
Identification of Key Prognostic Genes of Triple Negative Breast Cancer by LASSO-Based Machine Learning and Bioinformatics Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
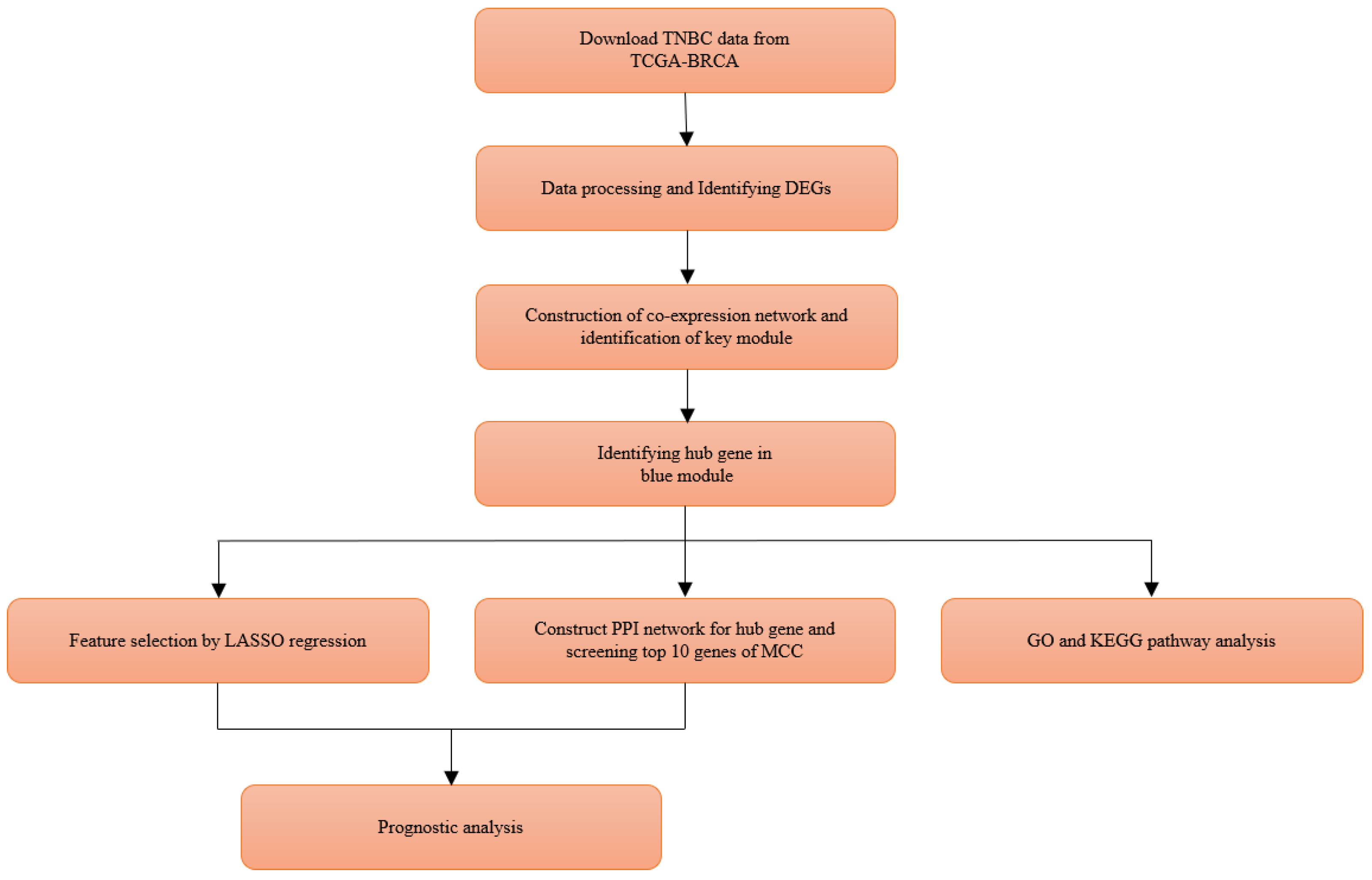
2. Materials and Methods
2.1. Data Collection from TCGA
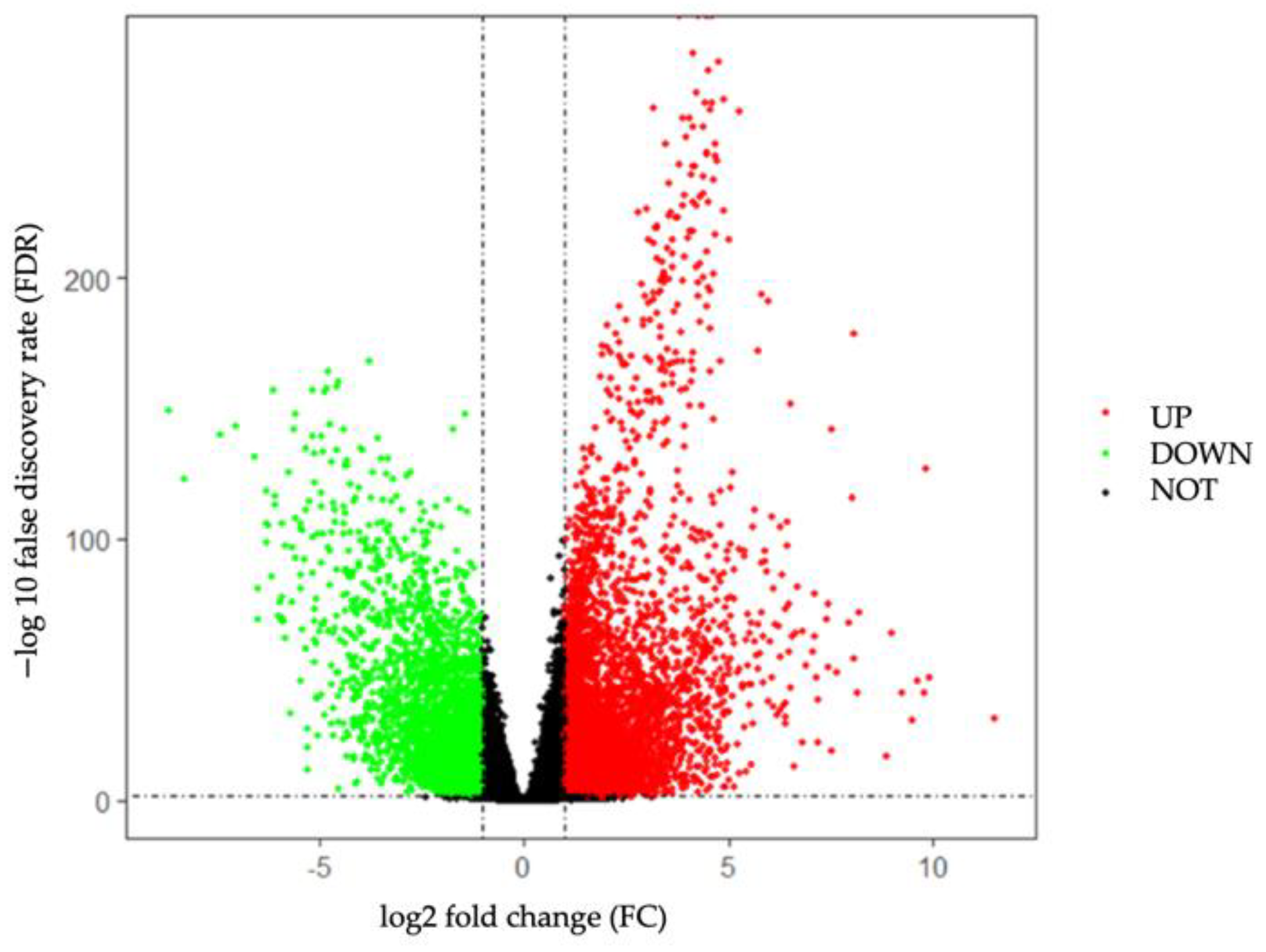
2.2. Screening for Differentially Expressed Genes
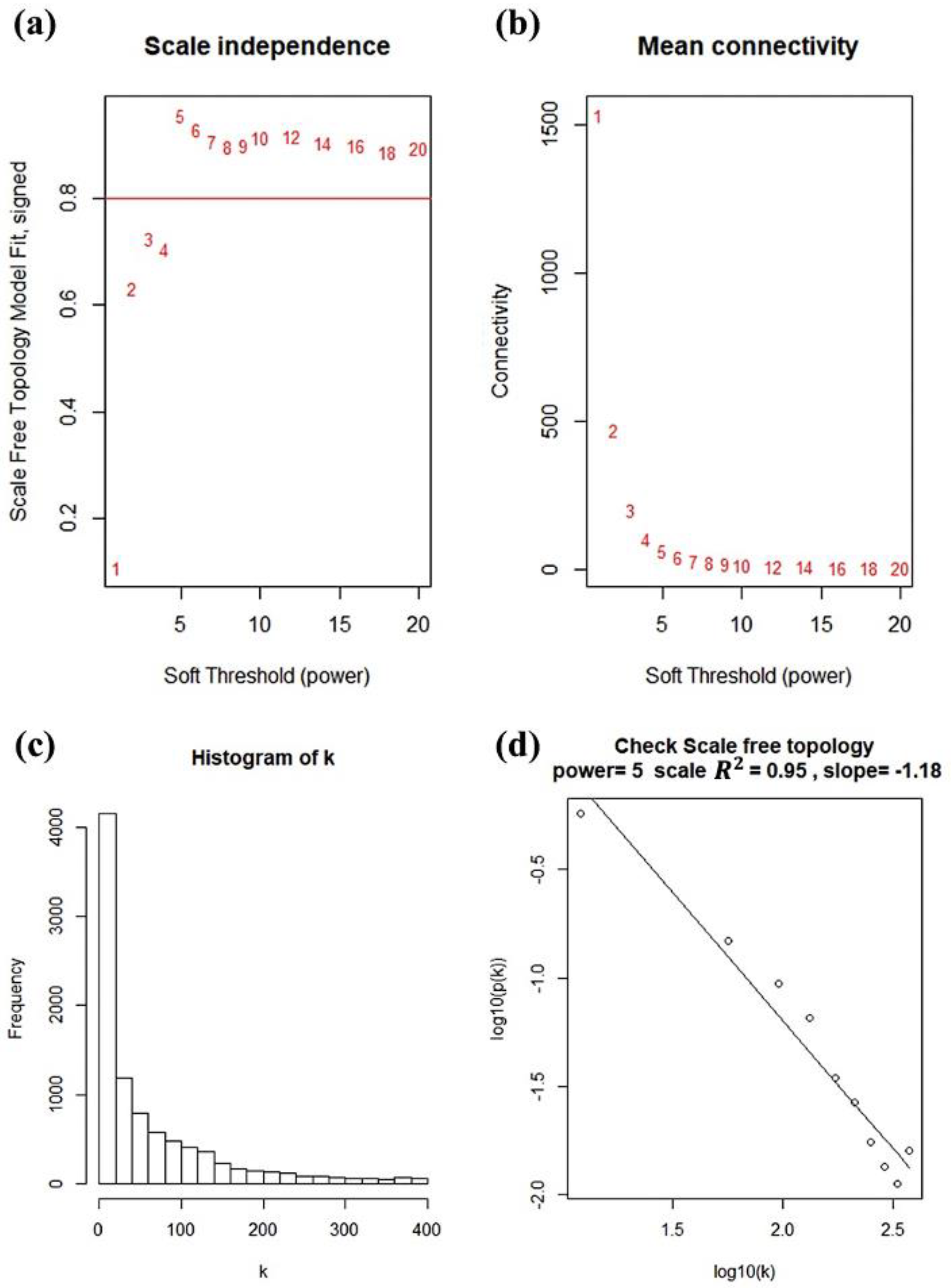
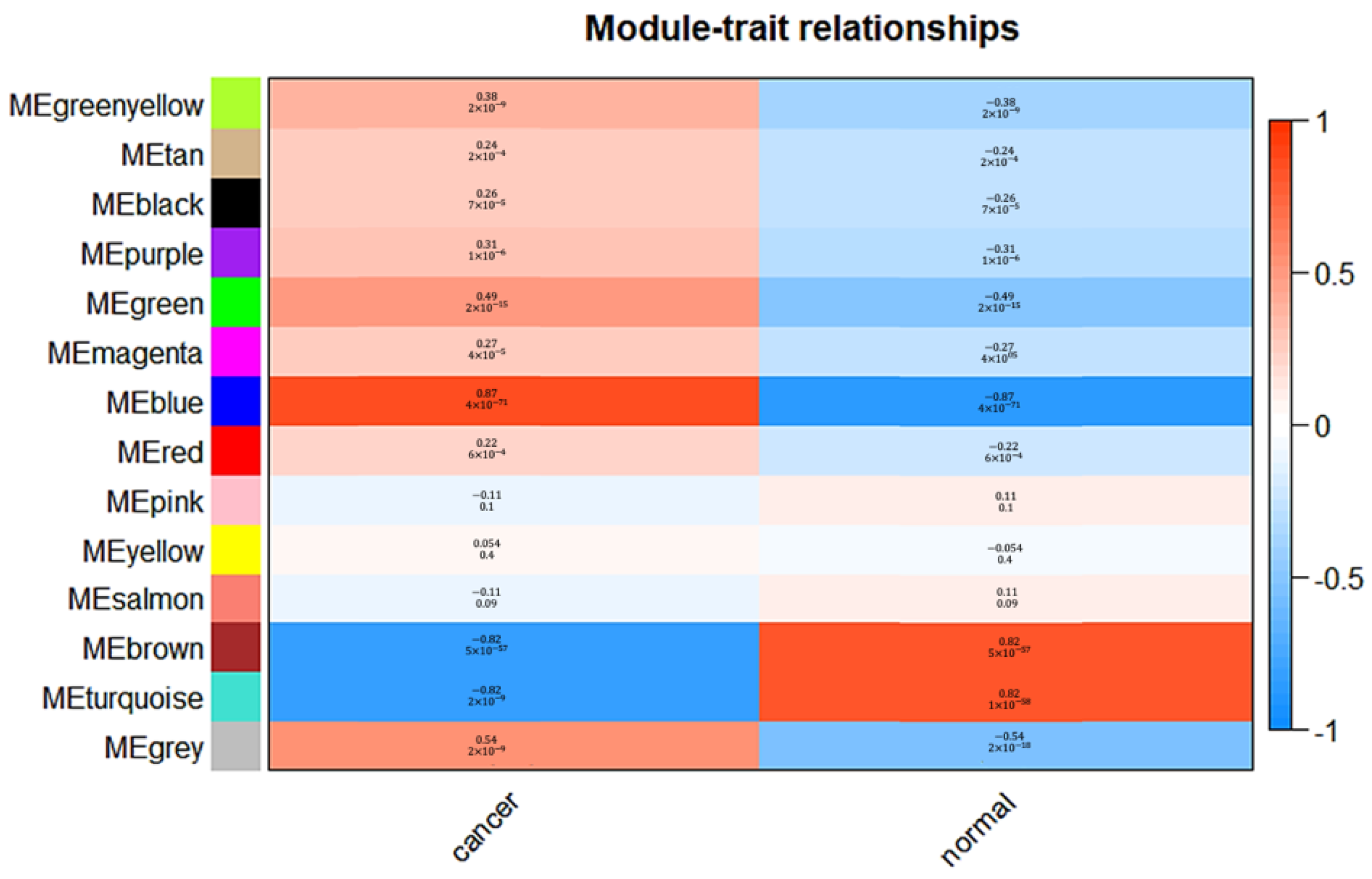
2.3. Construction of Co-Expression Network and Identificaion of Key Modules
2.4. Candidate Module Hub Genes Identification
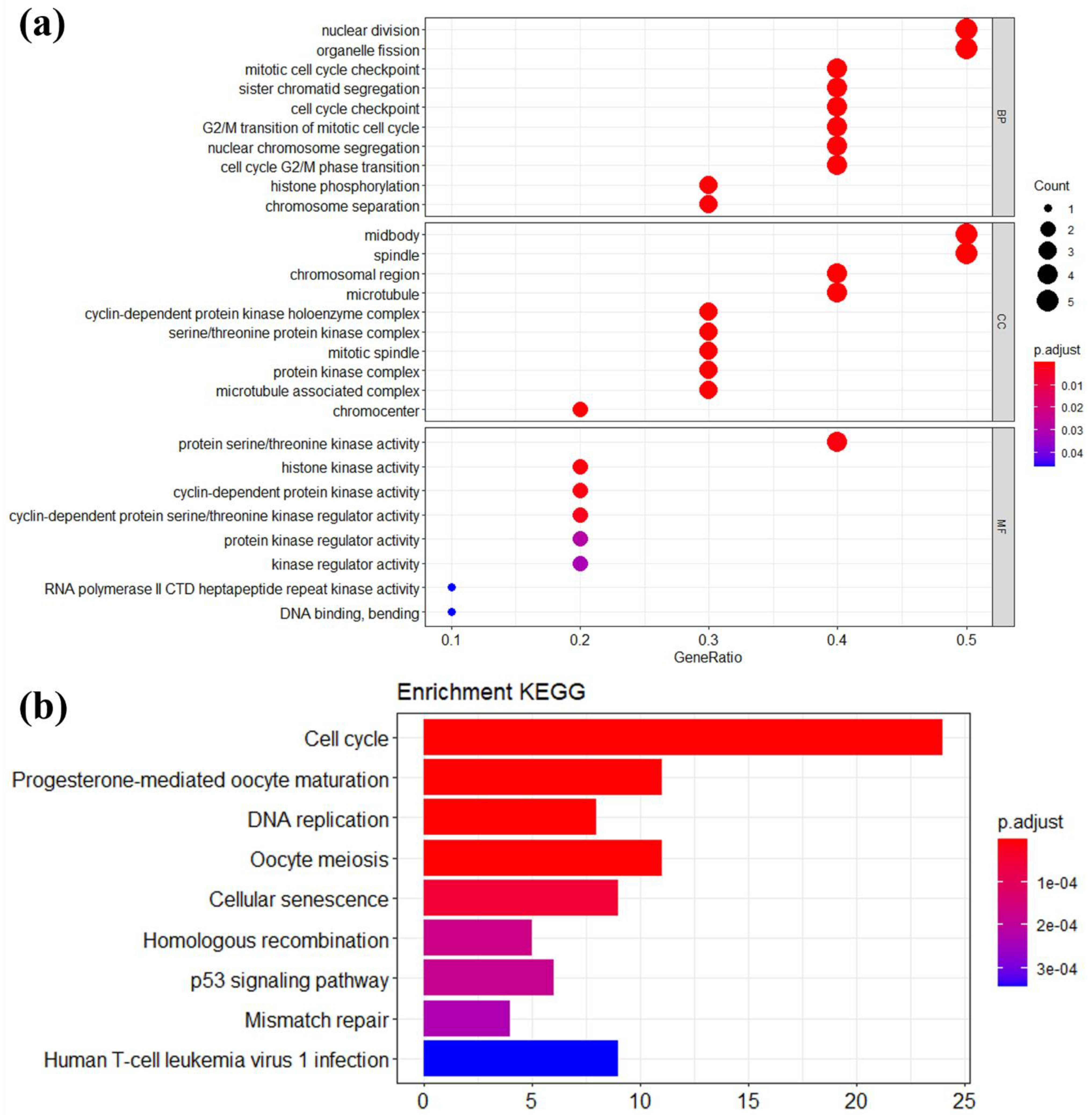
2.5. Functional and Pathway Enrichment Analyses
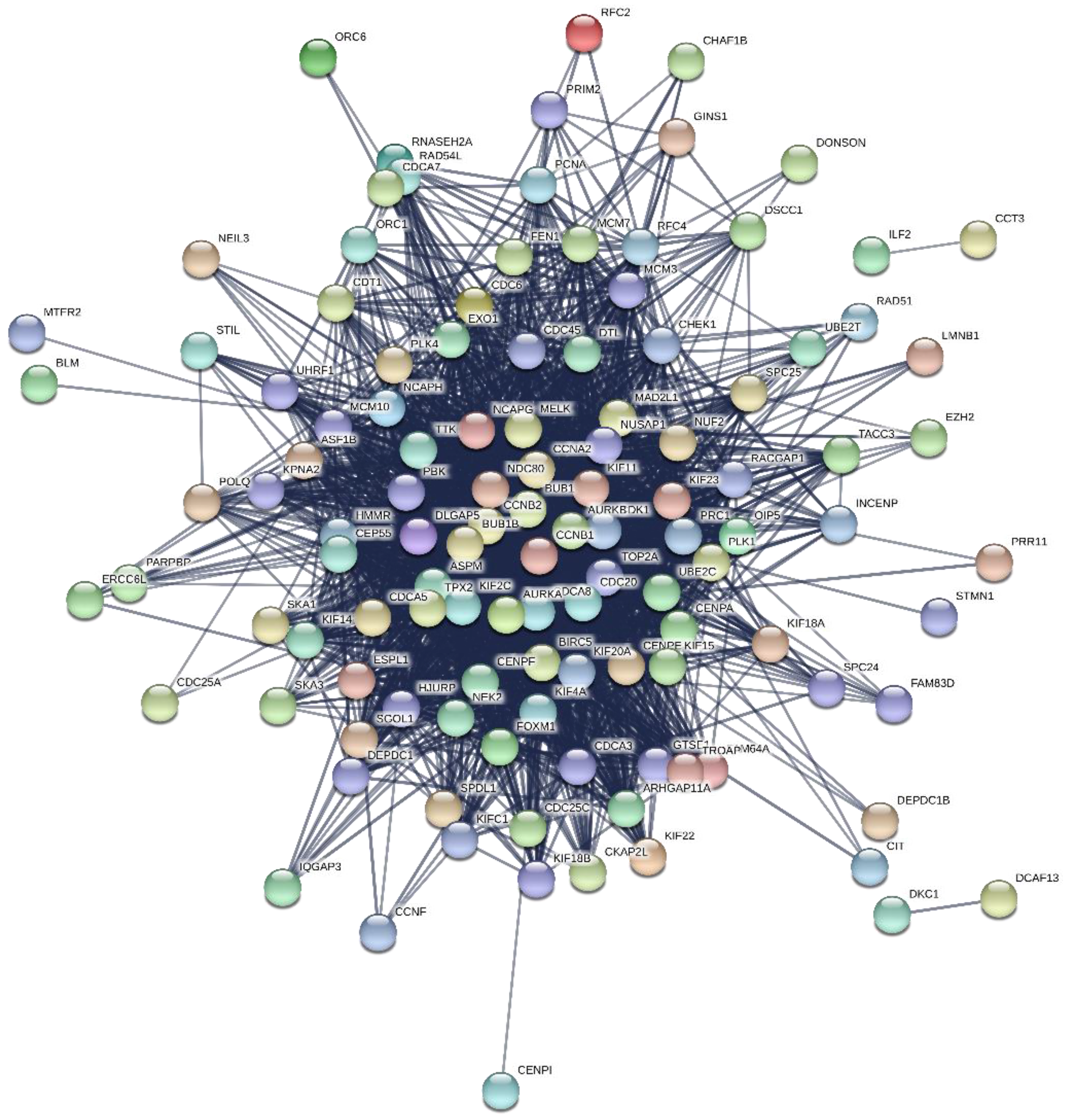
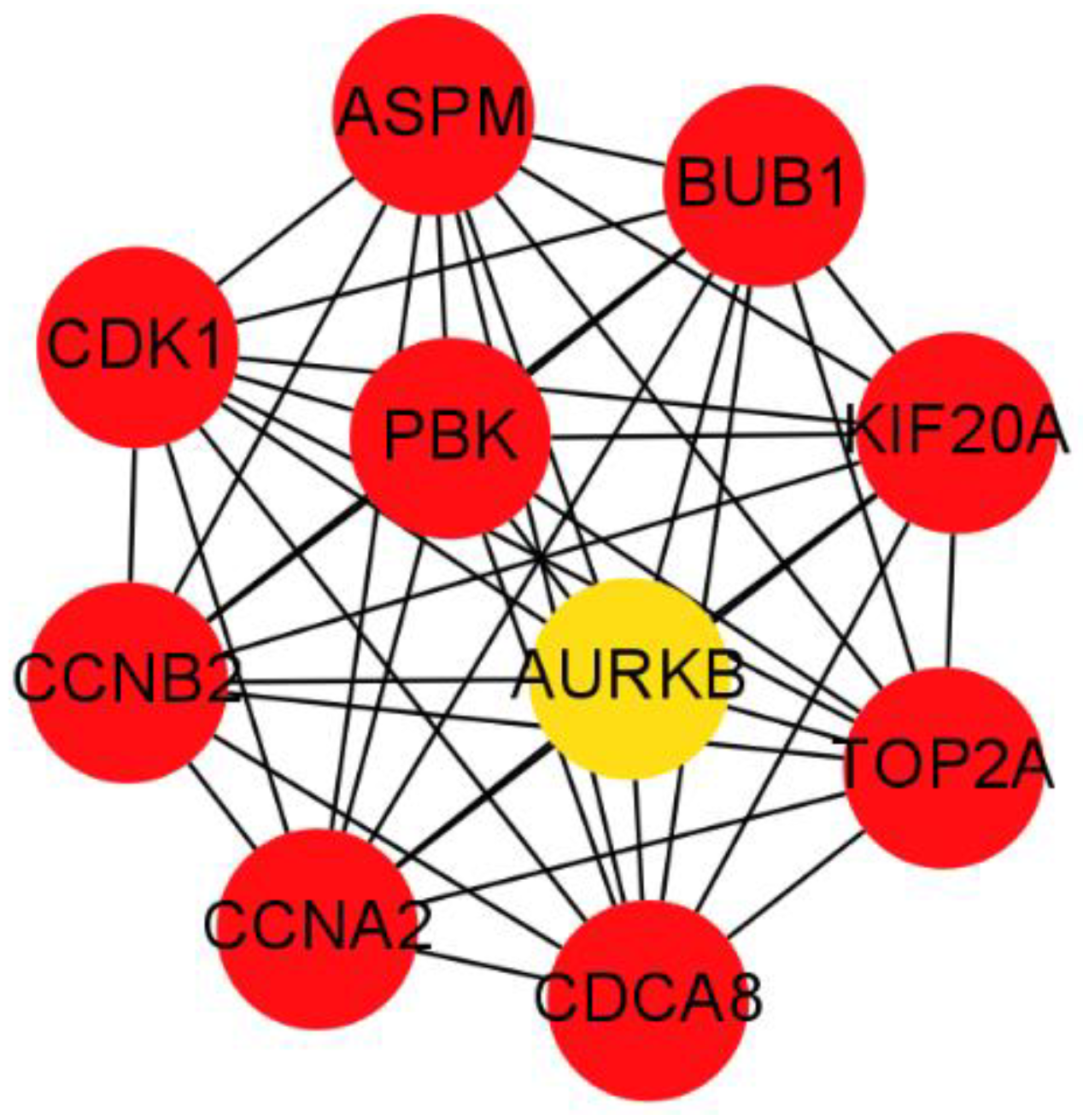
2.6. PPI Network Construction and Modules Selection
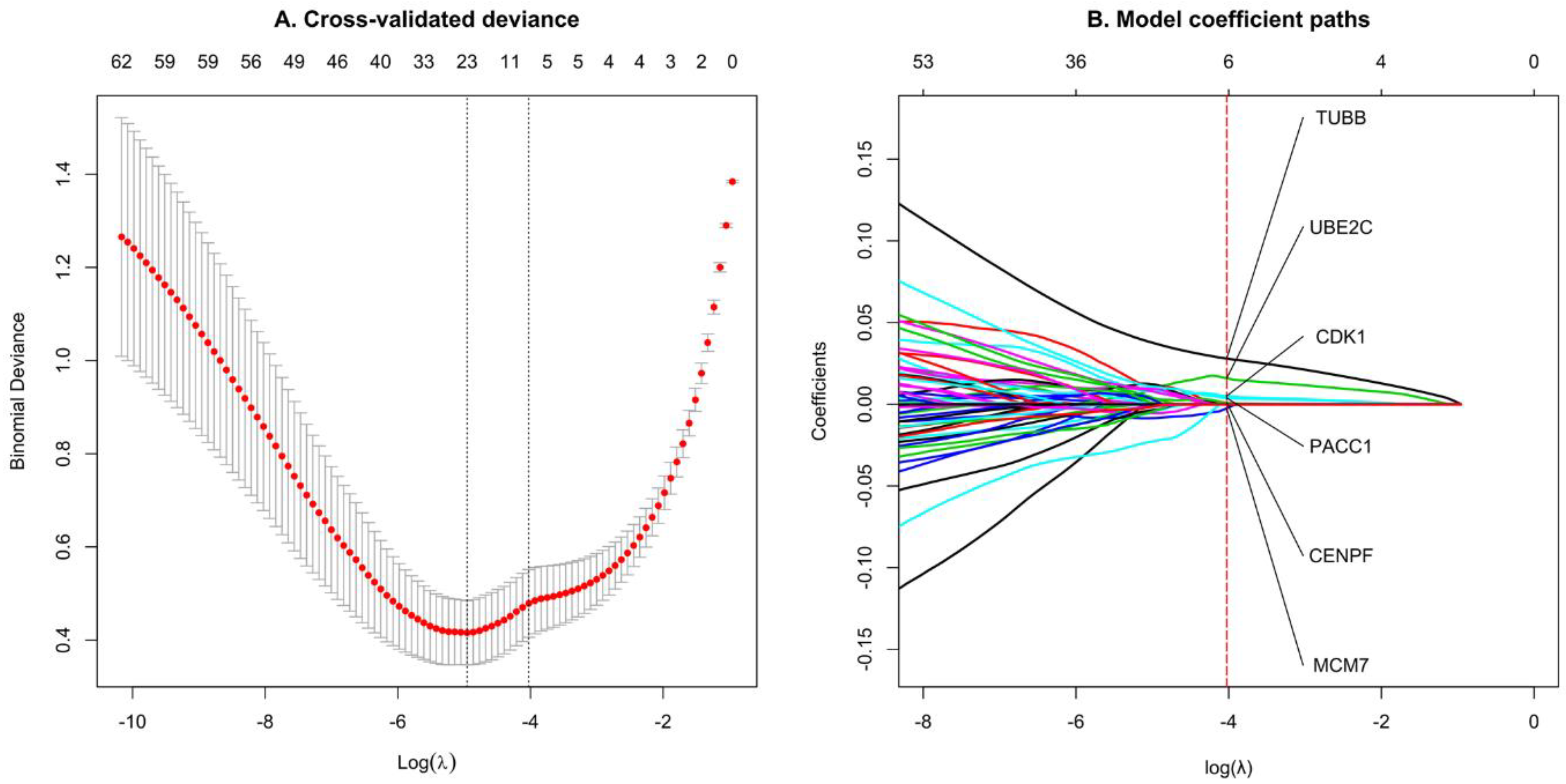
2.7. Feature Selection by LASSO Regression
2.8. Verification of Prognostic Value of Hub Genes
3. Results
3.1. Identification of DEGs in TNBC from TCGA
3.2. Weighted Co-Expression Network Construction and Gene Modules Identification
3.3. Module Genes Identification and Functional Enrichment Analysis
3.4. PPI Network Construction and Hub Genes Identification
3.5. Select Significant TNBC Genes Using LASSO Regression Model
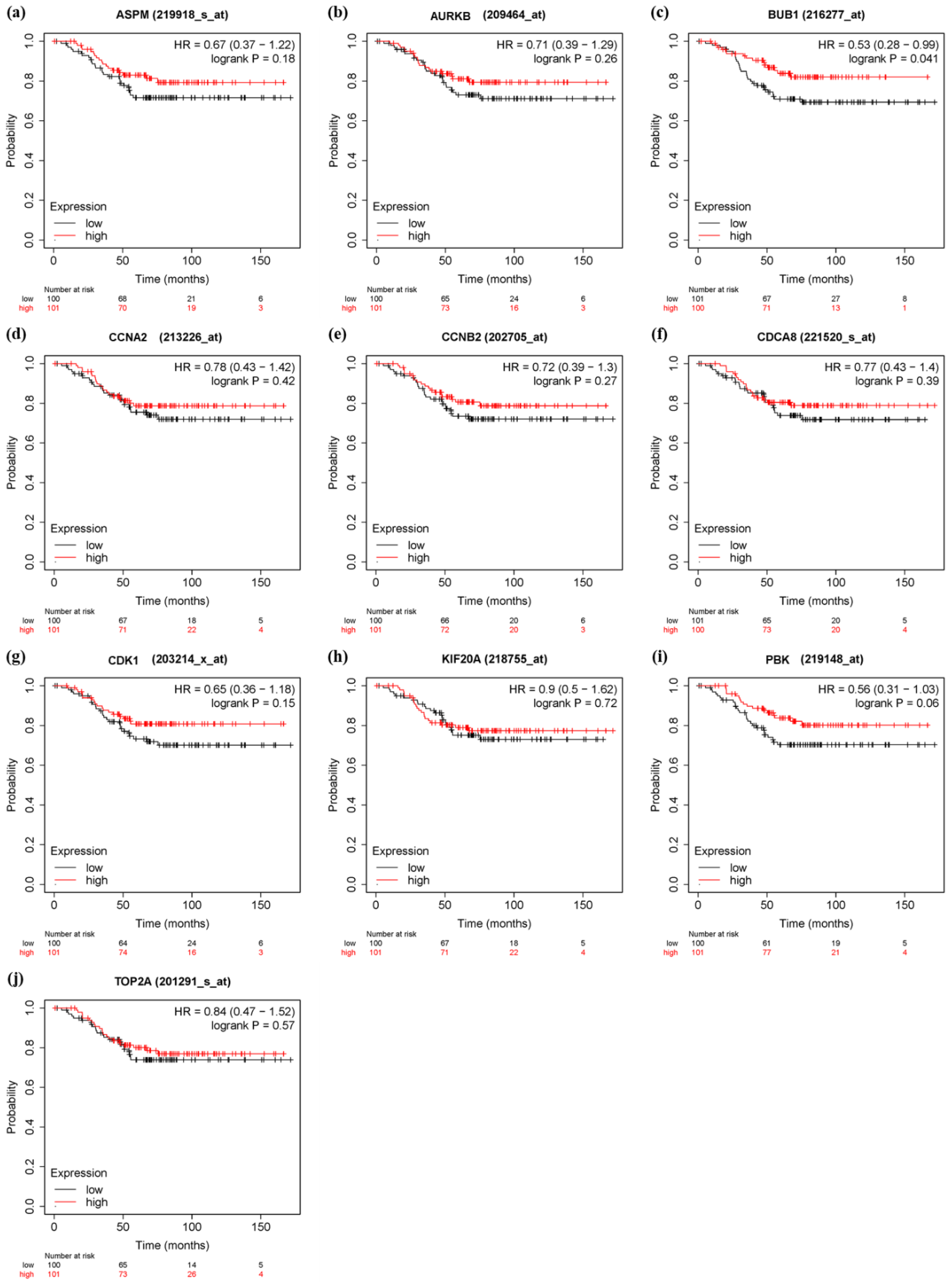
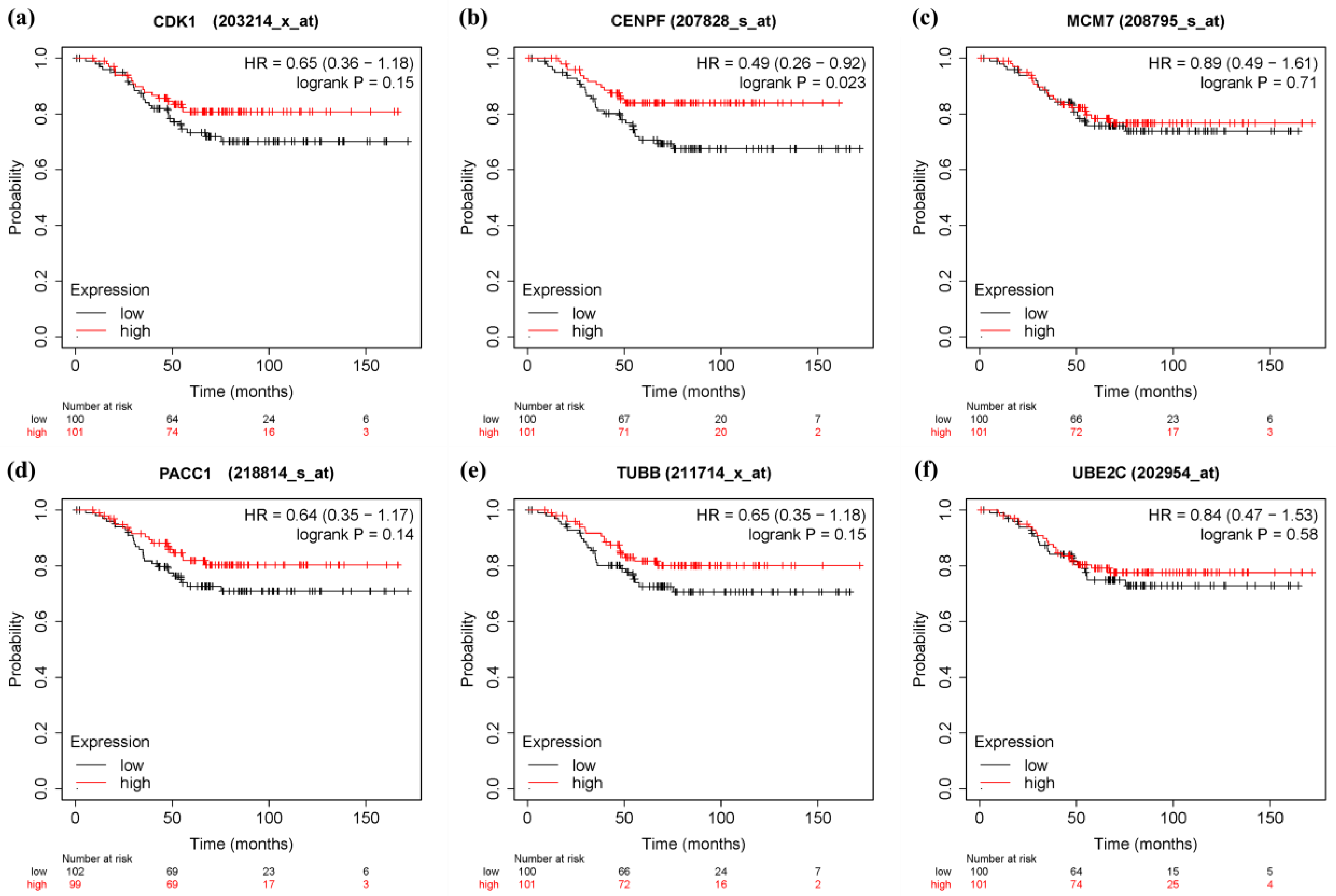
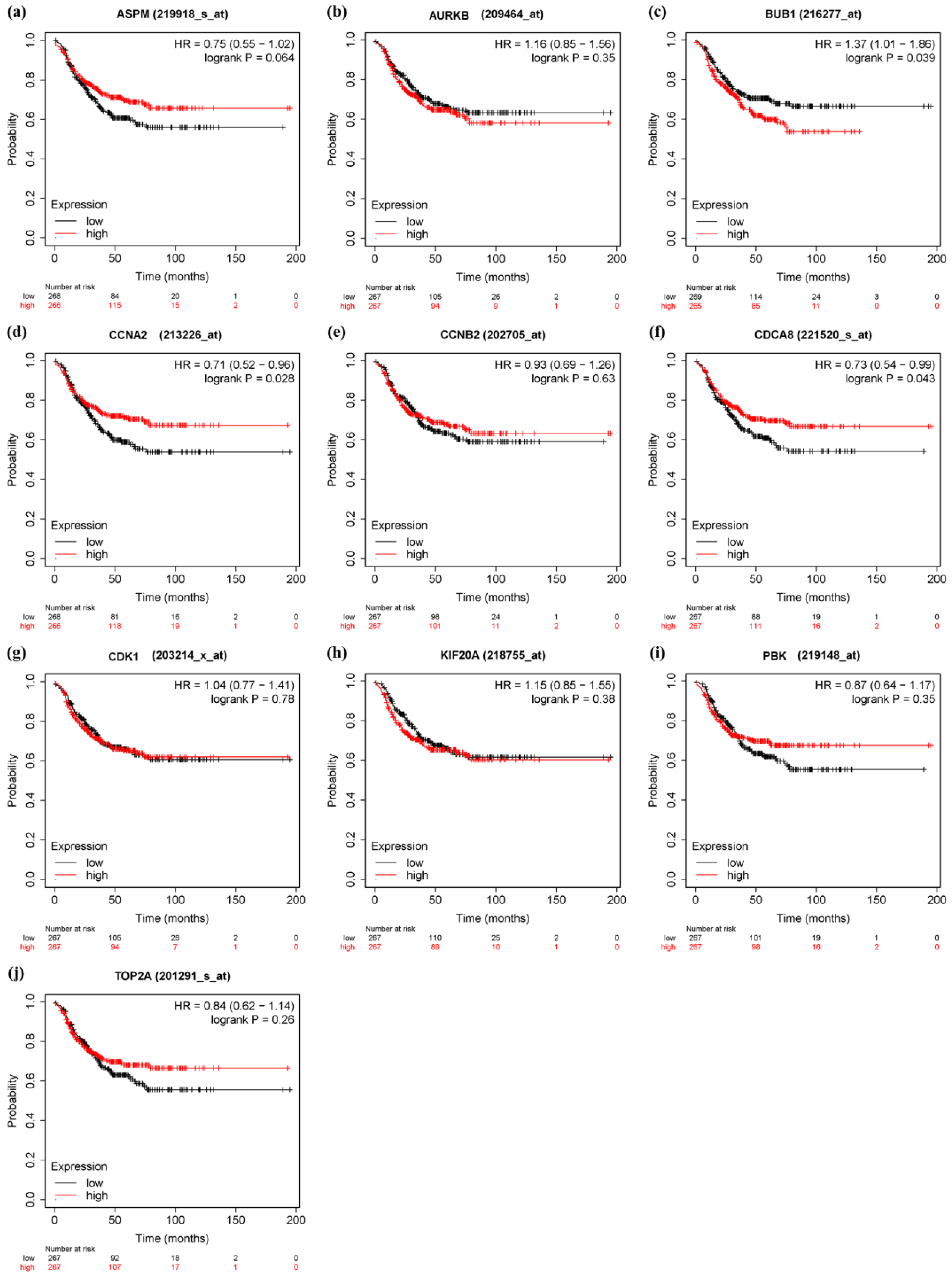
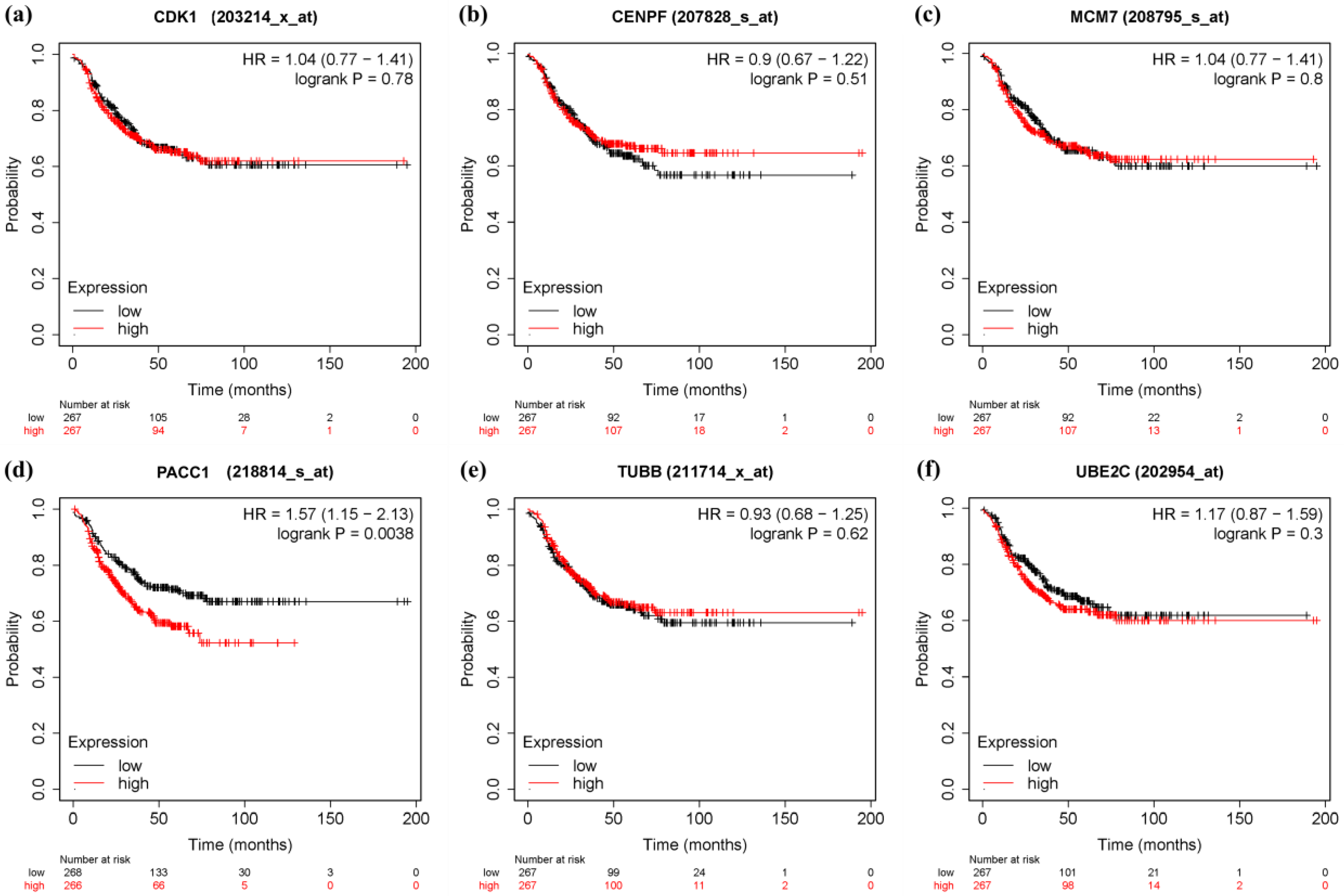
3.6. Survival Analysis and Prognostic Value Verification of Key Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, C.L.; Olin, J.L. Triple negative breast cancer: A brief review of its characteristics and treatment options. J. Pharm. Pract. 2012, 25, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Oakman, C.; Viale, G.; Di Leo, A. Management of triple negative breast cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Butte, A. The use and analysis of microarray data. Nat. Rev. Drug Discov. 2002, 1, 951–960. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Muthukrishnan, R.; Rohini, R. LASSO: A feature selection technique in predictive modeling for machine learning. In Proceedings of the 2016 IEEE International Conference on Advances in Computer Applications (ICACA), Coimbatore, India, 24–24 October 2016. [Google Scholar]
- Xue, F.; Yang, L.; Dai, B.; Xue, H.; Zhang, L.; Ge, R.; Sun, Y. Bioinformatics profiling identifies seven immune-related risk signatures for hepatocellular carcinoma. PeerJ 2020, 8, e8301. [Google Scholar] [CrossRef]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Reiner, A.; Yekutieli, D.; Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [Green Version]
- Ogata, H.; Goto, S.; Fujibuchi, W.; Kanehisa, M. Computation with the KEGG pathway database. Biosystems 1998, 47, 119–128. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016, 45, D362–D368. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Melkumova, L.; Shatskikh, S.Y. Comparing Ridge and LASSO estimators for data analysis. Procedia Eng. 2017, 201, 746–755. [Google Scholar] [CrossRef]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.A.; Grissom, P.M.; Arzhanik, V.K.; Zaytsev, A.V.; Renganathan, K.; McClure-Begley, T.; Old, W.M.; Ahn, N.; McIntosh, J.R. Centromere protein F includes two sites that couple efficiently to depolymerizing microtubules. J. Cell Biol. 2015, 209, 813–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manalo, A.; Schroer, A.K.; Fenix, A.M.; Shancer, Z.; Coogan, J.; Brolsma, T.; Burnette, D.T.; Merryman, W.D.; Bader, D.M. Loss of CENP-F results in dilated cardiomyopathy with severe disruption of cardiac myocyte architecture. Sci. Rep. 2018, 8, 7546. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Huang, J.; Lan, J.; Zhou, K.; Gao, Y.; Song, Z.; Deng, Y.; Liu, L.; Dong, Y.; Liu, X. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019, 19, 264. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, Y.-J.; Xi, M.; Wan, Y.-P.; Hua, W.; Liu, Y.-L.; Wan, S.; Zhou, Y.-L.; Luo, H.-W.; Wu, S.-L.; Zhong, W.-D. Enhanced expression of centromere protein F predicts clinical progression and prognosis in patients with prostate cancer. Int. J. Mol. Med. 2015, 35, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Wu, F.; Xu, H.; Wei, G.; Ding, M.; Xu, F.; Deivasigamani, A.; Zhou, G.; Hui, K.M.; Xia, H. Centromere protein F promotes progression of hepatocellular carcinoma through ERK and cell cycle-associated pathways. Cancer Gene Ther. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Yuan, B.; Xu, Y.; Woo, J.-H.; Wang, Y.; Bae, Y.K.; Yoon, D.-S.; Wersto, R.P.; Tully, E.; Wilsbach, K.; Gabrielson, E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin. Cancer Res. 2006, 12, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Myrie, K.A.; Percy, M.J.; Azim, J.N.; Neeley, C.K.; Petty, E.M. Mutation and expression analysis of human BUB1 and BUB1B in aneuploid breast cancer cell lines. Cancer Lett. 2000, 152, 193–199. [Google Scholar] [CrossRef]
- Takagi, K.; Miki, Y.; Shibahara, Y.; Nakamura, Y.; Ebata, A.; Watanabe, M.; Ishida, T.; Sasano, H.; Suzuki, T. BUB1 immunolocalization in breast carcinoma: Its nuclear localization as a potent prognostic factor of the patients. Horm. Cancer 2013, 4, 92–102. [Google Scholar] [CrossRef]
- Han, J.Y.; Han, Y.K.; Park, G.-Y.; Kim, S.D.; Geun Lee, C. Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Sci. Rep. 2015, 5, 15993. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Katsaros, D.; Shen, Y.; Fu, Y.; Canuto, E.M.; Benedetto, C.; Lu, L.; Chu, W.M.; Risch, H.A.; Yu, H. Biological and Clinical Significance of MAD2L1 and BUB1, Genes Frequently Appearing in Expression Signatures for Breast Cancer Prognosis. PLoS ONE 2015, 10, e0136246. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.J.; Pan, Y.; Chen, X.Y.; Hou, P.F. BUB1 promotes proliferation of liver cancer cells by activating SMAD2 phosphorylation. Oncol. Lett. 2020, 19, 3506–3512. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Huang, F.; Zhang, H.; Chen, Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci. Rep. 2019, 39, BSR20182306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, Y.; Li, Y.; Li, T.; Shu, G.; Yin, G. CCNA2 acts as a novel biomarker in regulating the growth and apoptosis of colorectal cancer. Cancer Manag. Res. 2018, 10, 5113. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhong, Q.; Li, Z.; Lin, Z.; Chen, H.; Wang, P. Integrated Profiling Identifies CCNA2 as a Potential Biomarker of Immunotherapy in Breast Cancer. OncoTargets Ther. 2021, 14, 2433. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, G.; Xiao, Y.; Zhang, T.; Su, F.; Chang, R.; Ling, X.; Bai, Y. Upregulated cyclins may be novel genes for triple-negative breast cancer based on bioinformatic analysis. Breast Cancer 2020, 27, 903–911. [Google Scholar] [CrossRef]
- Lu, Y.; Su, F.; Yang, H.; Xiao, Y.; Zhang, X.; Su, H.; Zhang, T.; Bai, Y.; Ling, X. E2F1 transcriptionally regulates CCNA2 expression to promote triple negative breast cancer tumorigenicity. Cancer Biomark. 2022, 33, 57–70. [Google Scholar] [CrossRef]
- Hanley, M.L.; Yoo, T.Y.; Sonnett, M.; Needleman, D.J.; Mitchison, T.J. Chromosomal passenger complex hydrodynamics suggests chaperoning of the inactive state by nucleoplasmin/nucleophosmin. Mol. Biol. Cell 2017, 28, 1444–1456. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Chen, S.; Jiang, J.; Yao, J.; Wang, G.; Zhou, Q.; Li, S. CDCA8 expression and its clinical relevance in patients with bladder cancer. Medicine 2018, 97, e11899. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Xu, J.; Hou, X.; Zhan, H.; Zhen, Y. Double-targeting CDCA8 and E2F1 inhibits the growth and migration of malignant glioma. Cell Death Dis. 2021, 12, 146. [Google Scholar] [CrossRef]
- Dai, C.; Miao, C.-X.; Xu, X.-M.; Liu, L.-J.; Gu, Y.-F.; Zhou, D.; Chen, L.-S.; Lin, G.; Lu, G.-X. Transcriptional activation of human CDCA8 gene regulated by transcription factor NF-Y in embryonic stem cells and cancer cells. J. Biol. Chem. 2015, 290, 22423–22434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, D.; Lu, Z.; Qiao, J.; Yan, M.; Cui, S.; Liu, Z. Expression of CDCA8 correlates closely with FOXM1 in breast cancer: Public microarray data analysis and immunohistochemical study. Neoplasma 2015, 62, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, S.; Dal Maso, T.; Chen, J.-W.; Bury, M.; Wouters, J.; Michiels, C.; Le Calvé, B. Transmembrane (TMEM) protein family members: Poorly characterized even if essential for the metastatic process. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Zhao, J.; Zhu, D.; Zhang, X.; Zhang, Y.; Zhou, J.; Dong, M. TMEM206 promotes the malignancy of colorectal cancer cells by interacting with AKT and extracellular signal-regulated kinase signaling pathways. J. Cell. Physiol. 2019, 234, 10888–10898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, S.-Y.; Yang, X.; Wang, Y.-Q.; Cheng, Y.-X. TMEM206 is a potential prognostic marker of hepatocellular carcinoma. Oncol. Lett. 2020, 20, 174. [Google Scholar] [CrossRef] [PubMed]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.-L.; Cai, J.-H.; Wang, C.C.N. Identification of Key Prognostic Genes of Triple Negative Breast Cancer by LASSO-Based Machine Learning and Bioinformatics Analysis. Genes 2022, 13, 902. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050902
Chen D-L, Cai J-H, Wang CCN. Identification of Key Prognostic Genes of Triple Negative Breast Cancer by LASSO-Based Machine Learning and Bioinformatics Analysis. Genes. 2022; 13(5):902. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050902
Chicago/Turabian StyleChen, De-Lun, Jia-Hua Cai, and Charles C. N. Wang. 2022. "Identification of Key Prognostic Genes of Triple Negative Breast Cancer by LASSO-Based Machine Learning and Bioinformatics Analysis" Genes 13, no. 5: 902. https://0-doi-org.brum.beds.ac.uk/10.3390/genes13050902