
Case Review: Whole-Exome Sequencing Analyses Identify Carriers of a Known Likely Pathogenic Intronic BRCA1 Variant in Ovarian Cancer Cases Clinically Negative for Pathogenic BRCA1 and BRCA2 Variants

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. WES Analysis of BRCA1 and BRCA2 Loci
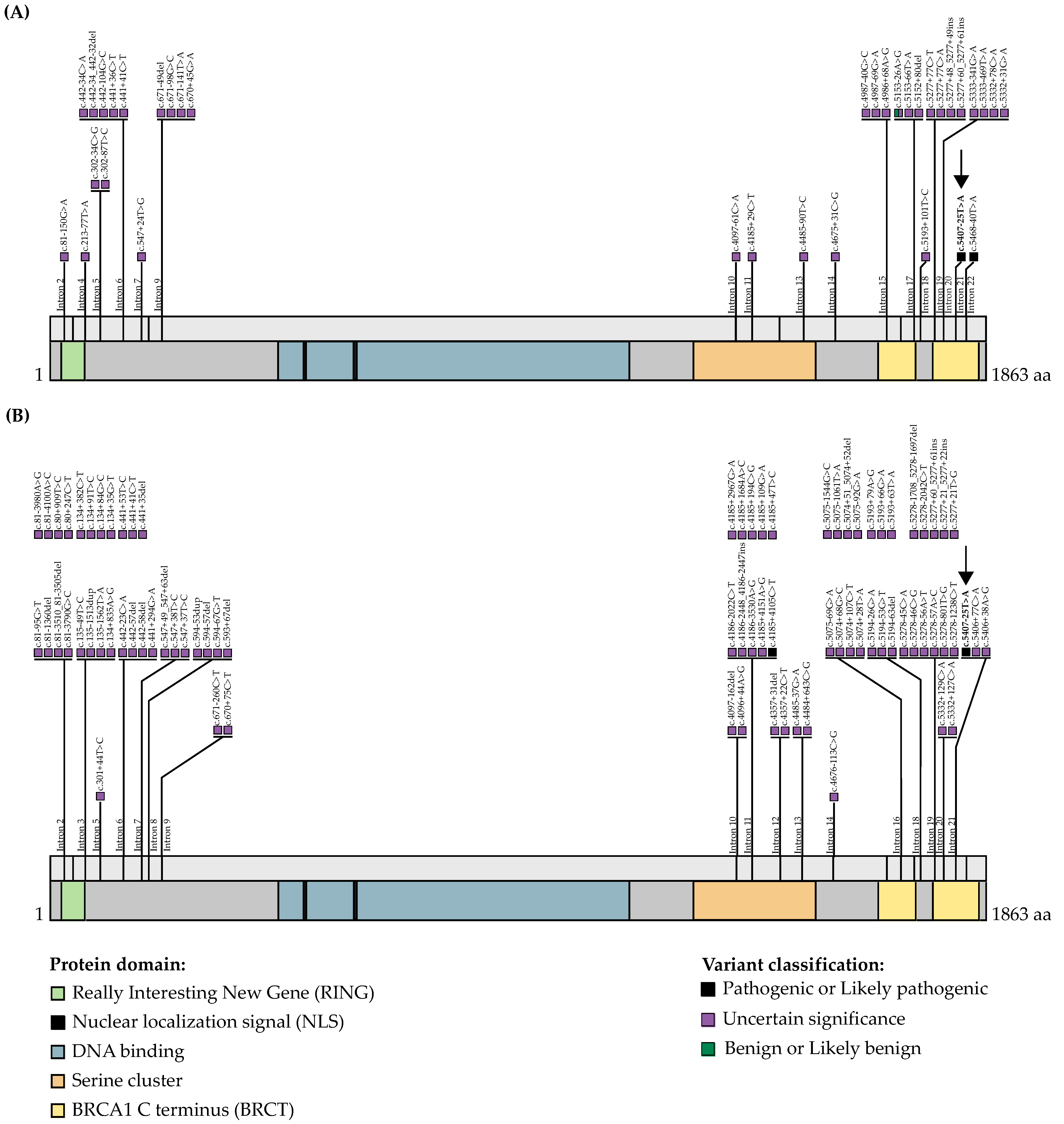
2.3. Databases and In Silico Tools for the Evaluation of BRCA1 and BRCA2 Variants
2.4. Surveying Carrier Frequencies in Other in-House Sequencing Data of OC Cases
2.5. Profiling Tumor DNA from BRCA1 c.5407-25T>A Variant Carriers
2.6. Characterization of BRCA1 c.5407-25T>A Carriers for Co-Occurring Pathogenic Variants in Other Known OC Risk Genes
3. Results
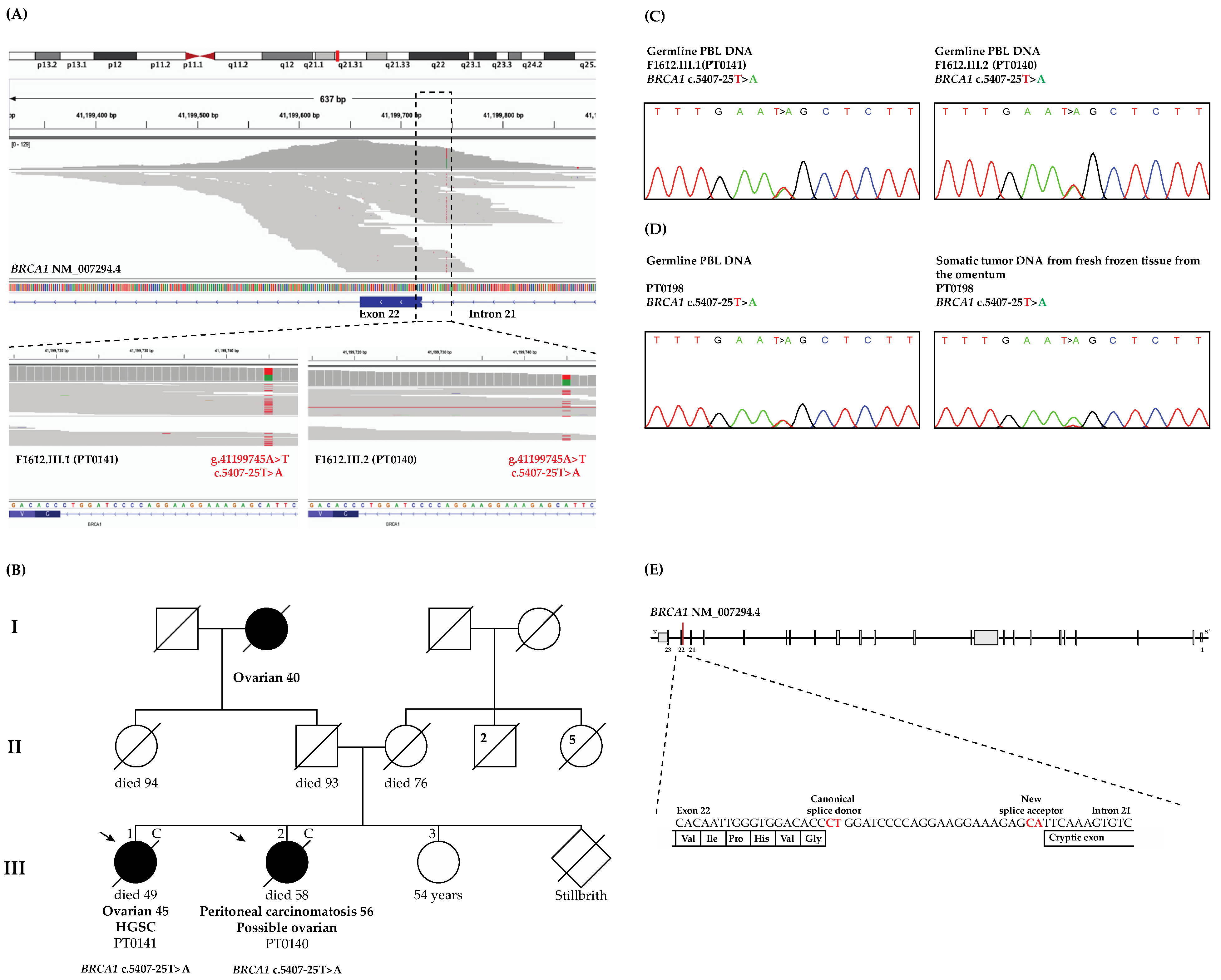
3.1. WES and Bioinformatics Analysis Identified BRCA1 c.5407-25T>A as a Candidate Pathogenic Variant
3.2. WES Analyses Identified Another OC Case Harboring BRCA1 c.5407-25T>A
3.3. WES Analyses of BRCA1 c.5407-25T>A Carriers Suggest That They Are Unlikely to Harbor Pathogenic Variants in the Other Known OC Risk Genes
3.4. LOH Analysis of the Tumor DNA from BRCA1 c.5407-25T>A Carrier Revealed Loss of the Wild-Type Allele
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. J. Am. Med. Assoc. 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- PDQ Adult Treatment Editorial Board. Ovarian Epithelial, Fallopian Tube, and Primary Peritoneal Cancer Treatment (PDQ®): Patient Version; National Cancer Institute at the National Institutes of Health: Bethesda, MD, USA, 2002; ISBN 0099-2240. [Google Scholar]
- Torre, L.A.; Trabert, B.; Desantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pavanello, M.; Chan, I.H.; Ariff, A.; Pharoah, P.D.; Gayther, S.A.; Ramus, S.J. Rare germline genetic variants and the risks of epithelial ovarian cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef]
- da Costa, P.J.; Menezes, J.; Romão, L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int. J. Biochem. Cell Biol. 2017, 91, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Wieme, G.; Kral, J.; Rosseel, T.; Zemankova, P.; Parton, B.; Vocka, M.; Van Heetvelde, M.; Kleiblova, P.; Blaumeiser, B.; Soukupova, J.; et al. Prevalence of germline pathogenic variants in cancer predisposing genes in czech and belgian pancreatic cancer patients. Cancers 2021, 13, 4430. [Google Scholar] [CrossRef] [PubMed]
- Casadei, S.; Gulsuner, S.; Shirts, B.H.; Mandell, J.B.; Kortbawi, H.M.; Norquist, B.S.; Swisher, E.M.; Lee, M.K.; Goldberg, Y.; O’Connor, R.; et al. Characterization of splice-altering mutations in inherited predisposition to cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 26798–26807. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Truong, T.T.N.; Weaver, J.E.; Bove, B.A.; Cattie, K.; Armstrong, B.A.; Daly, M.B.; Godwin, A.K. Intronic alterations in BRCA1 and BRCA2: Effect on mRNA splicing fidelity and expression. Hum. Mutat. 2006, 27, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Lionel, A.C.; Costain, G.; Monfared, N.; Walker, S.; Reuter, M.S.; Hosseini, S.M.; Thiruvahindrapuram, B.; Merico, D.; Jobling, R.; Nalpathamkalam, T.; et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 2017, 20, 435–443. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef]
- Montalban, G.; Bonache, S.; Bach, V.; Gisbert-Beamud, A.; Tenés, A.; Moles-Fernández, A.; López-Fernández, A.; Carrasco, E.; Balmaña, J.; Diez, O.; et al. BRCA1 and BRCA2 whole cDNA analysis in unsolved hereditary breast/ovarian cancer patients. Cancer Genet. 2021, 258, 10–17. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Munson, K.M.; Eng, M.; Mandell, J.B.; Gulsuner, S.; King, M.C. CRISPR–Cas9/long-read sequencing approach to identify cryptic mutations in BRCA1 and other tumour suppressor genes. J. Med. Genet. 2021, 58, 850–852. [Google Scholar] [CrossRef]
- Pirim, D.; Kaya, N.; Yıldırım, E.U.; Sag, Ş.O.; Temel, S.G. Characterization and in silico analyses of the BRCA1/2 variants identified in individuals with personal and/or family history of BRCA-related cancers. Int. J. Biol. Macromol. 2020, 162, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Montalban, G.; Bonache, S.; Moles-Fernández, A.; Gisbert-Beamud, A.; Tenés, A.; Bach, V.; Carrasco, E.; López-Fernández, A.; Stjepanovic, N.; Balmaña, J.; et al. Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J. Med. Genet. 2019, 56, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Gelli, E.; Colombo, M.; Pinto, A.M.; De Vecchi, G.; Foglia, C.; Amitrano, S.; Morbidoni, V.; Imperatore, V.; Manoukian, S.; Baldassarri, M.; et al. Usefulness and limitations of comprehensive characterization of mRNA splicing profiles in the definition of the clinical relevance of BRCA1/2 variants of uncertain significance. Cancers 2019, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Rivera, B.; Iorio, M.D.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally null RAD51D missense mutation associates strongly with ovarian carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef]
- Felicio, P.S.; Grasel, R.S.; Campacci, N.; de Paula, A.E.; Galvão, H.C.R.; Torrezan, G.T.; Sabato, C.S.; Fernandes, G.C.; Souza, C.P.; Michelli, R.D.; et al. Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 2021, 42, 290–299. [Google Scholar] [CrossRef]
- Fierheller, C.T.; Guitton-Sert, L.; Alenezi, W.M.; Revil, T.; Oros, K.K.; Gao, Y.; Bedard, K.; Arcand, S.L.; Serruya, C.; Behl, S.; et al. A functionally impaired missense variant identified in French Canadian families implicates FANCI as a candidate ovarian cancer-predisposing gene. Genome Med. 2021, 13, 186. [Google Scholar] [CrossRef]
- Shigemizu, D.; Momozawa, Y.; Abe, T.; Morizono, T.; Boroevich, K.A.; Takata, S.; Ashikawa, K.; Kubo, M.; Tsunoda, T. Performance comparison of four commercial human whole-exome capture platforms. Sci. Rep. 2015, 5, 12742. [Google Scholar] [CrossRef]
- Asan; Xu, Y.; Jiang, H.; Tyler-Smith, C.; Xue, Y.; Jiang, T.; Wang, J.; Wu, M.; Liu, X.; Tian, G.; et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011, 12, R95. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Khera, A.V.; Franci-Oli, L.C.; Gauthier, L.D.; Wang, H.; Watts, N.A.; et al. gnomAD-SV An open resource of structural variation for medical and population genetics The Genome Aggregation Database (gnomAD) Production Team 7, The gnomAD Consortium. bioRxiv 2019. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Brown, J.M.; Dashnow, H.; Wallace, A.D.; Velinder, M.; Tristani-Firouzi, M.; Schiffman, J.D.; Tvrdik, T.; Mao, R.; Best, D.H.; et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med. 2021, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature review of BARD1 as a cancer predisposing gene with a focus on breast and ovarian cancers. Genes 2020, 11, 856. [Google Scholar] [CrossRef] [PubMed]
- Fierheller, C.T.; Alenezi, W.M.; Tonin, P.N. The genetic analyses of French Canadians of Quebec facilitate the characterization of new cancer predisposing genes implicated in hereditary breast and/or ovarian cancer syndrome families. Cancers 2021, 13, 3406. [Google Scholar] [CrossRef]
- Liu, Y.L.; Breen, K.; Catchings, A.; Ranganathan, M.; Latham, A.; Goldfrank, D.J.; Grisham, R.N.; Long Roche, K.; Frey, M.K.; Chi, D.S.; et al. Risk-reducing bilateral salpingo-oophorectomy for ovarian cancer: A review and clinical guide for hereditary predisposition genes. JCO Oncol. Pract. 2021, 18, 201–209. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. Genetic/familial high-risk assessment: Breast, ovarian, and pancreatic, version 1.2020 featured updates to the NCCN guidelines. JNCCN J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.; Hunt, S.E.; Waddell, N.; Spurdle, A.B. A plugin for the Ensembl Variant Effect Predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Ponting, C.P. Biological function in the twilight zone of sequence conservation. BMC Biol. 2017, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2012, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- de Garibay, G.R.; Fernandez-Garcia, I.; Mazoyer, S.; de Calais, F.L.; Ameri, P.; Vijayakumar, S.; Martinez-Ruiz, H.; Damiola, F.; Barjhoux, L.; Thomassen, M.; et al. Altered regulation of BRCA1 exon 11 splicing is associated with breast cancer risk in carriers of BRCA1 pathogenic variants. Hum. Mutat. 2021, 42, 1488–1502. [Google Scholar] [CrossRef]
- Kanchi, K.L.; Johnson, K.J.; Lu, C.; McLellan, M.D.; Leiserson, M.D.M.; Wendl, M.C.; Zhang, Q.; Koboldt, D.C.; Xie, M.; Kandoth, C.; et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014, 5, 3156. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Chasin, L.A. Multiple splicing defects in an intronic false exon. Mol. Cell. Biol. 2000, 20, 6414–6425. [Google Scholar] [CrossRef]
- Qian, X.; Wang, J.; Wang, M.; Igelman, A.D.; Jones, K.D.; Li, Y.; Wang, K.; Goetz, K.E.; Birch, D.G.; Yang, P.; et al. Identification of deep-intronic splice mutations in a large cohort of patients with inherited retinal diseases. Front. Genet. 2021, 12, 647400. [Google Scholar] [CrossRef]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Köbel, M.; Kang, E.Y. The evolution of ovarian carcinoma subclassification. Cancers 2022, 14, 416. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Eccles, D.M.; Rahman, N.; Young, K.; Bulman, M.; Amir, E.; Shenton, A.; Howell, A.; Lalloo, F. A new scoring system for the chances of identifying a BRCA1/2 mutation outperforms existing models including BRCAPRO. J. Med. Genet. 2004, 41, 474–480. [Google Scholar] [CrossRef]
- Evans, D.G.; Lalloo, F.; Wallace, A.; Rahman, N. Update on the Manchester Scoring System for BRCA1 and BRCA2 testing. J. Med. Genet. 2005, 42, e39. [Google Scholar] [CrossRef]
- Evans, D.G.; Harkness, E.F.; Plaskocinska, I.; Wallace, A.J.; Clancy, T.; Woodward, E.R.; Howell, T.A.; Tischkowitz, M.; Lalloo, F. Pathology update to the Manchester Scoring System based on testing in over 4000 families. J. Med. Genet. 2017, 54, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Hamann, U.; Liu, X.; Bungardt, N.; Ulrich Ulmer, H.; Bastert, G.; Sinn, H.P. Similar contributions of BRCA1 and BRCA2 germline mutations to early-onset breast cancer in Germany. Eur. J. Hum. Genet. 2003, 11, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Høberg-Vetti, H.; Ognedal, E.; Buisson, A.; Vamre, T.B.A.; Ariansen, S.; Hoover, J.M.; Eide, G.E.; Houge, G.; Fiskerstrand, T.; Haukanes, B.I.; et al. The intronic BRCA1 c.5407-25T>A variant causing partly skipping of exon 23—A likely pathogenic variant with reduced penetrance? Eur. J. Hum. Genet. 2020, 28, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Høberg-Vetti, H.; Bjorvatn, C.; Fiane, B.E.; Aas, T.; Woie, K.; Espelid, H.; Rusken, T.; Eikesdal, H.P.; Listøl, W.; Haavind, M.T.; et al. BRCA1/2 testing in newly diagnosed breast and ovarian cancer patients without prior genetic counselling: The DNA-BONus study. Eur. J. Hum. Genet. 2016, 24, 881–888. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Important differences with common interests. Nat. Rev. Cancer 2012, 12, 372. [Google Scholar] [CrossRef]
- Konstantopoulou, I.; Tsitlaidou, M.; Fostira, F.; Pertesi, M.; Stavropoulou, A.V.; Triantafyllidou, O.; Tsotra, E.; Tsiftsoglou, A.P.; Tsionou, C.; Droufakou, S.; et al. High prevalence of BRCA1 founder mutations in Greek breast/ovarian families. Clin. Genet. 2014, 85, 36–42. [Google Scholar] [CrossRef]
- Heramb, C.; Wangensteen, T.; Grindedal, E.M.; Ariansen, S.L.; Lothe, S.; Heimdal, K.R.; Mæhle, L. BRCA1 and BRCA2 mutation spectrum—An update on mutation distribution in a large cancer genetics clinic in Norway. Hered. Cancer Clin. Pract. 2018, 16, 3. [Google Scholar] [CrossRef]
- Møller, P.; Dominguez-Valentin, M.; Rødland, E.A.; Hovig, E. Causes for frequent pathogenic BRCA1 variants include low penetrance in fertile ages, recurrent de-novo mutations and genetic drift. Cancers 2019, 11, 132. [Google Scholar] [CrossRef]
- Wappenschmidt, B.; Becker, A.A.; Hauke, J.; Weber, U.; Engert, S.; Köhler, J.; Kast, K.; Arnold, N.; Rhiem, K.; Hahnen, E.; et al. Analysis of 30 putative BRCA1 splicing mutations in hereditary breast and ovarian cancer families identifies exonic splice site mutations that escape in silico prediction. PLoS ONE 2012, 7, e50800. [Google Scholar] [CrossRef]
- Bonnet, C.; Krieger, S.; Vezain, M.; Rousselin, A.; Tournier, I.; Martins, A.; Berthet, P.; Chevrier, A.; Dugast, C.; Layet, V.; et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J. Med. Genet. 2008, 45, 438–446. [Google Scholar] [CrossRef]
- Ozcelik, H.; Shi, X.; Chang, M.C.; Tram, E.; Vlasschaert, M.; Nicola, N.D.; Kiselova, A.; Yee, D.; Goldman, A.; Dowar, M.; et al. Long-range PCR and next-generation sequencing of BRCA1 and BRCA2 in breast cancer. J. Mol. Diagn. 2012, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Velasco, E.; Infante, M.; Durán, M.; Pérez-Cabornero, L.; Sanz, D.J.; Esteban-Cardeñosa, E.; Miner, C. Heteroduplex analysis by capillary array electrophoresis for rapid mutation detection in large multiexon genes. Nat. Protoc. 2007, 2, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Landrith, T.; Li, B.; Cass, A.A.; Conner, B.R.; LaDuca, H.; McKenna, D.B.; Maxwell, K.N.; Domchek, S.; Morman, N.A.; Heinlen, C.; et al. Splicing profile by capture RNA-seq identifies pathogenic germline variants in tumor suppressor genes. NPJ Precis. Oncol. 2020, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Shi, J.; Cai, Q.; Shu, X.O.; He, J.; Wen, W.; Allen, J.; Pharoah, P.; Dunning, A.; Hunter, D.J.; et al. Use of deep whole-genome sequencing data to identify structure risk variants in breast cancer susceptibility genes. Hum. Mol. Genet. 2018, 27, 853–859. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Acedo, A.; Velasco, E.A. Identification of eight spliceogenic variants in BRCA2 Exon 16 by minigene assays. Front. Genet. 2018, 9, 188. [Google Scholar] [CrossRef]
- Acedo, A.; Sanz, D.J.; Durán, M.; Infante, M.; Pérez-Cabornero, L.; Miner, C.; Velasco, E.A. Comprehensive splicing functional analysis of DNA variants of the BRCA2 gene by hybrid minigenes. Breast Cancer Res. 2012, 14, R87. [Google Scholar] [CrossRef]
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alducci, E.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Danis, D.; Jacobsen, J.O.B.; Carmody, L.C.; Gargano, M.A.; McMurry, J.A.; Hegde, A.; Haendel, M.A.; Valentini, G.; Smedley, D.; Robinson, P.N. Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am. J. Hum. Genet. 2021, 108, 1564–1577. [Google Scholar] [CrossRef]
- Moles-Fernández, A.; Duran-Lozano, L.; Montalban, G.; Bonache, S.; López-Perolio, I.; Menéndez, M.; Santamariña, M.; Behar, R.; Blanco, A.; Carrasco, E.; et al. Computational tools for splicing defect prediction in breast/ovarian cancer genes: How efficient are they at predicting RNA alterations? Front. Genet. 2018, 9, 366. [Google Scholar] [CrossRef]
- Kurnit, K.C.; Fleming, G.F.; Lengyel, E. Updates and new options in advanced epithelial ovarian cancer treatment. Obstet. Gynecol. 2021, 137, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Peng, H.; Qi, X.; Wu, M.; Zhao, X. Targeted therapies in gynecological cancers: A comprehensive review of clinical evidence. Signal Transduct. Target. Ther. 2020, 5, 137. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Year Reported | Population 1 | Number of Carriers per Study Group | Cancer Type in Carriers | Study Group Investigated 2 | Reference |
|---|---|---|---|---|---|
| 2003 | Germany | 1/90 | Breast | Early-onset cases not selected for family history of cancer | [53] |
| 2014 | Greece | 1/473 | Breast | HBC and HBOC families | [57] |
| 2016 | Norway | 2/893 | Breast | Cases not selected for family history of cancer | [55] |
| 2018 | Norway | 9/669 | Breast | HBC and HBOC families | [58] |
| 2019 | Norway | 8/1914 | Breast or ovarian | Sporadic cases and families | [59] |
| 2020 | Norway, France, United States of America | 20 | Breast or ovarian | Selected HBC and HBOC families | [54] |
| 2022 | French Canadian, Ashkenazi Jewish, Austria, United Kingdom, Germany, Italy | 2/27 | Ovarian | Families with at least two OC case within first-, second- or third-degree relatives | This report |
| 2022 | French Canadian | 1/53 | Ovarian | Sporadic OC case with early onset of the disease not selected for family history of cancer | This report |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alenezi, W.M.; Fierheller, C.T.; Revil, T.; Serruya, C.; Mes-Masson, A.-M.; Foulkes, W.D.; Provencher, D.; El Haffaf, Z.; Ragoussis, J.; Tonin, P.N. Case Review: Whole-Exome Sequencing Analyses Identify Carriers of a Known Likely Pathogenic Intronic BRCA1 Variant in Ovarian Cancer Cases Clinically Negative for Pathogenic BRCA1 and BRCA2 Variants. Genes 2022, 13, 697. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040697
Alenezi WM, Fierheller CT, Revil T, Serruya C, Mes-Masson A-M, Foulkes WD, Provencher D, El Haffaf Z, Ragoussis J, Tonin PN. Case Review: Whole-Exome Sequencing Analyses Identify Carriers of a Known Likely Pathogenic Intronic BRCA1 Variant in Ovarian Cancer Cases Clinically Negative for Pathogenic BRCA1 and BRCA2 Variants. Genes. 2022; 13(4):697. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040697
Chicago/Turabian StyleAlenezi, Wejdan M., Caitlin T. Fierheller, Timothée Revil, Corinne Serruya, Anne-Marie Mes-Masson, William D. Foulkes, Diane Provencher, Zaki El Haffaf, Jiannis Ragoussis, and Patricia N. Tonin. 2022. "Case Review: Whole-Exome Sequencing Analyses Identify Carriers of a Known Likely Pathogenic Intronic BRCA1 Variant in Ovarian Cancer Cases Clinically Negative for Pathogenic BRCA1 and BRCA2 Variants" Genes 13, no. 4: 697. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13040697