
Co-Occurring Heterozygous CNOT3 and SMAD6 Truncating Variants: Unusual Presentation and Refinement of the IDDSADF Phenotype

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
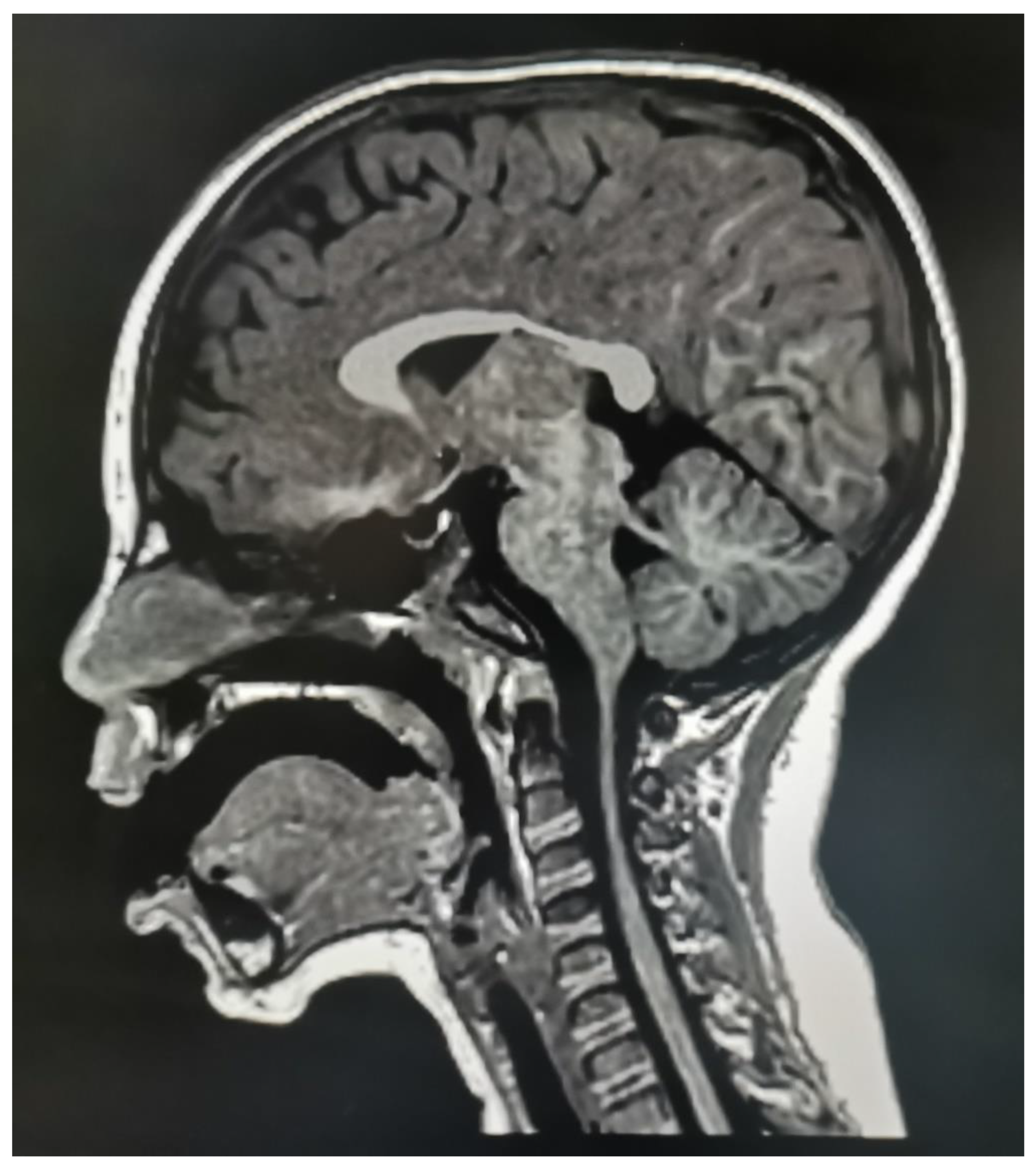
2.1. Clinical Case
2.2. Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Posey, J.E.; Harel, T.; Liu, P.; Pengfel, L.; Rosenfeld, J.; James, R.A.; Akdemir, Z.H.C.; Walklewitz, M.; Bi, W.; Xiao, R.; et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Blanco, K.; Sayan, S.A.; Hunter, J.M.; Shinde, D.N.; Wayburn, B.; Rossi, M.; Huang, J.; Stevens, C.A.; Muss, C.; et al. A retrospective review of multiple findings in diagnostic exome sequencing: Half are distinct and half are overlapping diagnoses. Genet. Med. 2019, 21, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, S.; Gazzo, A.; Versbraegen, N.; Nachtegael, C.; Aerts, J.; Moreau, Y.; Van Dooren, S.; Nowe, A.; Smits, G.; Lenaerts, T. Predicting disease-causing variant combinations. Proc. Natl. Acad. Sci. USA 2019, 116, 11878–11887. [Google Scholar] [CrossRef] [PubMed]
- Balci, T.B.; Hartley, T.; Xi, Y.; Dyment, D.A.; Beaulieu, C.L.; Bernier, F.P.; Dupuis, L.; Horvath, G.; Mendoza-Londono, R.; Prased, C.; et al. Debunking Occam’s razor: Diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin. Genet. 2017, 92, 281–289. [Google Scholar] [CrossRef]
- Karaca, E.; Posey, J.E.; Coban Akdemir, Z.; Pehlivan, D.; Harel, T.; Jhangiani, S.N.; Bayram, Y.; Song, X.; Bahrambeigi, V.; Yuregir, O.O.; et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet. Med. 2018, 20, 1528–1537. [Google Scholar] [CrossRef]
- Pizzo, L.; Jensen, M.; Polyak, A.; Resenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Krishnan, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes inindividuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef]
- Guo, T.; Chung, J.H.; Wang, T.; McDonald-McGinn, D.M.; Kates, W.R.; Hawuła, W.; Coleman, K.; Zackai, E.; Emanuel, B.S.; Morrow, B.E. Histone modifier genes alter conotruncal heart phenotypes in 22q11.2 deletion syndrome. Am. J. Hum. Genet. 2015, 97, 869–877. [Google Scholar] [CrossRef]
- Grillo, E.; Rizzo, C.L.; Bianciardi, L.; Bizzarri, V.; Baldassarri, M.; Spiga, O.; Furini, S.; De Felice, C.; Signorini, C.; Leoncini, S.; et al. Revealing the complexity of a monogenic disease: Rett syndrome exome sequencing. PLoS ONE 2013, 8, e56599. [Google Scholar] [CrossRef]
- Schuurs-Hoeijmakers, J.H.; Oh, E.C.; Vissers, L.E.; Swinkels, M.E.M.; Gilissen, C.; Willemsen, M.A.; Holvoet, M.; Steehouwer, M.; Veltman, J.A.; de Vries, B.B.A.; et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet. 2012, 91, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef]
- Martin, R.; Splitt, M.; Genevieve, D.; Aten, E.; Collins, A.; de Bie, C.I.; Faivre, L.; Foulds, N.; Giltay, J.; Ibitoye, R.; et al. De novo variants in CNOT3 cause a variable neurodevelopmental disorder. Eur. J. Hum. Genet. 2019, 27, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Begemann, M.; Demuth, S.; Kraft, F.; Dey, D.; Schuler, H.; Busse, S.; Hausler, M.; Zerres, K.; Kurth, I.; et al. Inherited cases of CNOT3-associated intellectual developmental disorder with speech delay, autism, and dysmorphic facies. Clin. Genet. 2020, 98, 408–412. [Google Scholar] [CrossRef]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β family signaling by inhibitory smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Glen, E.; Topf, A.; Hall, D.; O’Sullivan, J.; Sneddon, L.; Wren, C.; Avery, P.; Lewis, R.J.; ten Dijke, P.; et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum. Mutat. 2012, 33, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Zen, X.; Qi, H.; Chang, W.; Morton, S.; De Palma, S.R.; Halder, S.; et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef]
- Kloth, K.; Bierhals, T.; Johannsen, J.; Harms, F.L.; Juusola, J.; Johnson, M.C.; Grange, D.K.; Kutsche, K. Biallelic variants in SMAD6 are associated with a complex cardiovascular phenotype. Hum. Genet. 2019, 138, 625–634. [Google Scholar] [CrossRef]
- Gillis, E.; Kumar, A.A.; Luyckx, I.; Preuss, C.; Cannaerts, E.; van de Beek, G.; Wieschendorf, B.; Alaerts, M.; Bolar, N.; Vandeweyer, R.; et al. Candidate gene resequencing in a large bicuspid aortic valve-associated thoracic aortic aneurysm cohort: SMAD6 as an important contributor. Front. Physiol. 2017, 8, 730. [Google Scholar] [CrossRef]
- Luyckx, I.; MacCarrick, G.; Kempers, M.; Meester, J.; Geryl, C.; Rombouts, O.; Peeters, N.; Claes, C.; Boeckx, N.; Sakalihasan, N.; et al. Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. Eur. J. Hum. Genet. 2019, 27, 1044–1053. [Google Scholar] [CrossRef]
- Park, J.E.; Park, J.S.; Jang, S.Y.; Park, S.E.; Kim, J.W.; Ki, C.S.; Kim, D.K. A novel SMAD6 variant in a patient with severely calcified bicuspid aortic valve and thoracic aortic aneurysm. Mol. Genet. Genom. Med. 2019, 7, e620. [Google Scholar] [CrossRef]
- Timberlake, A.T.; Choi, J.; Zaidi, S.; Lu, Q.; Nelson-Williams, C.; Brooks, E.D.; Bilguvar, K.; Tikhonova, I.; Mane, S.; Yang, J.F.; et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. eLife 2016, 5, e20125. [Google Scholar] [CrossRef]
- Calpena, E.; Cuellar, A.; Bala, K.; Swagemakers, S.M.A.; Koelling, N.; McGowan, S.J.; Phipps, J.M.; Balasubramanian, M.; Cunningham, M.L.; Douzgou, S.; et al. SMAD6 variants in craniosynostosis: Genotype and phenotype evaluation. Genet. Med. 2020, 22, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, Y.; Li, W.; Li, L.; Tu, M.; Zhao, L.; Mei, H.; Zhu, G.; Zhu, Y. SMAD6 is frequently mutated in nonsyndromic radioulnar synostosis. Genet. Med. 2019, 21, 2577–2585. [Google Scholar] [CrossRef]
- Bauer, C.K.; Calligari, P.; Radio, F.C.; Caputo, V.; Dentici, M.L.; Falah, N.; High, F.; Pantaleoni, F.; Barresi, S.; Ciolfi, A.; et al. Mutations in KCNK4 that Affect Gating Cause a Recognizable Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 621–630. [Google Scholar] [CrossRef]
- Flex, E.; Martinelli, S.; Van Dijck, A.; Ciolfi, A.; Cecchetti, S.; Coluzzi, E.; Pannone, L.; Andreoli, C.; Radio, F.C.; Pizzi, S.; et al. Aberrant Function of the C-terminal Tail of HIST1H1E Accelerates Cellular Senescence and Causes Premature Aging. Am. J. Hum. Genet. 2019, 105, 493–508. [Google Scholar] [CrossRef]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v1. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118, iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A database of human nonsynonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Jagadeesh, K.; Wenger, A.; Berger, M.; Guturu, H.; Stenson, P.; Cooper, D.; Bernstein, J.; Bejerano, G. M-CAP eliminates a majority of variants with uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–2805. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Paggi, J.M.; Ye, J.S.; Stenson, P.D. S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. Nat. Genet. 2019, 51, 755–763. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Robinson, P.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Boland, A.; Chen, Y.; Raisch, T.; Jonas, S.; Kuzuoğlu-Öztürk, D.; Wohlbold, L.; Weichenrieder, O.; Izaurralde, E. Structure and assembly of the NOT module of the human CCR4–NOT complex. Nat. Struct. Mol. Biol. 2013, 20, 1289–1297. [Google Scholar] [CrossRef]
- Wahle, E.; Winkler, G.S. RNA decay machines: Deadenylation by the Ccr4–Not and Pan2–Pan3 complexes. Biochim. Biophys. Acta 2013, 1829, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Suzuki, T.; Sato, T.; Takahashi, A.; Watanabe, H.; Kadowaki, A.; Natsui, M.; Inagaki, H.; Arakawa, S.; Nakaoka, S.; et al. The CCR4-NOT deadenylase complex controls Atg7-dependent cell death and heart function. Sci. Signal. 2018, 11, eaan3638. [Google Scholar] [CrossRef]
- Kruk, J.A.; Dutta, A.; Fu, J.; Gilmour, D.S.; Reese, J.C. The multifunctional Ccr4-Not complex directly promotes transcription elongation. Genes Dev. 2011, 25, 581–593. [Google Scholar] [CrossRef]
- Webster, M.W.; Chen, Y.H.; Stowell, J.A.W.; Alhusaini, N.; Sweet, T.; Graveley, B.R.; Coller, J.; Passmore, L.A. mRNA deadenylation is coupled to translation rates by the differential activities of Ccr4-Not nucleases. Mol. Cell 2018, 70, 1089–1100.e8. [Google Scholar] [CrossRef]
- Collart, M.A. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 2016, 7, 438–454. [Google Scholar] [CrossRef]
- Collart, M.A.; Panasenko, O.O. The Ccr4-Not complex: Architecture and structural insights. Subcell. Biochem. 2017, 83, 349–379. [Google Scholar] [CrossRef] [PubMed]
- Elmen, L.; Volpato, C.B.; Kervadec, A.; Pineda, S.; Kalvakuri, S.; Alayari, N.N.; Foco, L.; Pramstaller, P.P.; Ocorr, K.; Rossini, A.; et al. Silencing of CCR4-NOT complex subunits affects heart structure and function. Dis. Model. Mech. 2020, 13, dmm044727. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://gnomad.broadinstitute.org/ (accessed on 24 May 2021).
- Pinard, A.; Guey, S.; Guo, D.; Cecchi, A.C.; Kharas, N.; Wallace, S.; Regalado, E.S.; Hostetler, E.M.; Sharrief, A.Z.; Bergametti, F.; et al. The pleiotropy associated with de novo variants in CHD4, CNOT3, and SETD5 extends to moyamoya angiopathy. Genet. Med. 2020, 22, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Ren, Z.; Yao, F.; Ma, J.; Song, J.; Bennet, B.; Zhen, Y.; Wang, L.; Hu, G.; et al. Cnot3 enhances human embryonic cardiomyocyte proliferation by promoting cell cycle inhibitor mRNA degradation. Sci. Rep. 2017, 7, 1500. [Google Scholar] [CrossRef] [PubMed]
- Neely, G.G.; Kuba, K.; Cammarato, A.; Isobe, K.; Amann, S.; Zhang, L.; Murata, M.; Elmén, L.; Gupta, V.; Arora, S.; et al. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 2010, 141, 142–153. [Google Scholar] [CrossRef]


{kind=link}
| Martin et al., 2019 [11] | Meyer et al., 2020 [12] | Present Case | Total | |
|---|---|---|---|---|
| PATIENTS/FEATURES | ||||
| Sex | 6 F/10 M | 5 F | M | 11F/11M |
| Type of variant | 8MS/8TR | 2MS/3TR | TR | 10MS/12TR |
| Low birth weight (≤5th cent) | 4/16 SGA * | 2/5 SGA | 5th cent | |
| Low birth height (≤5th cent) | NR | 4/5 ≤ 5th cent | 10th cent | |
| birth head circumference | NR | 5/5 normal range | 3rd cent | |
| Weight | 3/15 ≤ 2nd cent | 5/5 Normal range | 75th cent | |
| Height | 13/16 ≤ 25thcent | 4/5 ≤ 10th cent | 25th cent | |
| Head circumference | 14/16 cases normal | 5/5 normal range | 60th cent | |
| DEVELOPMENT | ||||
| Developmental delay | 16/16 | 5/5 | + | 22/22 (100%) |
| Muscular hypotonia | 10/16 | 2/3 | + | 13/20 (65%) |
| Speech delay | 15/16 | 4/4 | + | 20/21 (95%) |
| Behavior anomalies (BA)/autism (A) | 11/16 (5 BA/7A) | 1/5 (BA) | + (BA) | 13/22 (59%) |
| EYES | ||||
| Vision | 8/15 (4/16 strabism) | 4/5 (strabism) | myopia/ astigmatism | 13/21 (62%) |
| BRAIN MRI ANOMALIES | 7/12 | 1/1 | + | 9/14 (64%) |
| CC hypoplasia | 3/12 | 1/1 | + | 5/14 (36%) (55%TOT) |
| Seizures/EEGabnormalities | 4/16 | 1/5 | (1 critic episode) | 5/21 (24%) |
| FACIAL FEATURES | ||||
| Prominent forehead | 5/16 ° | 5/5 | + | 11/22 (50%) |
| Flat/high nasal bridge | 10/16 | nr | + | 11/17 (65%) |
| Upslanted palpebral fissures | 8/16 # | nr | + | 9/17 (53%) |
| Low set ears | nr | 2/5 | + | 3/6 (50%) |
| Flabby cheeks/nasolabial sulci prominence | 10/10 @ | 2/2 @ | + | 13/13 (100%) |
| Broad nasal tip | 8/16 | 3/5 | + | 12/22 (55%) |
| Anteverted nares | 11/16 | nr | + | 12/17 (71%) |
| Smooth philtrum | 9/16 | 2/2 | + | 12/19 (63%) |
| Long philtrum | 6/16 | 2/2 | + | 9/19 (47%) |
| Thin upper lip | 9/16 | 2/2 | + | 12/19 (63%) |
| Lower lip prominence with prominent labiomental groove | 7/10 ^ | 2/2 ^ | + | 10/13 (77%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priolo, M.; Radio, F.C.; Pizzi, S.; Pintomalli, L.; Pantaleoni, F.; Mancini, C.; Cordeddu, V.; Africa, E.; Mammì, C.; Dallapiccola, B.; et al. Co-Occurring Heterozygous CNOT3 and SMAD6 Truncating Variants: Unusual Presentation and Refinement of the IDDSADF Phenotype. Genes 2021, 12, 1009. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071009
Priolo M, Radio FC, Pizzi S, Pintomalli L, Pantaleoni F, Mancini C, Cordeddu V, Africa E, Mammì C, Dallapiccola B, et al. Co-Occurring Heterozygous CNOT3 and SMAD6 Truncating Variants: Unusual Presentation and Refinement of the IDDSADF Phenotype. Genes. 2021; 12(7):1009. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071009
Chicago/Turabian StylePriolo, Manuela, Francesca Clementina Radio, Simone Pizzi, Letizia Pintomalli, Francesca Pantaleoni, Cecilia Mancini, Viviana Cordeddu, Emilio Africa, Corrado Mammì, Bruno Dallapiccola, and et al. 2021. "Co-Occurring Heterozygous CNOT3 and SMAD6 Truncating Variants: Unusual Presentation and Refinement of the IDDSADF Phenotype" Genes 12, no. 7: 1009. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071009