
Regulation of BLM Nucleolar Localization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cloning
- 5′ external primer: CAGCTTTTGGCCTACTTTGGT
- 3′ external primer: TTCCTTTTCGGTTAGAGCGGA
- 3′ 1332-1349 deletion internal primer: CCACTGGAAGCAGTTCTGGTTTTACTTGCAAAGTAGTGG
- 5′ 1332-1349 deletion internal primer: CAAGTAAAACCAGAACTGCTTCCAGTGGTTCCAA
- 3′ S1342A/S1345A internal primer: CTCTTAGCCCTTTGGGCGGCTGG
- 5′ S1342A/S1345A internal primer: CCAGCCGCCCAAAGGGCTAAGAG
- 3′ S1342D/S1345D internal primer: CTCTTATCCCTTTGGTCGGCTGGC
- 5′ S1342D/S1345D internal primer: GCCAGCCGACCAAAGGGATAAGAG
- 3′ S1342A internal primer: CTCTTAGACCTTTGGGCGGCTGG
- 5′ S1342A internal primer: CCAGCCGCCCAAAGGTCTAAGAG
- 3′ S1345A internal primer: CTCTTAGCCCTTTGGGAGGCTGG
- 5′ S1345A internal primer: CCAGCCTCCCAAAGGGCTAAGAG
- 3′ S1342D internal primer: CTCTTAGACCTTTGGTCGGCTGGC
- 5′ S1342D internal primer: GCCAGCCGACCAAAGGTCTAAGAG
- 3′ S1345D internal primer: CTCTTATCCCTTTGGGAGGCTGGC
- 5′ S1345D internal primer: GCCAGCCTCCCAAAGGGATAAGAG
- 5′ TOP1 primer: CGTAGAGCTCGGATCCCTAACCATGCACCACCACCACCACCACAGTGGGGACCACCTC
- 3′ TOP1 primer: TACGCTCGAGTCAAAACTCATAGTCTTCATCAGCC
2.3. In Vitro Transcription and Translation
2.4. Protein Purification
2.5. Helicase Assays
- DNA20: CGCTAGCAATATTCTGCAGC
- RNA20: CGCUAGCAAUAUUCUGCAGC
- DNA33: GCTGCAGAATATTGCTAGCGGGAATTCGGCGCG
- TTTTTTTTTTTTTTTTTAGGGTTAGGGCATGCACTAC
- GTAGTGCATGCCCTAACCCTAATTTTTTTTTTTTTTT
2.6. Alignment and in Silico Phosphorylation Prediction
2.7. Immunofluorescence
3. Results
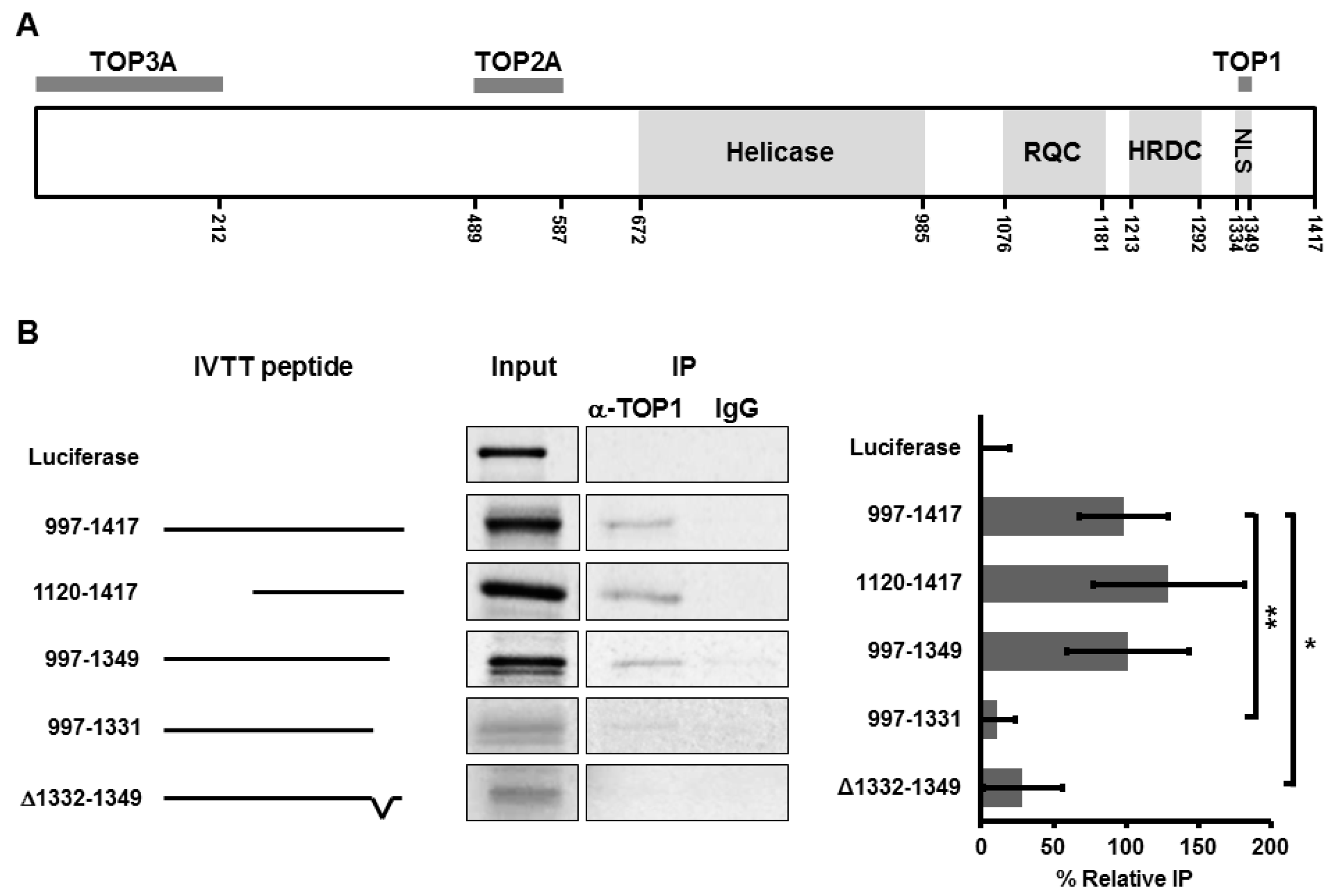
3.1. BLM Amino Acids 1332–1349 Are Required for Binding to TOP1
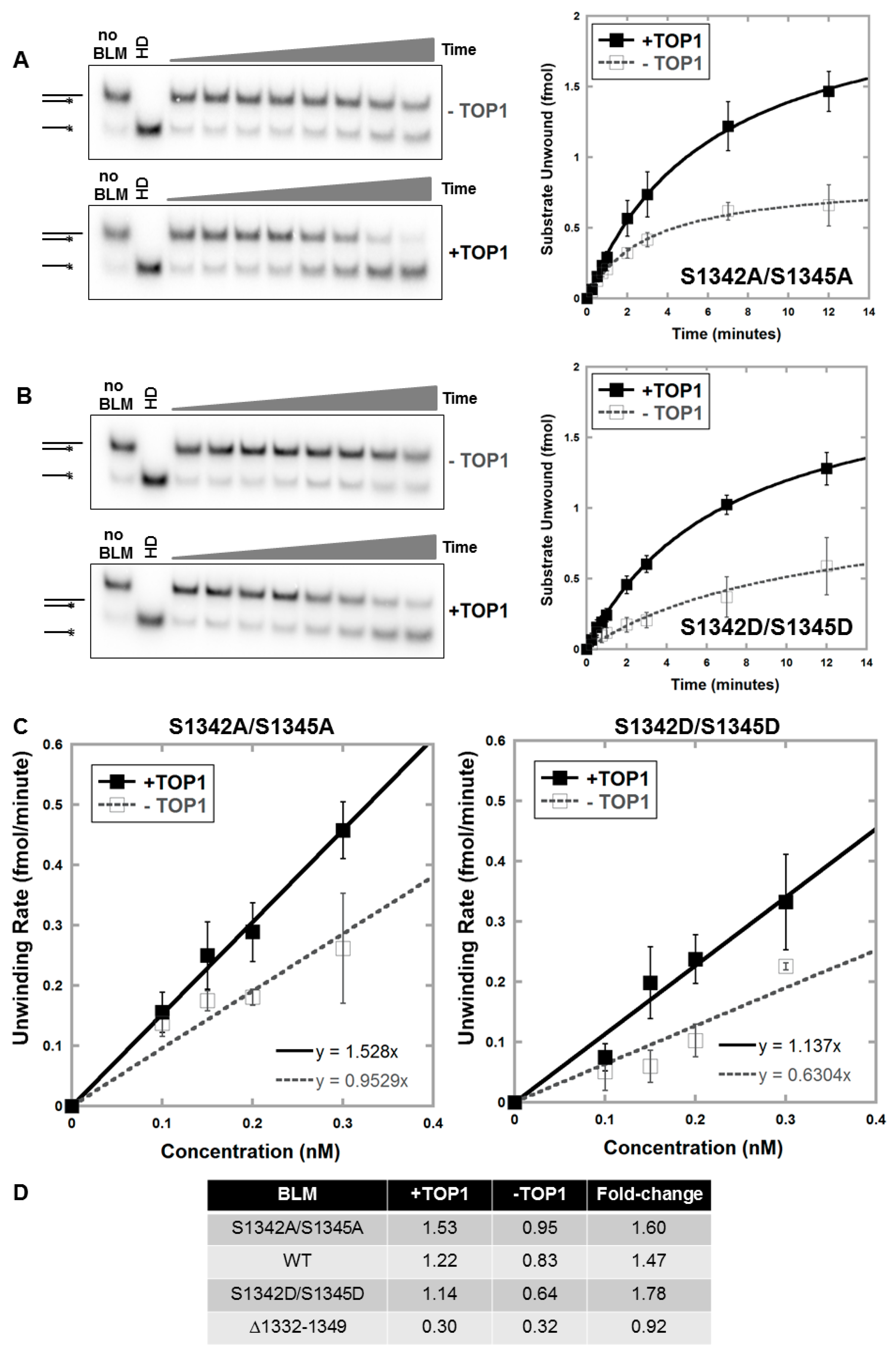
3.2. Deletion of Amino Acids 1332–1349 in BLM Abolishes TOP1-Mediated Unwinding of RNA:DNA Substrate
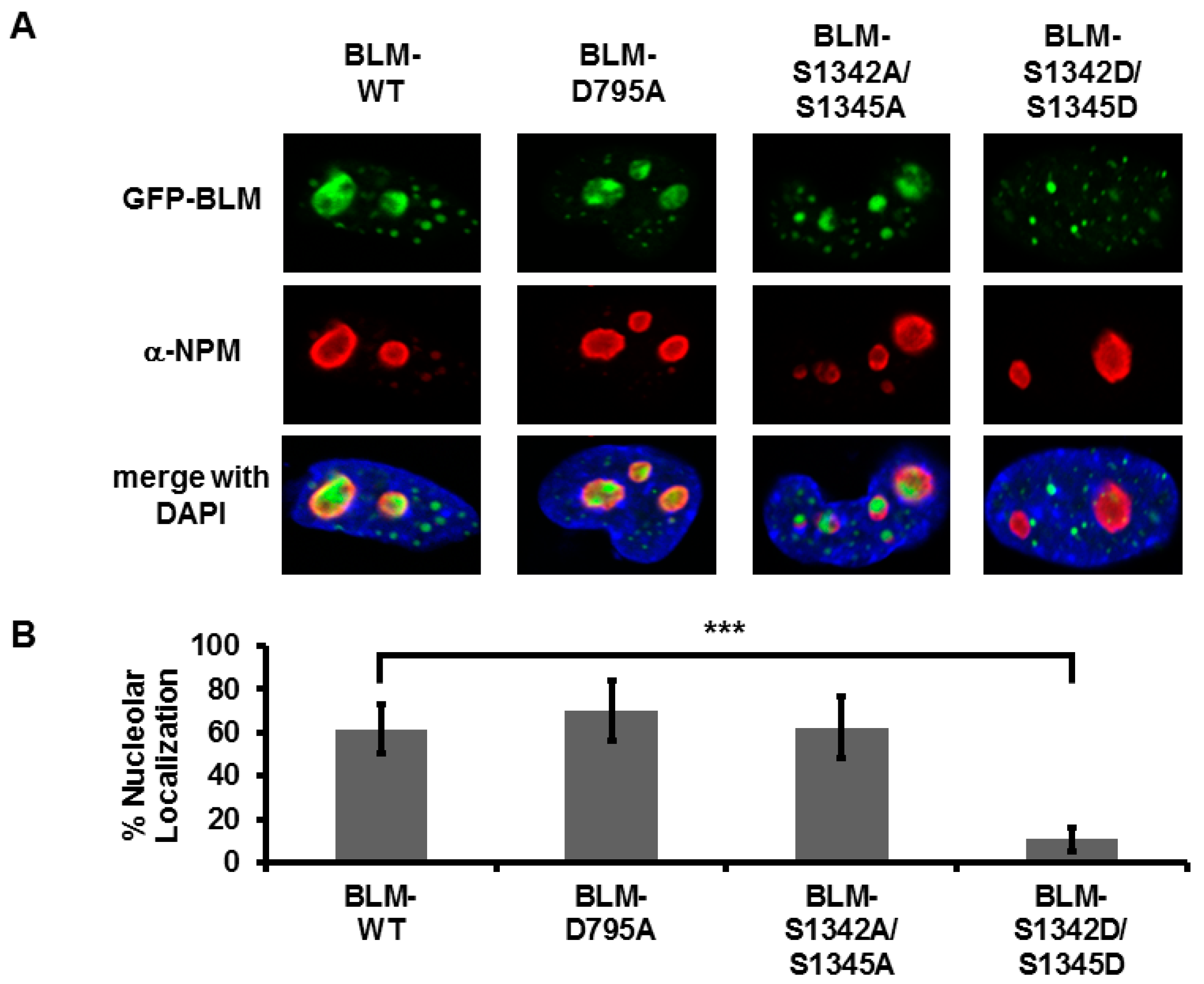
3.3. Amino Acids in the Region 1332–1349 Are Critical for Nucleolar Targeting of BLM
3.4. Alanine or Aspartic acid Mutagenesis of S1342 and S1345 Does Not Alter the Functional Interaction of BLM and TOP1
4. Discussion and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shaw, P.J.; Jordan, E.G. The nucleolus. Annu. Rev. Cell Dev. Biol. 1995, 11, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Raska, I.; Shaw, P.J.; Cmarko, D. Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell Biol. 2006, 18, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; van Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.L.; Zomerdijk, J. Structure and function of ribosomal RNA gene chromatin. Biochem. Soc. Trans. 2008, 36, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Shore, D. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Hurt, E.; Bassler, J. Driving ribosome assembly. Biochim. Biophys. Acta 2010, 1803, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Schwarzacher, H.G.; Wachtler, F. The nucleolus. Anat. Embryol. 1993, 188, 515–536. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.O.J.; Dundr, M.; Szebeni, A. The nucleolus: An old factory with unexpected capabilities. Trends. Cell Biol. 2000, 10, 189–196. [Google Scholar] [CrossRef]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Gazda, H.T. Ribosomopathies: How a common root can cause a tree of pathologies. Dis. Model. Mech. 2015, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Rudra, D.; Warner, J.R. What better measure than ribosome synthesis? Genes Dev. 2004, 18, 2431–2436. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhou, Y.; Casanova, E.; Chai, M.; Kiss, E.; Grone, H.J.; Schutz, G.; Grummt, I. Genetic inactivation of the transription factor Tif-1a leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 2005, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Cellular stress and nucleolar function. Cell Cycle 2005, 4, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear rna polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed]
- Kirn-Safran, C.B.; Oristian, D.S.; Focht, R.J.; Parker, S.G.; Vivian, J.L.; Carson, D.D. Global growth deficiencies in mice lacking the ribosomal protein HIP/RPL29. Dev. Dyn. 2007, 236, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Zhang, Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-MDM2-p53 pathway. Oncogene 2010, 29, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta 2014, 1842, 802–816. [Google Scholar] [CrossRef] [PubMed]
- German, J. Bloom’s syndrome. I. Genetical and clinical observations in the first twenty-seven patients. Am. J. Hum. Genet. 1969, 21, 196–227. [Google Scholar] [PubMed]
- Lechner, J.F.; Kaighn, M.E.; Jetten, A.M.; Groden, J.; German, J. Bloom’s syndrome cells have an abnormal serum growth response. Exp. Cell Res. 1983, 145, 381–388. [Google Scholar] [CrossRef]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef]
- German, J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet. Cytogenet. 1997, 93, 100–106. [Google Scholar] [CrossRef]
- Risinger, M.A.; Groden, J. Crosslinks and crosstalk: Human cancer syndromes and DNA repair defects. Cancer Cell 2004, 6, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.; Vogiatzi, M.G.; Sanz, M.M.; German, J. Evaluation of short stature, carbohydrate metabolism and other endocrinopathies in Bloom’s syndrome. Horm. Res. 2006, 66, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Sanij, E.; Rothblum, L.I.; Hannan, R.D.; Pearson, R.B. Dysregulation of RNA polymerase I transcription during disease. Biochim. Biophys. Acta 2013, 1829, 342–360. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.; Jones, N.C.; Sandell, L.L.; Jayasinghe, S.M.; Crane, J.; Rey, J.-P.; Dixon, M.J.; Trainor, P.A. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 13403–13408. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.C.; Lynn, M.L.; Gaudenz, K.; Sakai, D.; Aoto, K.; Rey, J.-P.; Glynn, E.F.; Ellington, L.; Du, C.; Dixon, J.; et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008, 14, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Chester, N.; Kuo, F.; Kozak, C.; O’Hara, C.D.; Leder, P. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 1998, 12, 3382–3393. [Google Scholar] [CrossRef] [PubMed]
- Therman, E.; Otto, P.G.; Shahidi, N.T. Mitotic recombination and segregation of satellites in Bloom’s syndrome. Chromosoma 1981, 82, 627–636. [Google Scholar] [CrossRef] [PubMed]
- German, J. Bloom syndrome: A mendelian prototype of somatic mutational disease. Medicine 1993, 72, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Schawalder, J.; Paric, E.; Neff, N.F. Telomere and ribosomal DNA repeats are chromosomal targets of the Bloom syndrome DNA helicase. BMC Cell Biol. 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yankiwski, V.; Marciniak, R.A.; Guarente, L.; Neff, N.F. Nuclear structure in normal and Bloom syndrome cells. Proc. Natl. Acad. Sci. USA 2000, 97, 5214–5219. [Google Scholar] [CrossRef] [PubMed]
- Yankiwski, V.; Noonan, J.P.; Neff, N.F. The C-terminal domain of the Bloom syndrome DNA helicase is essential for genomic stability. BMC Cell Biol. 2001. [Google Scholar] [CrossRef] [Green Version]
- Killen, M.W.; Stults, D.M.; Adachi, N.; Hanakahi, L.; Pierce, A.J. Loss of bloom syndrome protein destabilizes human gene cluster architecture. Hum. Mol. Genet. 2009, 18, 3417–3428. [Google Scholar] [CrossRef] [PubMed]
- Grierson, P.M.; Lillard, K.; Behbehani, G.K.; Combs, K.A.; Bhattacharyya, S.; Acharya, S.; Groden, J. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum. Mol. Genet. 2012, 21, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Grierson, P.M.; Acharya, S.; Groden, J. Collaborating functions of BLM and DNA topoisomerase I in regulating human rDNA transcription. Mutat. Res. 2013, 743, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, J.C.; Liu, L.F. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc. Natl. Acad. Sci. USA 1988, 85, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Hraiky, C.; Raymond, M.A.; Drolet, M. Rnase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA topoisomerase I in Escherichia coli. J. Biol. Chem. 2000, 275, 11257–11263. [Google Scholar] [CrossRef] [PubMed]
- Hage, A.E.; French, S.L.; Beyer, A.L.; Tollervey, D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010, 24, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Johnson, R.E.; Yu, S.L.; Prakash, L.; Prakash, S. Requirement of yeast SGS1 and SRS2 genes for replication and transcription. Science 1999, 286, 2339–2342. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.D.; Wang, L.; Youssoufian, H.; Martin, G.M.; Oshima, J. Werner helicase is localized to transcriptionally active nucleoli of cycling cells. Exp. Cell Res. 1998, 242, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, M.; Suzuki, T.; Itoh, C.; Goto, M.; Furuichi, Y.; Matsumoto, T. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I-associated complex. Oncogene 2002, 21, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Keirsey, J.; Russell, B.; Kavecansky, J.; Lillard-Wetherall, K.; Tahmaseb, K.; Turchi, J.J.; Groden, J. Telomerase-associated protein 1, HSP90, and Topoisomerase II-alpha associate directly with the BLM helicase in immortalized cells using ALT and modulate its helicase activity using telomeric DNA substrates. J. Biol. Chem. 2009, 284, 14966–14977. [Google Scholar] [CrossRef] [PubMed]
- Russell, B.; Bhattacharyya, S.; Keirsey, J.; Sandy, A.; Grierson, P.; Perchiniak, E.; Kavecansky, J.; Acharya, S.; Groden, J. Chromosome breakage is regulated by the interaction of the BLM helicase and Topoisomerase II-alpha. Cancer Res. 2011, 71, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Kaul, Z.; Gocha, A.S.; Martinez, A.R.; Harris, J.; Parvin, J.D.; Groden, J. Association of BLM and BRCA1 during telomere maintenance in ALT cells. PLoS ONE 2014, 9, e103819. [Google Scholar] [CrossRef] [PubMed]
- Gocha, A.S.; Acharya, S.; Groden, J. WRN loss induces switching of telomerase-independent mechanisms of telomere elongation. PLoS ONE 2014, 9, e93991. [Google Scholar] [CrossRef] [PubMed]
- Karow, J.K.; Chakraverty, R.K.; Hickson, I.D. The Bloom’s syndrome gene product is a 3′–5′ DNA helicase. J. Biol. Chem. 1997, 272, 30611–30614. [Google Scholar] [CrossRef] [PubMed]
- Lillard-Wetherell, K.; Machwe, A.; Langland, G.T.; Combs, K.A.; Behbehani, G.K.; Schonberg, S.A.; German, J.; Turchi, J.J.; Orren, D.K.; Groden, J. Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum. Mol. Genet. 2004, 13, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.-H.; Lee, T.-Y.; Liang, H.-K.; Huang, C.-M.; Wang, T.-Y.; Yang, Y.-H.; Chu, C.-H.; Huang, H.-D.; Ko, M.-T.; Hwang, J.-K. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucl. Acids Res. 2007, 35, W588–W594. [Google Scholar]
- Horn, H.; Schoof, E.M.; Kim, J.; Robin, X.; Miller, M.L.; Diella, F.; Palma, A.; Cesareni, G.; Jensen, L.J.; Linding, R. KinomeXplorer: an integrated platform for kinome biology studies. Nat. Methods 2014, 11, 603–604. [Google Scholar] [CrossRef] [PubMed]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucl. Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Li, A.; Wang, L.; Feng, H.; Yao, X. PPSP: Prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinf. 2006. [Google Scholar] [CrossRef]
- Phosphonet Kinase Predictor. Available online: http://www.phosphonet.ca/kinasepredictor.aspx?uni=P31751&ps=S130 (accessed on 12 June 2016).
- Wagner, W.; Adamczak, R.; Porollo, A.; Meller, J. Linear Regression Models for Solvent Accessibility Prediction in Proteins. J. Computational. Biol. 2005, 12, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Davies, S.L.; North, P.S.; Goulaouic, H.; Riou, J.F.; Turley, H.; Gatter, K.C.; Hickson, I.D. The Bloom’s syndrome gene product interacts with Topoisomerase III. J. Biol. Chem. 2000, 275, 9636–9644. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Beresten, S.F.; van Brabant, A.J.; Pandolfi, P.P.; Johnson, F.B.; Guarente, L.; Ellis, N.A. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum. Mol. Genet. 2001, 10, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.B.; Lombard, D.B.; Neff, N.F.; Mastrangelo, M.A.; Dewolf, W.; Ellis, N.A.; Marciniak, R.A.; Yin, Y.; Jaenisch, R.; Guarente, L. Association of the Bloom syndrome protein with Topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res. 2000, 60, 1162–1167. [Google Scholar] [PubMed]
- Kaneko, H.; Orii, K.O.; Matsui, E.; Shimozawa, N.; Fukao, T.; Matsumoto, T.; Shimamoto, A.; Furuichi, Y.; Hayakawa, S.; Kasahara, K.; et al. BLM (the causative gene of Bloom syndrome) protein translocation into the nucleus by a nuclear localization signal. Biochem. Biophys. Res. Comm. 1997, 240, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Gharibyan, V.; Youssoufian, H. Localization of the Bloom syndrome helicase to punctate nuclear structures and the nuclear matrix and regulation during the cell cycle: Comparison with the Werner’s syndrome helicase. Mol. Carcinog. 1999, 26, 261–273. [Google Scholar] [CrossRef]
- Bohm, S.; Bernstein, K.A. The role of post-translational modifications in fine tuning BLM helicase function during DNA repair. DNA Repair 2014, 22, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.S.; Boisvert, F.M.; McDowall, M.D.; Lamond, A.I.; Barton, G.J. Characterization and prediction of protein nucleolar localization sequences. Nucl. Acids Res. 2010, 38, 7388–7399. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Hiscox, J.A. Nucleolar targeting: The hub of the matter. EMBO Rep. 2009, 10, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Audas, T.E.; Jacob, M.D.; Lee, S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol. Cell 2012, 45, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Manthei, K.A.; Keck, J.L. The BLM dissolvasome in DNA replication and repair. Cell. Mol. Life Sci. 2013, 70, 4067–4084. [Google Scholar] [CrossRef] [PubMed]
- Ababou, M.; Dutertre, S.; Lecluse, Y.; Onclercq, R.; Chatton, B.; Amor-Gueret, M. ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene 2000, 19, 5955–5963. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Ababou, M.; Onclercq, R.; Delic, J.; Chatton, B.; Jaulin, C.; Amor-Gueret, M. Cell cycle regulation of the endogenous wild type Bloom’s syndrome DNA helicase. Oncogene 2000, 19, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Sekhri, R.; Tintignac, L.A.; Onclercq-Delic, R.; Chatton, B.; Jaulin, C.; Amor-Gueret, M. Dephosphorylation and subcellular compartment change of the mitotic Bloom’s syndrome DNA helicase in response to ionizing radiation. J. Biol. Chem. 2002, 277, 6280–6286. [Google Scholar] [CrossRef] [PubMed]
- Beamish, H.; Kedar, P.; Kaneko, H.; Chen, P.; Fukao, T.; Peng, C.; Beresten, S.; Gueven, N.; Purdie, D.; Lees-Miller, S.; et al. Functional link between BLM defective in Bloom’s syndrome and the ataxia-telangiectasia-mutated protein, ATM. J. Biol. Chem. 2002, 277, 30515–30523. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Kaminker, P.; Hansen, R.K.; Campisi, J. ATR and ATM-dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle 2004, 3, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.L.; North, P.S.; Dart, A.; Lakin, N.D.; Hickson, I.D. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol. Cell Biol. 2004, 24, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Robles, A.I.; Linke, S.P.; Sinogeeva, N.I.; Zhang, R.; Pedeux, R.; Ward, I.M.; Celeste, A.; Nussenzweig, A.; Chen, J.; et al. Functional interaction between BLM helicase and 53BP1 in a CHK1-mediated pathway during S-phase arrest. J. Cell Biol. 2004, 166, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Eladad, S.; Ye, T.Z.; Hu, P.; Leversha, M.; Beresten, S.; Matunis, M.J.; Ellis, N.A. Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum. Mol. Genet. 2005, 14, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.A.; Fan, A.M.; Meng, L.; Doe, C.F.; North, P.S.; Hickson, I.D.; Pommier, Y. Phosphorylation of BLM, dissociation from topoisomerase IIIalpha, and colocalization with gamma-H2AX after topoisomerase i-induced replication damage. Mol. Cell Biol. 2005, 25, 8925–8937. [Google Scholar] [CrossRef] [PubMed]
- Bayart, E.; Dutertre, S.; Jaulin, C.; Guo, R.B.; Xi, X.G.; Amor-Gueret, M. The Bloom syndrome helicase is a substrate of the mitotic Cdc2 kinase. Cell Cycle 2006, 5, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Leng, M.; Chan, D.W.; Luo, H.; Zhu, C.; Qin, J.; Wang, Y. MPS1-dependent mitotic BLM phosphorylation is important for chromosome stability. Proc. Natl. Acad. Sci. USA 2006, 103, 11485–11490. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Kaur, S.; Sengupta, S. Phosphorylation-dependent interactions of BLM and 53BP1 are required for their anti-recombinogenic roles during homologous recombination. Carcinogesis 2008, 29, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, S.; Guzzo, C.M.; Ellis, N.A.; Sung, K.S.; Choi, C.Y.; Matunis, M.J. Small ubiquitin-related modifier (SUMO) binding determines substrate recognition and paralog-selective SUMO modification. J. Biol. Chem. 2008, 283, 29405–29415. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, K.J.; Woo, L.L.; Zhu, J.; Huo, D.; Matunis, M.J.; Ellis, N.A. SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 2009, 7, e1000252. [Google Scholar] [CrossRef] [PubMed]
- Rog, O.; Miller, K.M.; Ferreira, M.G.; Cooper, J.P. Sumoylation of RecQ helicase controls the fate of dysfunctional telomeres. Mol. Cell 2009, 33, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Modi, P.; Srivastava, V.; Mudgal, R.; Tikoo, S.; Arora, P.; Mohanty, D.; Sengupta, S. Chk1-dependent constitutive phosphorylation of BLM helicase at serine 646 decreases after DNA damage. Mol. Cancer Res. 2010, 8, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, K.J.; Yagle, M.K.; Matunis, M.J.; Ellis, N.A. BLM sumoylation regulates ssDNA accumulation at stalled replication forks. Front. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Gong, Z. TopBP1 controls BLM level to maintain genomic stability. Mol. Cell 2013, 52, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, S.; Madhavan, V.; Hussain, M.; Miller, E.S.; Arora, P.; Zlatanou, A.; Modi, P.; Townsend, K.; Stewart, G.S.; Sengupta, S. Ubiquitin-dependent recruitment of the Bloom syndrome helicase upon replication stress is required to suppress homologous recombination. EMBO J. 2013, 32, 1778–1792. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.S. DNA replication initiation patterns and spatial dynamics of the human ribosomal RNA gene loci. J. Cell Sci. 2011, 124, 2743–2752. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, M.J.; Lee, E.Y.H.P.; Maizels, N. Localization and dynamic relocalization of mammalian Rad52 during the cell cycle and in response to DNA damage. Curr. Biol. 1999, 9, 975–978. [Google Scholar] [CrossRef]
- Torres-Rosell, J.; Sunjevaric, I.; De Piccoli, G.; Sacher, M.; Eckert-Boulet, N.; Reid, R.; Jentsch, S.; Rothstein, R.; Aragon, L.; Lisby, M. The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat. Cell Biol. 2007, 9, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Eckert-Boulet, N.; Lisby, M. Regulation of rDNA stability by sumoylation. DNA Repair 2009, 8, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Zheng, X.F.S. Opposing role of condensin and radiation-sensitive gene RAD52 in ribosomal DNA stability regulation. J. Biol. Chem. 2009, 284, 21908–21919. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tangeman, L.; McIlhatton, M.A.; Grierson, P.; Groden, J.; Acharya, S. Regulation of BLM Nucleolar Localization. Genes 2016, 7, 69. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7090069
Tangeman L, McIlhatton MA, Grierson P, Groden J, Acharya S. Regulation of BLM Nucleolar Localization. Genes. 2016; 7(9):69. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7090069
Chicago/Turabian StyleTangeman, Larissa, Michael A. McIlhatton, Patrick Grierson, Joanna Groden, and Samir Acharya. 2016. "Regulation of BLM Nucleolar Localization" Genes 7, no. 9: 69. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7090069