
Novel Mutation in CRYBB3 Causing Pediatric Cataract and Microphthalmia

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Clinical Evaluation
2.3. Genomic DNA Preparation
2.4. Library Preparation and Clinical Exome Sequencing
2.5. Bioinformatics Analysis
2.6. Sanger Sequencing
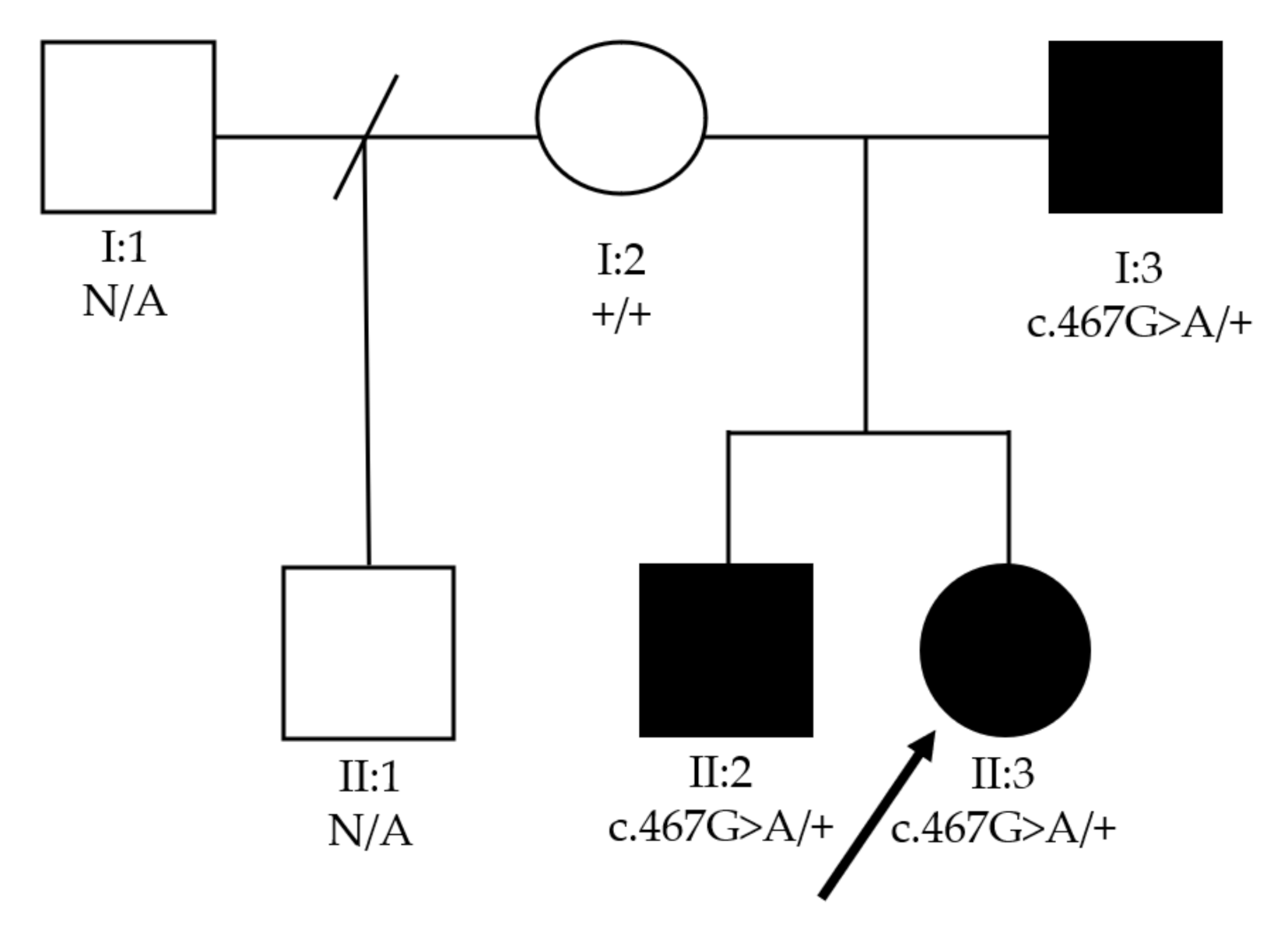
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hejtmancik, J.F. Congenital cataracts and their molecular genetics. Semin. Cell Dev. Biol. 2008, 19, 134–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheeladevi, S.; Lawrenson, J.; Fielder, A.R.; Suttle, C.M. Global prevalence of childhood cataract: A systematic review. Eye 2016, 30, 1160–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Fry, M.; Al-Samarraie, M.; Gilbert, C.; Steinkuller, P.G. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2012, 16, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Hejtmancik, J.F. Biology of Inherited Cataracts and Opportunities for Treatment. Annu. Rev. Vis. Sci. 2019, 5, 123–149. [Google Scholar] [CrossRef] [PubMed]
- Haargaard, B.; Wohlfahrt, J.; Fledelius, H.C.; Rosenberg, T.; Melbye, M. A nationwide Danish study of 1027 cases of congenital/infantile cataracts: Etiological and clinical classifications. Ophthalmology 2004, 111, 2292–2298. [Google Scholar] [CrossRef] [PubMed]
- Cat-Map Complete List. Available online: https://cat-map.wustl.edu/ (accessed on 31 March 2021).
- ABraOM: Brazilian Genomic Variants. Available online: http://abraom.ib.usp.br/ (accessed on 31 March 2021).
- Li, D.; Wang, S.; Ye, H.; Tang, Y.; Qiu, X.; Fan, Q.; Rong, X.; Liu, X.; Chen, Y.; Yang, J.; et al. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population. Mol. Vis. 2016, 22, 589–598. [Google Scholar] [PubMed]
- Sekeroglu, H.T.; Karaosmanoglu, B.; Taskiran, E.Z.; Kiper, P.O.S.; Alikasifoglu, M.; Boduroglu, K.; Coskun, T.; Utine, G.E. Molecular Etiology of Isolated Congenital Cataract Using Next-Generation Sequencing: Single Center Exome Sequencing Data from Turkey. Mol. Syndr. 2020, 11, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Messina-Baas, O.; Cuevas-Covarrubias, S.A. Inherited Congenital Cataract: A Guide to Suspect the Genetic Etiology in the Cataract Genesis. Mol. Syndr. 2017, 8, 58–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannabiran, C.; Balasubramanian, D. Molecular genetics of cataract. Indian J. Ophthalmol. 2000, 48, 5–13. [Google Scholar] [PubMed]
- Delaye, M.; Tardieu, A. Short-range order of crystallin proteins accounts for eye lens transparency. Nat. Cell Biol. 1983, 302, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Cao, Z.; Zhu, Y.; Liu, L.; Tong, Y.; Chen, X.; Wang, Y.; Lu, C.; Ma, X.; Yang, J. Mutation screening of crystallin genes in Chinese families with congenital cataracts. Mol. Vis. 2019, 25, 427–437. [Google Scholar] [PubMed]
- Li, J.; Chen, X.; Yan, Y.; Yao, K. Molecular genetics of congenital cataracts. Exp. Eye Res. 2020, 191, 107872. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.; Waiswo, M. The genetic and molecular basis of congenital cataract. Arq. Bras. Oftalmol. 2011, 74, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazuddin, S.A. Mutations in βB3-Crystallin Associated with Autosomal Recessive Cataract in Two Pakistani Families. Investig. Opthalmol. Vis. Sci. 2005, 46, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Self, J.E.; Taylor, R.; Solebo, A.L.; Biswas, S.; Parulekar, M.; Borman, A.D.; Ashworth, J.; McClenaghan, R.; Abbott, J.; O’Flynn, E.; et al. Cataract management in children: A review of the literature and current practice across five large UK centres. Eye 2020, 34, 2197–2218. [Google Scholar] [CrossRef] [PubMed]
- Chak, M.; Rahi, J. Incidence of and Factors Associated with Glaucoma after Surgery for Congenital Cataract: Findings from the British Congenital Cataract Study. Ophthalmology 2008, 115, 1013–1018.e2. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age at Time of Diagnosis (months) | Bilateral Cataract Phenotype | Other Ocular Abnormalities |
|---|---|---|---|---|
| I:3 | M | 36 | Unknown | Microphthalmia |
| II:2 | M | 18 | Unknown | Microphthalmia, aqueous misdirection |
| II:3 | F | 3 | Anterior polar | Microphthalmia, aphakic glaucoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zin, O.A.; Neves, L.M.; Motta, F.L.; Horovitz, D.D.G.; Guida, L.; Gomes, L.H.F.; Cunha, D.P.; Rodrigues, A.P.S.; Zin, A.A.; Sallum, J.M.F.; et al. Novel Mutation in CRYBB3 Causing Pediatric Cataract and Microphthalmia. Genes 2021, 12, 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071069
Zin OA, Neves LM, Motta FL, Horovitz DDG, Guida L, Gomes LHF, Cunha DP, Rodrigues APS, Zin AA, Sallum JMF, et al. Novel Mutation in CRYBB3 Causing Pediatric Cataract and Microphthalmia. Genes. 2021; 12(7):1069. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071069
Chicago/Turabian StyleZin, Olivia A., Luiza M. Neves, Fabiana L. Motta, Dafne D. G. Horovitz, Leticia Guida, Leonardo H. F. Gomes, Daniela P. Cunha, Ana Paula S. Rodrigues, Andrea A. Zin, Juliana M. F. Sallum, and et al. 2021. "Novel Mutation in CRYBB3 Causing Pediatric Cataract and Microphthalmia" Genes 12, no. 7: 1069. https://0-doi-org.brum.beds.ac.uk/10.3390/genes12071069