landin-Olsson, M.; Sundkvist, G.; Svanberg, L.; Lernmark, A. Islet autoantibodies in cord blood from children who developed type I (insulin-dependent) diabetes mellitus before 15 years of age. Diabetologia 1999, 42, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgren, M.; Lynch, K.; Larsson, C.; Elding Larsson, H.; Diabetes Prediction in Skåne study group. Cord blood insulinoma-associated protein 2 autoantibodies are associated with increased risk of type 1 diabetes in the population-based Diabetes Prediction in Skåne study. Diabetologia 2015, 58, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczwara, K.; Bonifacio, E.; Ziegler, A.G. Transmission of maternal islet antibodies and risk of autoimmune diabetes in offspring of mothers with type 1 diabetes. Diabetes 2004, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Warram, J.H.; Krolewski, A.S.; Gottlieb, M.S.; Kahn, C.R. Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N. Engl. J. Med. 1984, 311, 149–152. [Google Scholar] [CrossRef]
- Warram, J.H.; Krolewski, A.S.; Kahn, C.R. Determinants of IDDM and perinatal mortality in children of diabetic mothers. Diabetes 1988, 37, 1328–1334. [Google Scholar] [CrossRef]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S. Risk of IDDM in children of diabetic mothers decreases with increasing maternal age at pregnancy. Diabetes 1991, 40, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Dahlquist, G.; Blom, L.; Lönnberg, G. The Swedish Childhood Diabetes Study-a multivariate analysis of risk determinants for diabetes in different age groups. Diabetologia 1991, 34, 757–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pociot, F.; Nørgaard, K.; Hobolth, N.; Andersen, O.; Nerup, J.; Danish Study Group of Diabetes in Childhood. A nationwide population-based study of the familial aggregation of type 1 (insulin-dependent) diabetes mellitus in Denmark. Diabetologia 1993, 36, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Harjutsalo, V.; Reunanen, A.; Tuomilehto, J. Differential transmission of type 1 diabetes from diabetic fathers and mothers to their offspring. Diabetes 2006, 55, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Holm, B.C.; Svensson, J.; Akesson, C.; Arvastsson, J.; Ljungberg, J.; Lynch, K.; Ivarsson, S.A.; Lernmark, A.; Cilio, C.M.; Diabetes Prediction Study in Skåne (DiPiS). Evidence for immunological priming and increased frequency of CD4+ CD25+ cord blood T cells in children born to mothers with type 1 diabetes. Clin. Exp. Immunol. 2006, 146, 493–502. [Google Scholar] [CrossRef]
- Lentini, A.; Nestor, C.E. Mapping DNA methylation in mammals: The state of the Art. Methods Mol. Biol. 2021, 2198, 37–50. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustrating the proposed role of epigenetics as a link between genetic and environmental factors in the autoimmune destruction of the pancreatic beta cells.
Figure 1.
Schematic illustrating the proposed role of epigenetics as a link between genetic and environmental factors in the autoimmune destruction of the pancreatic beta cells.


{kind=link}
Table 1.
Studies on DNA methylation and type 1 diabetes.
| Reference/Year | Method | Sample | Results |
|---|---|---|---|
| [139]/2010 | Genome-wide DNA methylation | Whole blood | Association of 19 CpG sites with risk of diabetic nephropathy |
| [62]/2011 | Epigenome-wide association study (EWAS) | Monocytes | Presence of T1D-specific methylation variable positions in the T1D-affected co-twins |
| [140]/2012 | Methylation of specific genes | Whole blood | Association of CpG methylation at the INS locus with T1D |
| [141]/2013 | Methylation of specific genes | Peripheral blood | Effect of IL2RA risk alleles on T1D may be partially mediated through CpG methylation change |
| [142]/2014 | Methylation of specific genes | Peripheral blood | Decreased IGFBP1 DNA methylation levels are associated with T1D |
| [143]/2015 | Genome-wide DNA methylation | Whole blood | Subjects with T1D and proliferative diabetic retinopathy exhibit altered DNA methylation patterns in blood |
| [144]/2016 | Epigenome-wide association study | T cells B cells Monocytes | T1D-associated differentially variable CpG positions are located in genes involved in immune cell metabolism |
| [145]/2017 | Methylation of specific genes | Tissue, pancreatic islets, whole blood | Unmethylated glucokinase gene is more islet-specific than unmethylated INS DNA |
| [146]/2018 | Genome-wide DNA methylation | Whole blood | Methylation mediates T1D risk at five non-HLA loci mainly by influencing local gene expression. |
| [147]/2019 | Methylation of specific genes | Serum | A higher unmethylated INS ratio is associated with IAA levels at the time of T1D diagnosis |
| [148]/2020 | Methylation quantitative trait loci (mQTL) analyses | Peripheral blood | Identification of 10 single nucleotide polymorphism probe pairs significantly related to methylation levels prior to the development of T1D |
| [149]/2021 | Methylation of specific genes | Pancreatic islets | Pro-inflammatorycytokines and T1D genetic risk variants regulate CTSH transcription by differential DNA methylation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Barchetta, I.; Arvastsson, J.; Sarmiento, L.; Cilio, C.M. Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes. Genes 2021, 12, 887. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060887
AMA Style
Barchetta I, Arvastsson J, Sarmiento L, Cilio CM. Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes. Genes. 2021; 12(6):887. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060887
Chicago/Turabian StyleBarchetta, Ilaria, Jeanette Arvastsson, Luis Sarmiento, and Corrado M. Cilio. 2021. "Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes" Genes 12, no. 6: 887. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12060887
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.