
Visual Function and Ophthalmological Findings in CHARGE Syndrome: Revision of Literature, Definition of a New Clinical Spectrum and Genotype Phenotype Correlation

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Methods
2.3. Ophthalmologic Evaluation
- 0 = no retinal abnormalities (N);
- 1 = mild (optic nerve pallor (ONP) or small optic nerve coloboma (SONC));
- 2 = moderate (small chorioretinal coloboma (SCCR));
- 3 = severe (large chorioretinal coloboma (LCCR) or large optic nerve coloboma (LONC));
- 4 = extremely severe (chorioretinal coloboma with macular involvement (CCRM)/giant coloboma (GC)/microphthalmos (Microph)/retinal detachment (RD)).
2.4. Visual Function Behavioral Assessment
2.5. VISIOCHARGE Questionnaire
2.6. Statistical Analysis
3. Results
3.1. Ophthalmological Evaluation
3.2. Visual Function Behavioral Assessment
3.3. VISIOCHARGE Questionnaire
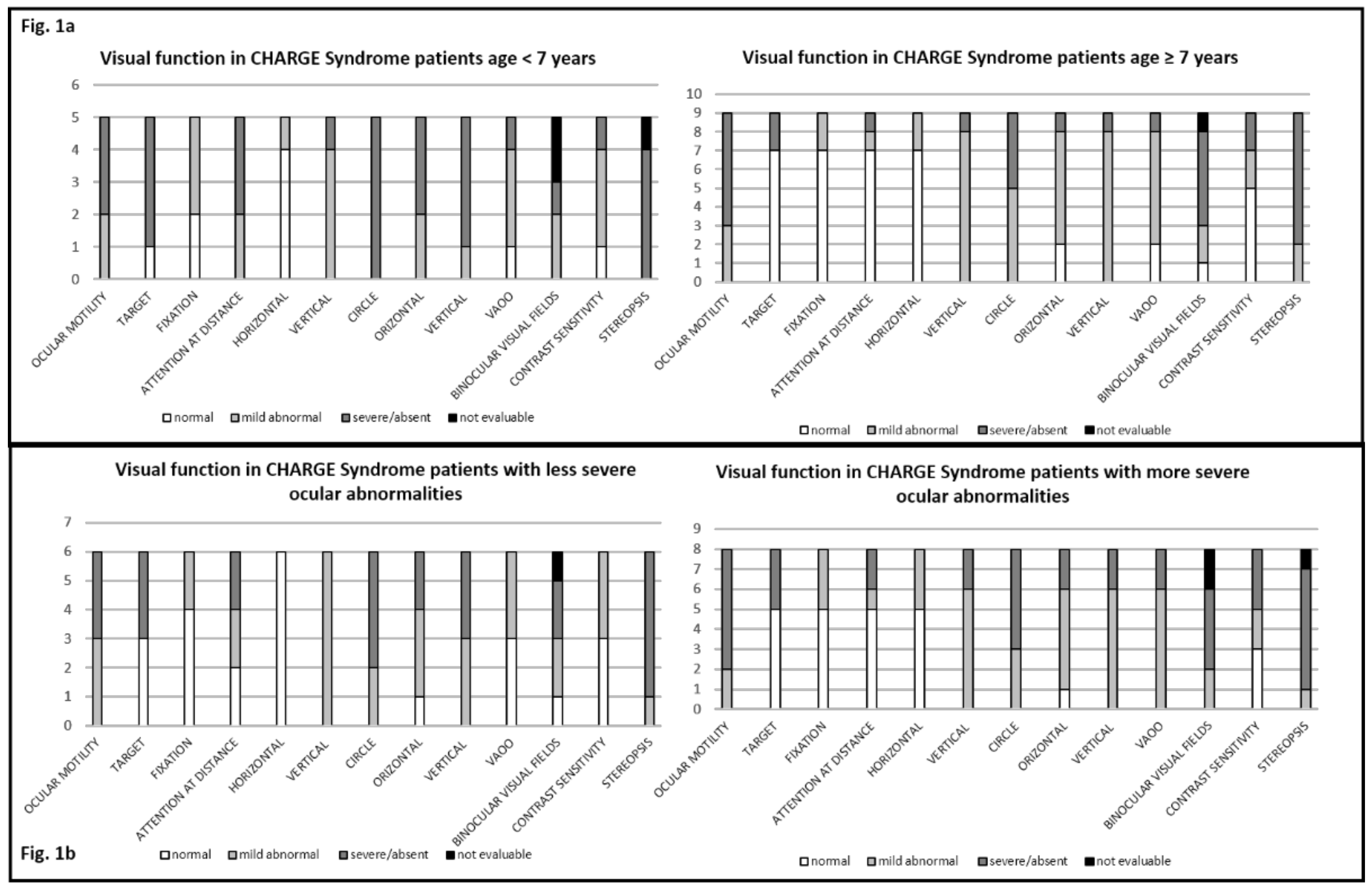
3.4. Visual Function Behaviural Assessment and Age at Assessment
3.5. Ocular Abnormalities and Visual Function Behavioral Assessment
3.6. Ocular Abnormalities and VISIOCHARGE
3.7. Genotype–Ocular-Phenotype Correlation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
| Category | Answer: n (%) Current study | Answer: n (%) Martin et al. [13] |
|---|---|---|
| Global Vision | ||
| Parental evaluation of global vision of their child | ||
| Normal or abnormal but without inconvenience Slightly bothered by his/her visual impairment Moderately bothered by his/her visual impairment Severely bothered by his/her visual impairment | 4 (28.5) 3 (21.4) 3 (21.4) 4 (28.5) | 10 (28) 7 (19) 8 (22) 11 (31) |
| Distance Vision | ||
| My child can watch television: | 13 (93) | 31 (86) |
| If yes, the television is placed at a distance of: | ||
| >2 m 50 cm to 2 m < 50 cm | 4/13 (31) 5/13 (38.5) 4/13 (31) | 7/31 (23) 17/31 (54) 7/31 (23) |
| My child can recognize a familiar face at a maximum distance of: | ||
| >10 m 2–10 m <2 m IDK | 2 (14.3) 6 (42.8) 6 (42.8) 0 | 9 (25) 13 (36) 12 (33) 2 (6) |
| When looking at the sky, my child can see: | ||
| The moon, at night A plane, at daytime | 9 (64.3) 6/13 (46.2), (1 IDK) | 19 (53), (9 IDK) 19 (53), (5 IDK) |
| The visual impairment of my child bothers him/her in moving: | ||
| Indoors, in a known place Indoors, in an unknown place Outdoors | 3 (21.4) 7 (50) 9 (64.3) | 3 (8), (4 IDK) 9 (25), (5 IDK 19 (53), (2 IDK) |
| Distance vision score mean ± SD | 0.49 ± 0.24 | 0.62 ± 0.30 |
| Near Vision | ||
| My child can use a tablet/PC | 12 (86%) | 32 (89%) |
| If yes, he/she uses it: | ||
| Normally With specific adjustments (e.g., character magnification) | 5/12 (42%) 7/12 (58%) | 28/32 (88%) 4/32 (12%) |
| My child can use a smartphone | 11 (78.6%) | 32 (89%) (1 IDK) |
| My child can read a text or identify a drawing of a minimum size of (at a distance of 40 cm): | ||
| Arial 8 Arial 10 Arial 18 Arial 28 Arial 48 >Arial 48 My child cannot perform this test | 1 (7) 5 (36) 2 (14.2) 1 (7) 4 (35.7) 0 1 (7) | 12 (33) 4 (11) 3 (8) 4 (11) 6 (17) 2 (6) 5 (14) |
| During lunch time, my child can see on table in front of him/her: | ||
| A grain of rice An olive An apricot/plum/strawberry An apple/orange | 12 (86) 14 (100) 14 (100) 14 (100) | 29 (81) 35 (97) 35 (97) 36 (100) |
| My child is able to see and catch a strand of hair | 9 (64.3) | 22 (61), (2 IDK) |
| Near vision score mean ± SD | 0.60 ± 0.17 | 0.73 ± 0.23 |
| Legend: IDK, I do not know | ||
References
- Pagon, R.A.; Graham, J.M., Jr.; Zonana, J.; Yong, S.L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 1981, 99, 223–227. [Google Scholar] [CrossRef]
- Verloes, A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am. J. Med. Genet. Part A 2005, 133, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.E.; Janssen, N.; Hoefsloot, L.H.; Jongmans, M.C.; Hofstra, R.M.; van Ravenswaaij-Arts, C.M.A. CHD7 mutations and CHARGE syndrome: The clinical implications of an expanding phenotype. J. Med. Genet. 2011, 48, 334–342. [Google Scholar] [CrossRef]
- Trider, C.L.; Arra-Robbar, A.; van Ravenswaaij-Arts, C.M.A.; Blake, K. Developing a CHARGE syndrome checklist: Health supervision across the lifespan (from head to toe). Am. J. Med. Genet. Part A 2017, 173, 684–691. [Google Scholar] [CrossRef] [PubMed]
- de Geus, C.M.; Free, R.H.; Verbist, B.M.; Sival, D.A.; Blake, K.D.; Meiners, L.C.; van Ravenswaaij-Arts, C.M. Guidelines in CHARGE syndrome and the missing link: Cranial imaging. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 450–464. [Google Scholar]
- Hsu, P.; Ma, A.; Wilson, M.; Williams, G.; Curotta, J.; Munns, C.F.; Mehr, S. CHARGE syndrome: A review. J. Paediatr. Child Health 2014, 50, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Wineland, A.; Menezes, M.D.; Shimony, J.S.; Shinawi, M.S.; Hullar, T.E.; Hirose, K. Prevalence of Semicircular Canal Hypoplasia in Patients With CHARGE Syndrome: 3C Syndrome. JAMA Otolaryngol. Head Neck Surg. 2017, 143, 168–177. [Google Scholar] [CrossRef]
- Russell-Eggitt, I.M.; Blake, K.D.; Taylor, D.S.; Wyse, R.K. The eye in the CHARGE association. Br. J. Ophthalmol. 1990, 74, 421–426. [Google Scholar] [CrossRef] [Green Version]
- McMain, K.; Robitaille, J.; Smith, I.; Johnson, J.; Wood, E.; Tremblay, F.; Blake, K. Ocular features of CHARGE syndrome. J. AAPOS 2008, 12, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Nishina, S.; Kosaki, R.; Yagihashi, T.; Azuma, N.; Okamoto, N.; Hatsukawa, Y.; Kurosawa, K.; Yamane, T.; Mizuno, S.; Tsuzuki, K.; et al. Ophthalmic features of CHARGE syndrome with CHD7 mutations. Am. J. Med. Genet. Part A 2012, 158, 514–518. [Google Scholar] [CrossRef]
- Strömland, K.; Sjögreen, L.; Johansson, M.; Ekman Joelsson, B.M.; Miller, M.; Danielsson, S.; Billstedt, E.; Gillberg, C.; Jacobsson, C.; Norinder, J.A.; et al. CHARGE association in Sweden: Malformations and functional deficits. Am. J. Med. Genet. Part A 2005, 133, 331–339. [Google Scholar] [CrossRef]
- Tellier, A.L.; Cormier-Daire, V.; Abadie, V.; Amiel, J.; Sigaudy, S.; Bonnet, D.; de Lonlay-Debeney, P.; Morrisseau-Durand, M.P.; Hubert, P.; Michel, J.L.; et al. CHARGE syndrome: Report of 47 cases and review. Am. J. Med. Genet. 1998, 76, 402–409. [Google Scholar] [CrossRef]
- Martin, G.C.; Robert, M.P.; Challe, G.; Trinh, N.T.; Attié-Bitach, T.; Brémond-Gignac, D.; Bodaghi, B.; Abadie, V. Functional Vision Analysis in Patients With CHARGE Syndrome. J. Pediatr. Ophthalmol. Strabismus 2020, 57, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Ricci, D.; Chieffo, D.; Battaglia, D.; Brogna, C.; Contaldo, I.; De Clemente, V.; Losito, E.; Dravet, C.; Mercuri, E.; Guzzetta, F. A prospective longitudinal study on visuo-cognitive development in Dravet syndrome: Is there a “dorsal stream vulnerability”? Epilepsy Res. 2015, 109, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Cesarini, L.; De Rose, P.; Ricci, D.; Selicorni, A.; Menghini, D.; Guzzetta, A.; Baranello, G.; Tinelli, F.; Mallardi, M.; et al. Visual processing in Noonan syndrome: Dorsal and ventral stream sensitivity. Am. J. Med. Genet. Part A 2011, 155, 2459–2464. [Google Scholar] [CrossRef]
- Rando, T.; Bancale, A.; Baranello, G.; Bini, M.; De Belvis, A.G.; Epifanio, R.; Frisone, M.F.; Guzzetta, A.; La Torre, G.; Ricci, D.; et al. Visual function in infants with West syndrome: Correlation with EEG patterns. Epilepsia 2004, 45, 781–786. [Google Scholar] [CrossRef]
- Hyvarinen, L.; Nasanen, R.; Laurinen, P. New visual acuity test for pre-school children. Acta Ophthalmol. 1980, 58, 507–511. [Google Scholar] [CrossRef]
- Teller, D.Y.; McDonald, M.A.; Preston, K.; Sebris, S.L.; Dobson, V. Assessment of visual acuity in infants and children: The acuity card procedure. Dev. Med. Child Neurol. 1986, 28, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Mohn, G.; Duin, J.V.H.V.; Fetter, W.P.F.; De Groot, L.; Hage, M. Acuity assessment of non-verbal infants and children: Clinical experience with the acuity card procedure. Dev. Med. Child Neurol. 1988, 30, 232–244. [Google Scholar] [CrossRef]
- Bailey, I.L.; Lovie-Kitchin, J.E. Visual acuity testing. From the laboratory to the clinic. Vision Res. 2013, 90, 2–9. [Google Scholar] [CrossRef]
- Carlson, N.B.; Kurtz, D. Clinical Procedures for Ocular Examination; Mc Graw-Hill: New York, NY, USA, 2016. [Google Scholar]
- Van Hof-van Duin, J.; Heersema, D.J.; Groenendaal, F.; Baerts, W.; Fetter, W.P.F. Visual field and grating acuity development in low-risk preterm infants during the first 2 1/2 years after term. Behav. Brain Res. 1992, 31, 115–122. [Google Scholar] [CrossRef]
- Bibliography of Developmental Medicine and Child Neurology: Selected Books and Articles Received in 1989; Mac Keith Press in Association with Blackwell Scientific Publications: London, UK; Lippincott: Philadelphia, PA, USA, 1990.
- Beaton, D.E.; Bombardier, C.; Guillemin, F.; Ferraz, M.B. Guidelines for the process of cross-cultural adaptation of self-report measures. Spine 2000, 25, 3186–3191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramaki, M.; Udaka, T.; Kosaki, R.; Makita, Y.; Okamoto, N.; Yoshihashi, H.; Oki, H.; Nanao, K.; Moriyama, N.; Oku, S.; et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006, 148, 410–414. [Google Scholar] [CrossRef]
- Lalani, S.R.; Safiullah, A.M.; Fernbach, S.D.; Harutyunyan, K.G.; Thaller, C.; Peterson, L.E.; McPherson, J.D.; Gibbs, R.A.; White, L.D.; Hefner, M.; et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 2006, 78, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongmans, M.C.J.; Admiraal, R.J.; Van der Donk, K.P.; Vissers, L.E.L.M.; Baas, A.F.; Kapusta, L.; van Hagen, J.M.; Donnai, D.; De Ravel, T.J.; Veltman, J.A.; et al. CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. J. Med. Genet. 2006, 43, 306–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, N.; Bergman, J.E.; Swertz, M.A.; Tranebjaerg, L.; Lodahl, M.; Schoots, J.; Hofstra, R.M.; van Ravenswaaij-Arts, C.M.; Hoefsloot, L.H. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum. Mutat. 2012, 33, 1149–1160. [Google Scholar] [CrossRef]

| Patients (Number) 14 | ||
|---|---|---|
| Demographics | ||
| Age (years) | 10.98 ± 7.6 | |
| Gender (M) | 8 | |
| Genetics | ||
| CHD7 mutation | 14 | 100 |
| Frameshift | 4 | 28.6 |
| Nonsense | 5 | 35.7 |
| Missense | 1 | 7 |
| Splicing | 4 | 28.6 |
| Clinical features | n | Percentage (%) |
| Major characteristics | ||
| Cranial nerve dysfunction | 7 | 50 |
| Choanal anomaly | 3 | 21.4 |
| Hearing loss | 14 | 100 |
| Sensorineural | 12 | 85.7 |
| Conductive | 2 | 14.3 |
| Hearing severity | ||
| Normal | 0 | 0 |
| Mild/moderate hearing loss | 3 | 21.4 |
| Severe to total hearing loss | 11 | 78.6 |
| Ocular abnormalities | 14 | 100 |
| Posterior coloboma | ||
| Unilateral | 4 | 28 |
| Bilateral | 9 | 65 |
| Iris coloboma | ||
| Unilateral | 2 | 14 |
| Bilateral | 4 | 28 |
| Optic nerve pallor | ||
| Unilateral | 0 | 0 |
| Bilateral | 1 | 7 |
| Microphthalmos | ||
| Unilateral | 2 | 14 |
| Bilateral | 0 | 0 |
| Retinal detachment | ||
| Unilateral | 1 | 7 |
| Bilateral | 0 | 0 |
| Retinal dystrophy | ||
| Unilateral | 1 | 7 |
| Bilateral | 0 | 0 |
| Minor characteristics | ||
| Cardiovascular malformation | 8 | 57.1 |
| Genital hypoplasia | 4 | 28.6 |
| Orofacial cleft | 2 | 14.3 |
| Tracheoesophageal fistula | 4 | 28.6 |
| Developmental delay | 14 | 100 |
| Growth deficiency | 3 | 21.4 |
| Distinctive CHARGE facies | 14 | 100 |
| Pt. ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 2 | 4 | 5 | 5 | 6 | 7 | 7 | 8 | 8 | 12 | 13 | 19 | 26 | 26 |
| Stereopsis | Absent | Absent | Absent | Absent | NE | Absent | Absent | Absent | Normal | Absent | Absent | Absent | Absent | Absent |
| Contrast sensitivity | Normal | Reduced | Reduced | Reduced | Reduced | Reduced | Reduced | Normal | Normal | Reduced | Normal | Normal | Reduced | Normal |
| Binocular visual field | 60° bilateral | 50° bilateral | 50° right 20° left | NE | NE | 40° right 60° left | 10° right 60° left | 30° bilateral | 90° bilateral | 50° right 60° left | 10° right 50° left | 50° bilateral | NE | 60° right 5° left |
| Vaoo Logmatr | 0.3 | 0.7 | 0.7 | 1 | 1.6 | 1.3 | 0.55 | 0.1 | 0.1 | 0.7 | 0.55 | 0.55 | 1 | 0 |
| Vertical saccades | Absent | HC | Absent | Absent | Absent | HC | HC | HC | HC | HC | HC | HC | Absent | HC |
| Horizontal saccades | Absent | HC | Absent | HC | Absent | Normal | HC | HC | Normal | HC | HC | HC | Absent | HC |
| Circle | Absent | Absent | Absent | Absent | Absent | Absent | Absent | HC | HC | Absent | HC | HC | Absent | HC |
| Vertical tracking | HC | HC | HC | HC | Absent | HC | HC | HC | HC | HC | HC | HC | Absent | HC |
| Horizontal tracking | Complete | Complete | Complete | Complete | HC | Complete | Complete | HC | Complete | Complete | Complete | Complete | HC | Complete |
| Attention at distance | 1 m | 2 m | 2.5 m | 1 m | 50 cm | 1.5 m | 3 m | 3 m | 3 m | 3 m | 3 m | 3 m | 50c m | 3 m |
| Fixation | Unstable | Stable | Stable | Unstable | Unstable | Stable | Unstable | Stable | Stable | Stable | Stable | Stable | Unstable | Stable |
| Ophth. Staging LE | 1 | 0 | 4 | 1 | 4 | 3 | 4 | 4 | 2 | 4 | 3 | 3 | 4 | 4 |
| Ophth. Staging RE | 1 | 1 | 1 | 1 | 3 | 3 | 4 | 4 | 0 | 4 | 4 | 2 | 4 | 0 |
| LE fundus | SONC | N | CCRM + Microph. | ONP | RD | LONC | CCRM | CCRM | SCCR | CCRM | LCCR | CCR | CCRM | GC |
| RE fundus | SONC | SONC | SONC | ONP | LCCR | LONC | CCRM | CCRM | N | CCRM | CCRM + Microph. | SCCR | CCRM | N |
| CHD7 nucleotide change predicted amino acid change | c.4795C>T p.Gln1599Ter | c.2957+5G>A (spl) a | c.5782C>T p.Gln1928Ter | c.2442+5G>A (spl) b | c.5722_5723delAC p.Thr1908ProfsTer17 | c.3004C>T p.n1001Ter | c.969-975delAACAA p.Val323TyrfsTer11 | c.2509_2512delCATT p.His837ValfsTer5 | c.7803C>G p.Tyr2601Ter | c.1163C>G p.Ser230Ter | c.6936+2T>A (spl) c | c.3156T>A p.Ser1052Arg | c.1774delC p.Gln592SerfsTer16 | c.7165-4A>G (spl) d |
| Review of Anatomical Defects in CHARGE Syndrome | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ocular Defect | 1990 | 1998 | 2006 | 2006 | 2008 | 2012 | 2020 | 2020 |
| Russel-Eggitt (n = 50) | Tellier (n = 47) | Aramaki (n = 17) | Jongmans (n = 47) | McMain (n = 9) | Nishina (n = 19) | Martin (n = 83) | Current Study (n = 14) | |
| Coloboma | 86% | 79% | 88% | 70% | 89% | 95% | 83% | 93% |
| Unilateral | 16% | 57% | N/S | 2% | 11% | 5% | 17% | 29% |
| Bilateral | 64% | 21% | N/S | 68% | 78% | 89% | 66% | 64% |
| Retinochoroidal | 80% | 43% | 58% | N/S | 89% | 95% | N/S | 71% |
| Optic disk | 74% | 17% | 30% | N/S | N/S | 95% | N/S | 36% |
| Macula | N/S | N/S | N/S | N/S | N/S | 68% | N/S | 43% |
| Iris | 26% | 6% | 12% | 19% | 11% | 89% | 14% | 36% |
| Eyelid | 2% | N/S | N/S | N/S | 0 | 0 | N/S | 0 |
| Microphthalmos | 42% | 34% | N/S | 21% | N/S | 26% | 34% | 14% |
| Unilateral | 26% | N/S | N/S | N/S | 11% | 10% | 30% | 14% |
| Bilateral | 16% | N/S | N/S | N/S | N/S | 16% | 3% | 0 |
| Microcornea | N/S | N/S | N/S | N/S | 11% | 21% | N/S | 0 |
| Cataract | 2% | N/S | N/S | N/S | 11% | 5% | 5% | 7% |
| Retinal detachment | 2% | N/S | N/S | N/S | 0 | 0 | 7% | 7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onesimo, R.; Ricci, D.; Agazzi, C.; Leone, S.; Petrianni, M.; Orazi, L.; Amore, F.; Salerni, A.; Leoni, C.; Chieffo, D.; et al. Visual Function and Ophthalmological Findings in CHARGE Syndrome: Revision of Literature, Definition of a New Clinical Spectrum and Genotype Phenotype Correlation. Genes 2021, 12, 972. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12070972
Onesimo R, Ricci D, Agazzi C, Leone S, Petrianni M, Orazi L, Amore F, Salerni A, Leoni C, Chieffo D, et al. Visual Function and Ophthalmological Findings in CHARGE Syndrome: Revision of Literature, Definition of a New Clinical Spectrum and Genotype Phenotype Correlation. Genes. 2021; 12(7):972. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12070972
Chicago/Turabian StyleOnesimo, Roberta, Daniela Ricci, Cristiana Agazzi, Simona Leone, Maria Petrianni, Lorenzo Orazi, Filippo Amore, Annabella Salerni, Chiara Leoni, Daniela Chieffo, and et al. 2021. "Visual Function and Ophthalmological Findings in CHARGE Syndrome: Revision of Literature, Definition of a New Clinical Spectrum and Genotype Phenotype Correlation" Genes 12, no. 7: 972. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12070972