
Molecular Correlates and Recent Advancements in the Diagnosis and Screening of FMR1-Related Disorders

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Fragile X Syndrome
2. Molecular Determinants of FXS
3. Molecular Determinants of FXTAS
4. Molecular Determinants of FXPOI
5. Repeat Instability of PM-Sized Alleles
6. Prevalence and Other PM-Associated Phenotypes
7. Potential Risks and Instability of Intermediate-Sized Alleles
8. Recommendation for FMR1 Diagnostic Testing
9. Diagnostic Tools for Characterizing the Multiple Molecular Facets of FMR1 Expansions
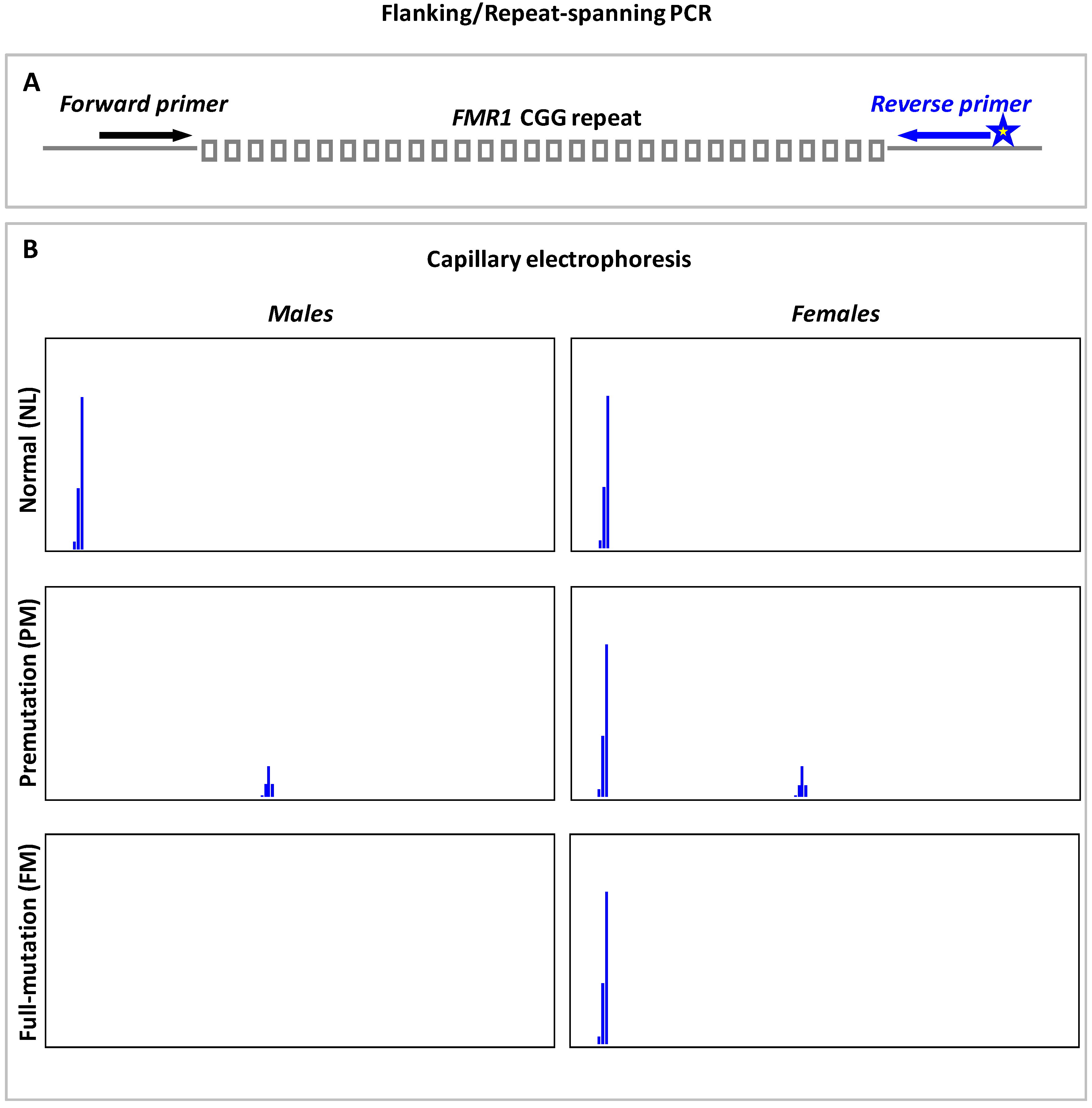
9.1. Flanking or Repeat-Spanning PCR
9.2. PCR-Based Assays for FMR1 Regulatory Region Methylation Analysis
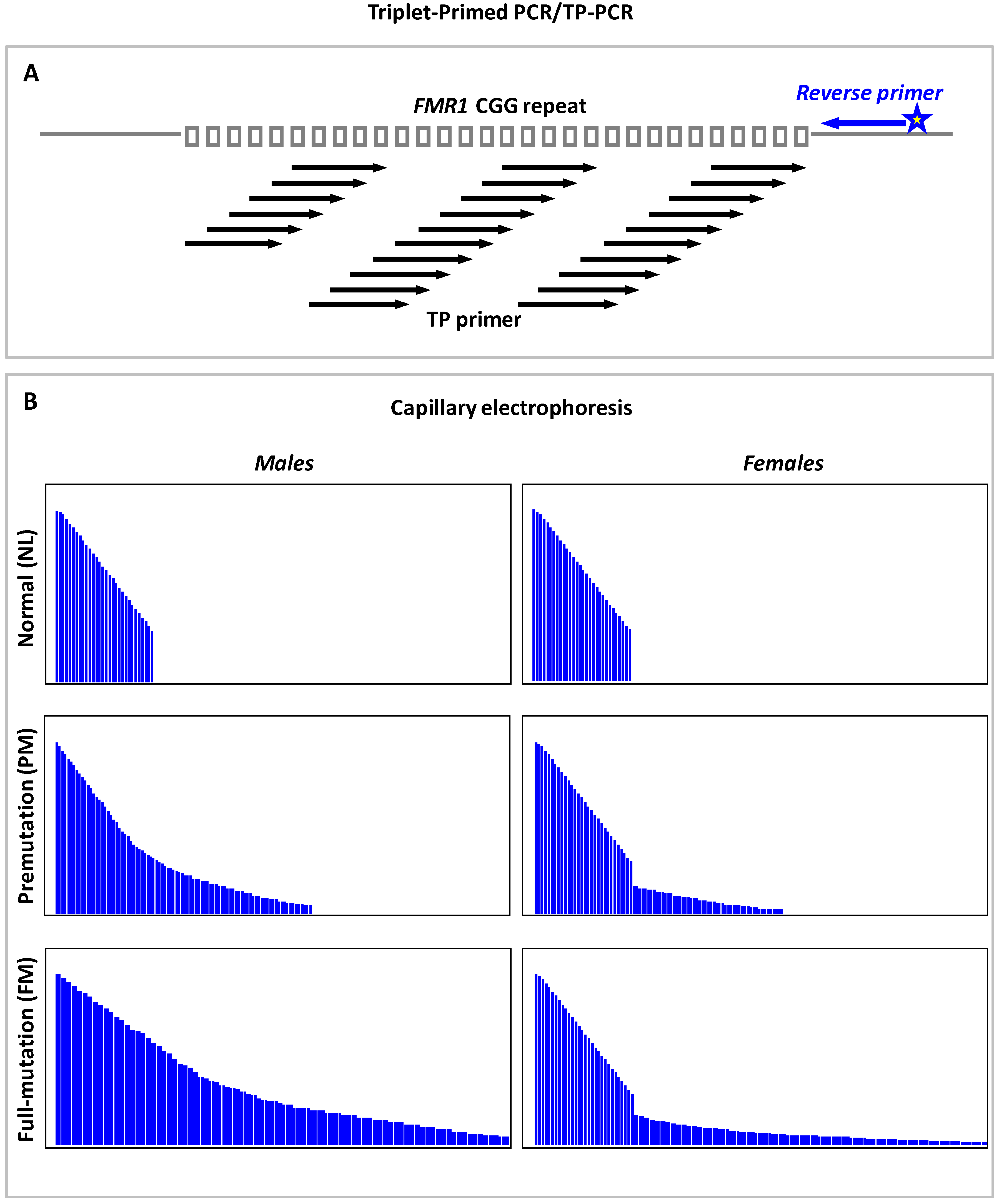
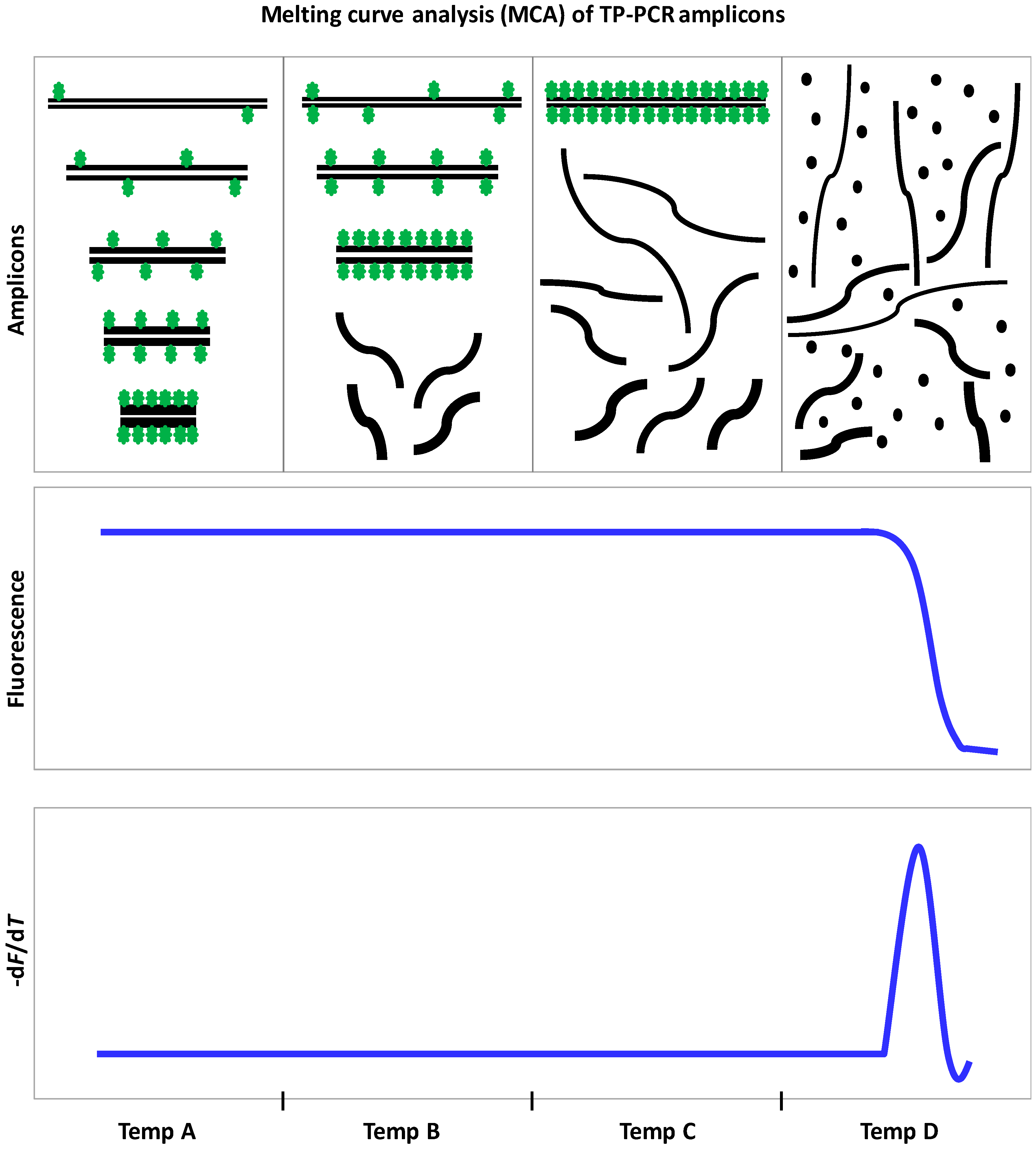
9.3. Triplet Repeat Primed PCR
10. Population-Based Screening for FMR1 Expansions
11. FMR1 Molecular Tests for Large-Scale Screening Applications
12. FMRP Antibody Tests for FXS Diagnosis and Screening
13. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Saul, R.A.; Tarleton, J.C. FMR1-Related Disorders. In GeneReviews(R); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Fong, C.T., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Usdin, K.; Hayward, B.E.; Kumari, D.; Lokanga, R.A.; Sciascia, N.; Zhao, X.N. Repeat-mediated genetic and epigenetic changes at the FMR1 locus in the Fragile X-related disorders. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Rajan, N.; Bagni, C. The FMRP regulon: From targets to disease convergence. Front. Neurosci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Ikeda, M.; Budimirovic, D.B.; Hjelm, L.N.; Kaufmann, W.E.; Warren, S.T. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: A case report and review of the literature. Am. J. Med. Genet. A 2008, 146A, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- De Boulle, K.; Verkerk, A.J.; Reyniers, E.; Vits, L.; Hendrickx, J.; Van Roy, B.; Van den Bos, F.; de Graaff, E.; Oostra, B.A.; Willems, P.J. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 1993, 3, 31–35. [Google Scholar] [CrossRef] [PubMed]
- De Graaff, E.; Rouillard, P.; Willems, P.J.; Smits, A.P.; Rousseau, F.; Oostra, B.A. Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum. Mol. Genet. 1995, 4, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Hammond, L.S.; Macias, M.M.; Tarleton, J.C.; Shashidhar Pai, G. Fragile X syndrome and deletions in FMR1: New case and review of the literature. Am. J. Med. Genet. 1997, 72, 430–434. [Google Scholar] [CrossRef]
- Handt, M.; Epplen, A.; Hoffjan, S.; Mese, K.; Epplen, J.T.; Dekomien, G. Point mutation frequency in the FMR1 gene as revealed by fragile X syndrome screening. Mol. Cell. Probes 2014, 28, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.R.; Chong, B.; Fawkner, M.; Lambiris, N.; Peters, H.; Kenneson, A.; Warren, S.T.; Love, D.R.; McGaughran, J. Microdeletion in the FMR-1 gene: An apparent null allele using routine clinical PCR amplification. J. Med. Genet. 2001, 38, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.K.; Archibald, A.D.; Cohen, J.; Metcalfe, S.A. A systematic review of population screening for fragile X syndrome. Genet. Med. 2010, 12, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, K.G.; Lyon, E.; Spector, E.B.; American College of Medical Genetics and Genomics. ACMG Standards and Guidelines for fragile X testing: A revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet. Med. 2013, 15, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.W.; Berry-Kravis, E. Autism and fragile X syndrome. Semin. Neurol. 2014, 34, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Saldarriaga, W.; Tassone, F.; Gonzalez-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X syndrome. Colomb. Med. 2014, 45, 190–198. [Google Scholar] [PubMed]
- Biancalana, V.; Glaeser, D.; McQuaid, S.; Steinbach, P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur. J. Hum. Genet. 2015, 23, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Heitz, D.; Biancalana, V.; Blumenfeld, S.; Kretz, C.; Boue, J.; Tommerup, N.; Van Der Hagen, C.; DeLozier-Blanchet, C.; Croquette, M.F.; et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 1991, 325, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.B.; Wiegers, A.M.; Smits, A.P.; Mohkamsing, S.; Duivenvoorden, H.J.; Fryns, J.P.; Curfs, L.M.; Halley, D.J.; Oostra, B.A.; van den Ouweland, A.M.; et al. Mental status of females with an FMR1 gene full mutation. Am. J. Hum. Genet. 1996, 58, 1025–1032. [Google Scholar] [PubMed]
- Pretto, D.; Yrigollen, C.M.; Tang, H.T.; Williamson, J.; Espinal, G.; Iwahashi, C.K.; Durbin-Johnson, B.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Houck, G.E., Jr.; Brown, W.T.; Dobkin, C.S. Mosaicism in fragile X affected males. Am. J. Med. Genet. 1994, 51, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, F.; Heitz, D.; Tarleton, J.; MacPherson, J.; Malmgren, H.; Dahl, N.; Barnicoat, A.; Mathew, C.; Mornet, E.; Tejada, I.; et al. A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: The first 2253 cases. Am. J. Hum. Genet. 1994, 55, 225–237. [Google Scholar] [PubMed]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Ligsay, A.; Hagerman, R.J. Fragile X syndrome: An aging perspective. Dev. Disabil. Res. Rev. 2013, 18, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.I.; Hunsaker, M.R.; Cunningham, C.L.; Greco, C.M.; Hagerman, R.J.; Noctor, S.C.; Hall, D.A.; Hagerman, P.J.; Tassone, F. Intranuclear inclusions in a fragile X mosaic male. Transl. Neurodegener. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Hull, C.E.; Safanda, J.F.; Carpenter, I.; Staley, L.W.; O’Connor, R.A.; Seydel, C.; Mazzocco, M.M.; Snow, K.; Thibodeau, S.N.; et al. High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 1994, 51, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Wohrle, D.; Salat, U.; Glaser, D.; Mucke, J.; Meisel-Stosiek, M.; Schindler, D.; Vogel, W.; Steinbach, P. Unusual mutations in high functioning fragile X males: Apparent instability of expanded unmethylated CGG repeats. J. Med. Genet. 1998, 35, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Loesch, D.Z.; Lachiewicz, A.; Taylor, A.K.; Hagerman, P.J. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am. J. Med. Genet. 2000, 94, 232–236. [Google Scholar] [CrossRef]
- Brouwer, J.R.; Mientjes, E.J.; Bakker, C.E.; Nieuwenhuizen, I.M.; Severijnen, L.A.; Van der Linde, H.C.; Nelson, D.L.; Oostra, B.A.; Willemsen, R. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated Fragile X full mutation. Exp. Cell. Res. 2007, 313, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Santa Maria, L.; Pugin, A.; Alliende, M.A.; Aliaga, S.; Curotto, B.; Aravena, T.; Tang, H.T.; Mendoza-Morales, G.; Hagerman, R.; Tassone, F. FXTAS in an unmethylated mosaic male with fragile X syndrome from Chile. Clin. Genet. 2014, 86, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Sherwell, S.; Kinsella, G.; Tassone, F.; Taylor, A.; Amor, D.; Sung, S.; Evans, A. Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin. Genet. 2012, 82, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Basuta, K.; Schneider, A.; Gane, L.; Polussa, J.; Woodruff, B.; Pretto, D.; Hagerman, R.; Tassone, F. High functioning male with fragile X syndrome and fragile X-associated tremor/ataxia syndrome. Am. J. Med. Genet. A 2015, 167A, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Revenga, L.; Madrigal, I.; Pagonabarraga, J.; Xuncla, M.; Badenas, C.; Kulisevsky, J.; Gomez, B.; Mila, M. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur. J. Hum. Genet. 2009, 17, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Coffey, S.M.; Cook, K.; Tartaglia, N.; Tassone, F.; Nguyen, D.V.; Pan, R.; Bronsky, H.E.; Yuhas, J.; Borodyanskaya, M.; Grigsby, J.; et al. Expanded clinical phenotype of women with the FMR1 premutation. Am. J. Med. Genet. A 2008, 146A, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Polussa, J.; Schneider, A.; Hagerman, R. Molecular advances leading to treatment implications for Fragile X premutation carriers. Brain Disord. Ther. 2014. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome—features, mechanisms and management. Nat. Rev. Neurol. 2016, 12, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Leehey, M.A.; Hagerman, R.J.; Beckett, L.A.; Hagerman, P.J. Size bias of fragile X premutation alleles in late-onset movement disorders. J. Med. Genet. 2006, 43, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Adams, J.; Berry-Kravis, E.M.; Cohen, S.S.; Brusco, A.; Leehey, M.A.; Li, L.; Hagerman, R.J.; Hagerman, P.J. CGG repeat length correlates with age of onset of motor signs of the fragile X-associated tremor/ataxia syndrome (FXTAS). Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Leehey, M.A.; Berry-Kravis, E.; Goetz, C.G.; Zhang, L.; Hall, D.A.; Li, L.; Rice, C.D.; Lara, R.; Cogswell, J.; Reynolds, A.; et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology 2008, 70, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Berman, R.F.; Martin, R.M.; Tassone, F.; Schwartz, P.H.; Chang, A.; Trapp, B.D.; Iwahashi, C.; Brunberg, J.; Grigsby, J.; et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006, 129, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Leavitt, B.R.; Farzin, F.; Jacquemont, S.; Greco, C.M.; Brunberg, J.A.; Tassone, F.; Hessl, D.; Harris, S.W.; Zhang, L.; et al. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am. J. Hum. Genet. 2004, 74, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.; Potanos, K.; Weinberg, D.; Zhou, L.; Goetz, C.G. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann. Neurol. 2005, 57, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Robertson-Dick, E.E.; O’Keefe, J.A.; Hadd, A.G.; Zhou, L.; Berry-Kravis, E. X-inactivation in the clinical phenotype of fragile X premutation carrier sisters. Neurol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Rodriguez-Revenga, L.; Feliu, A.; Badenas, C.; Madrigal, I.; Mila, M. Skewed X Inactivation in Women Carrying the FMR1 Premutation and Its Relation with Fragile-X-Associated Tremor/Ataxia Syndrome. Neurodegener. Dis. 2016, 16, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alegria, E.; Ibanez, B.; Minguez, M.; Poch, M.; Valiente, A.; Sanz-Parra, A.; Martinez-Bouzas, C.; Beristain, E.; Tejada, M.I. Analysis of FMR1 gene expression in female premutation carriers using robust segmented linear regression models. RNA 2007, 13, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.I.; Mendoza-Morales, G.; Lo, J.; Cao, R.; Hadd, A.; Latham, G.J.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 2014, 51, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.L. Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 2000, 97, 189–194. [Google Scholar] [CrossRef]
- Buijsen, R.A.; Visser, J.A.; Kramer, P.; Severijnen, E.A.; Gearing, M.; Charlet-Berguerand, N.; Sherman, S.L.; Berman, R.F.; Willemsen, R.; Hukema, R.K. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum. Reprod. 2016, 31, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.; Ward, D.; Murray, A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur. J. Hum. Genet. 2006, 14, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S.D.; Welt, C.; Sherman, S. FMR1 and the continuum of primary ovarian insufficiency. Semin. Reprod. Med. 2011, 29, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.G.; Sullivan, A.K.; Marcus, M.; Small, C.; Dominguez, C.; Epstein, M.P.; Charen, K.; He, W.; Taylor, K.C.; Sherman, S.L. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum. Reprod. 2007, 22, 2142–2152. [Google Scholar] [CrossRef] [PubMed]
- Tejada, M.I.; Garcia-Alegria, E.; Bilbao, A.; Martinez-Bouzas, C.; Beristain, E.; Poch, M.; Ramos-Arroyo, M.A.; Lopez, B.; Fernandez Carvajal, I.; Ribate, M.P.; et al. Analysis of the molecular parameters that could predict the risk of manifesting premature ovarian failure in female premutation carriers of fragile X syndrome. Menopause 2008, 15, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; Benedetti, S.; Goegan, M.; Menditto, I.; Marozzi, A.; Ferrari, M.; Toniolo, D. Skewed X-chromosome inactivation is not associated with premature ovarian failure in a large cohort of Italian patients. Am. J. Med. Genet. A 2006, 140, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Revenga, L.; Madrigal, I.; Badenas, C.; Xuncla, M.; Jimenez, L.; Mila, M. Premature ovarian failure and fragile X female premutation carriers: No evidence for a skewed X-chromosome inactivation pattern. Menopause 2009, 16, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Spath, M.A.; Nillesen, W.N.; Smits, A.P.; Feuth, T.B.; Braat, D.D.; van Kessel, A.G.; Yntema, H.G. X chromosome inactivation does not define the development of premature ovarian failure in fragile X premutation carriers. Am. J. Med. Genet. A 2010, 152A, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Yu, S.; Mulley, J.; Loesch, D.; Turner, G.; Donnelly, A.; Gedeon, A.; Hillen, D.; Kremer, E.; Lynch, M.; Pritchard, M.; et al. Fragile-X syndrome: Unique genetics of the heritable unstable element. Am. J. Hum. Genet. 1992, 50, 968–980. [Google Scholar] [PubMed]
- Nolin, S.L.; Lewis, F.A., 3rd.; Ye, L.L.; Houck, G.E., Jr.; Glicksman, A.E.; Limprasert, P.; Li, S.Y.; Zhong, N.; Ashley, A.E.; Feingold, E.; et al. Familial transmission of the FMR1 CGG repeat. Am. J. Hum. Genet. 1996, 59, 1252–1261. [Google Scholar] [PubMed]
- Ashley-Koch, A.E.; Robinson, H.; Glicksman, A.E.; Nolin, S.L.; Schwartz, C.E.; Brown, W.T.; Turner, G.; Sherman, S.L. Examination of factors associated with instability of the FMR1 CGG repeat. Am. J. Hum. Genet. 1998, 63, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, G.E., Jr.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Brondum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Rife, M.; Badenas, C.; Quinto, L.; Puigoriol, E.; Tazon, B.; Rodriguez-Revenga, L.; Jimenez, L.; Sanchez, A.; Mila, M. Analysis of CGG variation through 642 meioses in Fragile X families. Mol. Hum. Reprod. 2004, 10, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Durbin-Johnson, B.; Gane, L.; Nelson, D.L.; Hagerman, R.; Hagerman, P.J.; Tassone, F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 2012, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Mendoza-Morales, G.; Hagerman, R.; Tassone, F. Transmission of an FMR1 premutation allele in a large family identified through newborn screening: The role of AGG interruptions. J. Hum. Genet. 2013, 58, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Sah, S.; Glicksman, A.; Sherman, S.L.; Allen, E.; Berry-Kravis, E.; Tassone, F.; Yrigollen, C.; Cronister, A.; Jodah, M.; et al. Fragile X AGG analysis provides new risk predictions for 45–69 repeat alleles. Am. J. Med. Genet. A 2013, 161A, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E.; Holden, J.J.; Popovich, B.W.; Reiss, A.L.; Snow, K.; Thibodeau, S.N.; Richards, C.S.; Ward, P.A.; Nelson, D.L. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet. 1994, 8, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Latham, G.J.; Coppinger, J.; Hadd, A.G.; Nolin, S.L. The role of AGG interruptions in fragile X repeat expansions: A twenty-year perspective. Front. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Carvajal, I.; Lopez Posadas, B.; Pan, R.; Raske, C.; Hagerman, P.J.; Tassone, F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 2009, 11, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Iong, K.P.; Tong, T.H.; Lo, J.; Gane, L.W.; Berry-Kravis, E.; Nguyen, D.; Mu, L.Y.; Laffin, J.; Bailey, D.B.; et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Maenner, M.J.; Baker, M.W.; Broman, K.W.; Tian, J.; Barnes, J.K.; Atkins, A.; McPherson, E.; Hong, J.; Brilliant, M.H.; Mailick, M.R. FMR1 CGG expansions: Prevalence and sex ratios. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162B, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.M.; Baker, M.W.; Hong, J.; Maenner, M.; Greenberg, J.; Mandel, D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Newborn screening for fragile X syndrome. JAMA Neurol. 2014, 71, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146A, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, A.A.; Sukharev, D.; Campos, L.; Mu, Y.; Tassone, F.; Hessl, D.; Nguyen, D.V.; Loesch, D.; Hagerman, R.J. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Am. J. Med. Genet. A 2012, 158A, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Saito, N.; Reed, D.; Eldeeb, M.; Schneider, A.; Hessl, D.; Tassone, F.; Beckett, L.; Hagerman, R. Aging in Fragile X Premutation Carriers. Cerebellum 2016, 15, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Winarni, T.I.; Chonchaiya, W.; Sumekar, T.A.; Ashwood, P.; Morales, G.M.; Tassone, F.; Nguyen, D.V.; Faradz, S.M.; Van de Water, J.; Cook, K.; et al. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am. J. Med. Genet. A 2012, 158A, 2473–2481. [Google Scholar] [CrossRef] [PubMed]
- Leehey, M.A.; Legg, W.; Tassone, F.; Hagerman, R. Fibromyalgia in fragile X mental retardation 1 gene premutation carriers. Rheumatology 2011, 50, 2233–2236. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, A.; Liu, Y.; Nguyen, D.V.; Tassone, F.; Zhang, L.; Hagerman, R.J. Sleep apnea in fragile X premutation carriers with and without FXTAS. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156B, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Au, J.; Akins, R.S.; Berkowitz-Sutherland, L.; Tang, H.T.; Chen, Y.; Boyd, A.; Tassone, F.; Nguyen, D.V.; Hagerman, R. Prevalence and risk of migraine headaches in adult fragile X premutation carriers. Clin. Genet. 2013, 84, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Tassone, F.; Klepitskaya, O.; Leehey, M. Fragile X-associated tremor ataxia syndrome in FMR1 gray zone allele carriers. Mov. Disord. 2012, 27, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Winarni, T.I.; Zhang, L.; Tassone, F.; Hagerman, R.J. Fragile X-associated tremor/ataxia syndrome (FXTAS) in grey zone carriers. Clin. Genet. 2013, 84, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Bretherick, K.L.; Fluker, M.R.; Robinson, W.P. FMR1 repeat sizes in the gray zone and high end of the normal range are associated with premature ovarian failure. Hum. Genet. 2005, 117, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Bodega, B.; Bione, S.; Dalpra, L.; Toniolo, D.; Ornaghi, F.; Vegetti, W.; Ginelli, E.; Marozzi, A. Influence of intermediate and uninterrupted FMR1 CGG expansions in premature ovarian failure manifestation. Hum. Reprod. 2006, 21, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Streuli, I.; Fraisse, T.; Ibecheole, V.; Moix, I.; Morris, M.A.; de Ziegler, D. Intermediate and premutation FMR1 alleles in women with occult primary ovarian insufficiency. Fertil. Steril. 2009, 92, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.E.; Conway, G.S.; Macpherson, J.N.; Jacobs, P.A.; Murray, A. Intermediate sized CGG repeats are not a common cause of idiopathic premature ovarian failure. Hum. Reprod. 2010, 25, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Bui, Q.M.; Huggins, R.M.; Mitchell, R.J.; Hagerman, R.J.; Tassone, F. Transcript levels of the intermediate size or grey zone fragile X mental retardation 1 alleles are raised, and correlate with the number of CGG repeats. J. Med. Genet. 2007, 44, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Advanced technologies for the molecular diagnosis of fragile X syndrome. Expert Rev. Mol. Diagn. 2015, 15, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ding, X.; Ersalesi, N.; Brown, W.T.; Sherman, S.L.; Dobkin, C. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 2011, 31, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, A.; Juan, J.; Mila, M.; Guerrero, A. Expansion of an intermediate allele of the FMR1 gene in only two generations. Clin. Genet. 2005, 68, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, A.; Pomponi, M.G.; Marino, G.M.; Chiurazzi, P.; Rinaldi, M.M.; Dobosz, M.; Neri, G. Expansion to full mutation of a FMR1 intermediate allele over two generations. Eur. J. Hum. Genet. 2004, 12, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Finucane, B.; Abrams, L.; Cronister, A.; Archibald, A.D.; Bennett, R.L.; McConkie-Rosell, A. Genetic counseling and testing for FMR1 gene mutations: Practice guidelines of the national society of genetic counselors. J. Genet. Couns. 2012, 21, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.; Pletcher, B.A.; Driscoll, D.A. Fragile X syndrome: Diagnostic and carrier testing. Genet. Med. 2005, 7, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, T.F.; Dos Santos, J.M.; Goncalves, A.P.; Tassone, F.; Mendoza-Morales, G.; Ribeiro, M.G.; Kahn, E.; Boy, R.; Goncalves Pimentel, M.M.; Santos-Reboucas, C.B. Finding FMR1 mosaicism in Fragile X syndrome. Expert Rev. Mol. Diagn. 2016, 16, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hadd, A.; Sah, S.; Filipovic-Sadic, S.; Krosting, J.; Sekinger, E.; Pan, R.; Hagerman, P.J.; Stenzel, T.T.; Tassone, F.; et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J. Mol. Diagn. 2010, 12, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hadd, A.G.; Sah, S.; Houghton, J.F.; Filipovic-Sadic, S.; Zhang, W.; Hagerman, P.J.; Tassone, F.; Latham, G.J. High-resolution methylation polymerase chain reaction for fragile X analysis: Evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet. Med. 2011, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Filipovic-Sadic, S.; Sah, S.; Chen, L.; Krosting, J.; Sekinger, E.; Zhang, W.; Hagerman, P.J.; Stenzel, T.T.; Hadd, A.G.; Latham, G.J.; et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 2010, 56, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hantash, F.M.; Goos, D.G.; Tsao, D.; Quan, F.; Buller-Burckle, A.; Peng, M.; Jarvis, M.; Sun, W.; Strom, C.M. Qualitative assessment of FMR1 (CGG)n triplet repeat status in normal, intermediate, premutation, full mutation, and mosaic carriers in both sexes: Implications for fragile X syndrome carrier and newborn screening. Genet. Med. 2010, 12, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Rajan-Babu, I.S.; Law, H.Y.; Yoon, C.S.; Lee, C.G.; Chong, S.S. Simplified strategy for rapid first-line screening of fragile X syndrome: Closed-tube triplet-primed PCR and amplicon melt peak analysis. Expert Rev. Mol. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rajan-Babu, I.S.; Teo, C.R.; Lian, M.; Lee, C.G.; Law, H.Y.; Chong, S.S. Single-tube methylation-specific duplex-PCR assay for rapid and accurate diagnosis of Fragile X Mental Retardation 1-related disorders. Expert Rev. Mol. Diagn. 2015, 15, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Hecimovic, S.; Barisic, I.; Muller, A.; Petkovic, I.; Baric, I.; Ligutic, I.; Pavelic, K. Expand Long PCR for fragile X mutation detection. Clin. Genet. 1997, 52, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Law, H.Y.; Boehm, C.D.; Yoon, C.S.; Cutting, G.R.; Ng, I.S.; Chong, S.S. Robust fragile X (CGG)n genotype classification using a methylation specific triple PCR assay. J. Med. Genet. 2004. [Google Scholar] [CrossRef]
- Zhou, Y.; Lum, J.M.; Yeo, G.H.; Kiing, J.; Tay, S.K.; Chong, S.S. Simplified molecular diagnosis of fragile X syndrome by fluorescent methylation-specific PCR and GeneScan analysis. Clin. Chem. 2006, 52, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.S.; Eichler, E.E.; Nelson, D.L.; Hughes, M.R. Robust amplification and ethidium-visible detection of the fragile X syndrome CGG repeat using Pfu polymerase. Am. J. Med. Genet. 1994, 51, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Larsen, L.A.; Gronskov, K.; Norgaard-Pedersen, B.; Brondum-Nielsen, K.; Hasholt, L.; Vuust, J. High-throughput analysis of fragile X (CGG)n alleles in the normal and premutation range by PCR amplification and automated capillary electrophoresis. Hum. Genet. 1997, 100, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Lemonnier, A.; Gerard, M.; Chauve, C.; Tredano, M.; de Villemeur, T.B.; Aymard, P.; Bonnefont, J.P.; Feldmann, D. Improved fluorescent PCR-based assay for sizing CGG repeats at the FRAXA locus. Clin. Chem. Lab. Med. 1999, 37, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, H.; Tynan, J.A.; Fenwick, R.A.; Leon, J.A. Automated Detection of Trinucleotide Repeats in Fragile X Syndrome. Mol. Diagn. 1997, 2, 259–269. [Google Scholar] [CrossRef]
- Tzeng, C.C.; Lin, S.J.; Chen, Y.J.; Kuo, P.L.; Jong, Y.J.; Tsai, L.P.; Chen, R.M. An effective strategy of using molecular testing to screen mentally retarded individuals for fragile X syndrome. Diagn. Mol. Pathol. 2001, 10, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Saluto, A.; Brussino, A.; Tassone, F.; Arduino, C.; Cagnoli, C.; Pappi, P.; Hagerman, P.; Migone, N.; Brusco, A. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the fragile X mental retardation 1 gene. J. Mol. Diagn. 2005, 7, 605–612. [Google Scholar] [CrossRef]
- Khaniani, M.S.; Kalitsis, P.; Burgess, T.; Slater, H.R. An improved Diagnostic PCR Assay for identification of Cryptic Heterozygosity for CGG Triplet Repeat Alleles in the Fragile X Gene (FMR1). Mol. Cytogenet. 2008. [Google Scholar] [CrossRef] [PubMed]
- Todorov, T.; Todorova, A.; Georgieva, B.; Mitev, V. A unified rapid PCR method for detection of normal and expanded trinucleotide alleles of CAG repeats in huntington chorea and CGG repeats in fragile X syndrome. Mol. Biotechnol. 2010, 45, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Curtis-Cioffi, K.M.; Rodrigueiro, D.A.; Rodrigues, V.C.; Cicarelli, R.M.; Scarel-Caminaga, R.M. Comparison between the polymerase chain reaction-based screening and the Southern blot methods for identification of fragile X syndrome. Genet. Test. Mol. Biomark. 2012, 16, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Grasso, M.; Boon, E.M.; Filipovic-Sadic, S.; van Bunderen, P.A.; Gennaro, E.; Cao, R.; Latham, G.J.; Hadd, A.G.; Coviello, D.A. A novel methylation PCR that offers standardized determination of FMR1 methylation and CGG repeat length without southern blot analysis. J. Mol. Diagn. 2014, 16, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Chastain, P.D., 2nd; Eichler, E.E.; Kang, S.; Nelson, D.L.; Levene, S.D.; Sinden, R.R. Anomalous rapid electrophoretic mobility of DNA containing triplet repeats associated with human disease genes. Biochemistry 1995, 34, 16125–16131. [Google Scholar] [CrossRef] [PubMed]
- Kiba, Y.; Baba, Y. Unusual capillary electrophoretic behavior of triplet repeat DNA. J. Biochem. Biophys. Methods 1999, 41, 143–151. [Google Scholar] [CrossRef]
- O’Connell, C.D.; Atha, D.H.; Jakupciak, J.P.; Amos, J.A.; Richie, K. Standardization of PCR amplification for fragile X trinucleotide repeat measurements. Clin. Genet. 2002, 61, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kubota, T.; Song, M.; Daniel, R.; Berry-Kravis, E.M.; Prior, T.W.; Popovich, B.; Rosser, L.; Arinami, T.; Ledbetter, D.H. Methylation analysis of the fragile X syndrome by PCR. Genet. Test. 1997, 1, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Lassen, C.; Kristoffersson, U.; Aman, P. A methylation PCR approach for detection of fragile X syndrome. Hum. Mutat. 1999, 14, 71–79. [Google Scholar] [CrossRef]
- Weinhausel, A.; Haas, O.A. Evaluation of the fragile X (FRAXA) syndrome with methylation-sensitive PCR. Hum. Genet. 2001, 108, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C.; Gronskov, K.; Larsen, L.A.; Guldberg, P.; Brondum-Nielsen, K. A homogeneous assay for analysis of FMR1 promoter methylation in patients with fragile X syndrome. Clin. Chem. 2007, 53, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.G.; Hussein, I.R.; Abuzenadah, A.; Gari, M.; Bassiouni, R.; Sogaty, S.; Lary, S.; Al-Quaiti, M.; Al Balwi, M.; Al Qahtani, M. Molecular diagnosis of fragile X syndrome using methylation sensitive techniques in a cohort of patients with intellectual disability. Pediatr. Neurol. 2014, 50, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Gatta, V.; Gennaro, E.; Franchi, S.; Cecconi, M.; Antonucci, I.; Tommasi, M.; Palka, G.; Coviello, D.; Stuppia, L.; Grasso, M. MS-MLPA analysis for FMR1 gene: Evaluation in a routine diagnostic setting. BMC Med. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Nygren, A.O.; Lens, S.I.; Carvalho, R. Methylation-specific multiplex ligation-dependent probe amplification enables a rapid and reliable distinction between male FMR1 premutation and full-mutation alleles. J. Mol. Diagn. 2008, 10, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Tassone, F.; Loesch, D.Z.; Taylor, A.K.; Gehling, F.; Hagerman, R.J.; Burgess, T.; Ganesamoorthy, D.; Hennerich, D.; Gordon, L.; et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010, 19, 1618–1632. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Schwartz, C.E.; Bui, Q.M.; Li, X.; Skinner, C.; Field, M.; Wotton, T.; Hagerman, R.J.; Francis, D.; Amor, D.J.; et al. Early detection of fragile X syndrome: Applications of a novel approach for improved quantitative methylation analysis in venous blood and newborn blood spots. Clin. Chem. 2014, 60, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Godler, D.E.; Slater, H.R.; Bui, Q.M.; Ono, M.; Gehling, F.; Francis, D.; Amor, D.J.; Hopper, J.L.; Hagerman, R.; Loesch, D.Z. FMR1 intron 1 methylation predicts FMRP expression in blood of female carriers of expanded FMR1 alleles. J. Mol. Diagn. 2011, 13, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.P.; Barron, L.H.; Goudie, D.; Kelly, K.; Dow, D.; Fitzpatrick, D.R.; Brock, D.J. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J. Med. Genet. 1996, 33, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Lyon, E.; Laver, T.; Yu, P.; Jama, M.; Young, K.; Zoccoli, M.; Marlowe, N. A simple, high-throughput assay for Fragile X expanded alleles using triple repeat primed PCR and capillary electrophoresis. J. Mol. Diagn. 2010, 12, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Juusola, J.S.; Anderson, P.; Sabato, F.; Wilkinson, D.S.; Pandya, A.; Ferreira-Gonzalez, A. Performance evaluation of two methods using commercially available reagents for PCR-based detection of FMR1 mutation. J. Mol. Diagn. 2012, 14, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Abrams, L.; Cronister, A.; Brown, W.T.; Tassone, F.; Sherman, S.L.; Finucane, B.; McConkie-Rosell, A.; Hagerman, R.; Kaufmann, W.E.; Picker, J.; et al. Newborn, carrier, and early childhood screening recommendations for fragile X. Pediatrics 2012, 130, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Skinner, D.; Davis, A.M.; Whitmarsh, I.; Powell, C. Ethical, legal, and social concerns about expanded newborn screening: Fragile X syndrome as a prototype for emerging issues. Pediatrics 2008, 121, e693–e704. [Google Scholar] [CrossRef] [PubMed]
- Ross, L.F.; Acharya, K. Policy considerations in designing a fragile X population screening program. Genet. Med. 2008, 10, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.F.; Bajaj, K.; Klugman, S.D. Prenatal screening for fragile x: Carriers, controversies, and counseling. Rev. Obstet. Gynecol. 2013, 6, e1–e7. [Google Scholar] [PubMed]
- Ross, L.F. Ethical and policy issues in newborn screening of children for neurologic and developmental disorders. Pediatr. Clin. North Am. 2015, 62, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.L.; Gane, L.W.; Yarborough, M.; Hagerman, R.J.; Tassone, F. Newborn screening and cascade testing for FMR1 mutations. Am. J. Med. Genet. A 2013, 161A, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.C.; Chen, D.J.; Lin, C.N.; Chiu, C.Y.; Huang, C.B.; Chiu, P.C.; Lin, C.H.; Lin, S.J.; Tzeng, C.C. Feasibility of blood spot PCR in large-scale screening of fragile X syndrome in southern Taiwan. J. Formos. Med. Assoc. 2003, 102, 12–16. [Google Scholar] [PubMed]
- Levesque, S.; Dombrowski, C.; Morel, M.L.; Rehel, R.; Cote, J.S.; Bussieres, J.; Morgan, K.; Rousseau, F. Screening and instability of FMR1 alleles in a prospective sample of 24,449 mother-newborn pairs from the general population. Clin. Genet. 2009, 76, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Rife, M.; Mallolas, J.; Badenas, C.; Tazon, B.; Miguelez, M.R.; Pampols, T.; Sanchez, A.; Mila, M. Pilot study for the neonatal screening of fragile X syndrome. Prenat. Diagn. 2002, 22, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Saul, R.A.; Friez, M.; Eaves, K.; Stapleton, G.A.; Collins, J.S.; Schwartz, C.E.; Stevenson, R.E. Fragile X syndrome detection in newborns-pilot study. Genet. Med. 2008, 10, 714–719. [Google Scholar] [PubMed]
- Skinner, D.; Sparkman, K.L.; Bailey, D.B., Jr. Screening for Fragile X Syndrome: Parent attitudes and perspectives. Genet. Med. 2003, 5, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, S.; Ormond, K.E.; Kim, K.; Ross, L.F. Attitudes of genetic counselors towards expanding newborn screening and offering predictive genetic testing to children. Am. J. Med. Genet. A 2006, 140, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.; Ross, L.F. Fragile X screening: Attitudes of genetic health professionals. Am. J. Med. Genet. A 2009, 149A, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Kemper, A.R.; Bailey, D.B., Jr. Pediatricians’ knowledge of and attitudes toward fragile X syndrome screening. Acad. Pediatr. 2009, 9, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Levenson, D. A majority of parents accept newborn screening for fragile X. Am. J. Med. Genet. A 2011. [Google Scholar] [CrossRef]
- Skinner, D.; Choudhury, S.; Sideris, J.; Guarda, S.; Buansi, A.; Roche, M.; Powell, C.; Bailey, D.B., Jr. Parents’ decisions to screen newborns for FMR1 gene expansions in a pilot research project. Pediatrics 2011, 127, e1455–e1463. [Google Scholar] [CrossRef] [PubMed]
- Christie, L.; Wotton, T.; Bennetts, B.; Wiley, V.; Wilcken, B.; Rogers, C.; Boyle, J.; Turner, C.; Hansen, J.; Hunter, M.; et al. Maternal attitudes to newborn screening for fragile X syndrome. Am. J. Med. Genet. A 2013, 161A, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.; Schindler, A. Developmental and behavioral pediatricians’ attitudes toward screening for fragile X. Am. J. Intellect. Dev. Disabil. 2013, 118, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Musci, T.J.; Caughey, A.B. Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am. J. Obstet. Gynecol. 2005, 192, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Toledano-Alhadef, H.; Basel-Vanagaite, L.; Magal, N.; Davidov, B.; Ehrlich, S.; Drasinover, V.; Taub, E.; Halpern, G.J.; Ginott, N.; Shohat, M. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am. J. Hum. Genet. 2001, 69, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Zlotogora, J.; Grotto, I.; Kaliner, E.; Gamzu, R. The Israeli national population program of genetic carrier screening for reproductive purposes. Genet. Med. 2016, 18, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Bishop, E.; Raspa, M.; Skinner, D. Caregiver opinions about fragile X population screening. Genet. Med. 2012, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008, 10, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Teo, C.R.; Law, H.Y.; Lee, C.G.; Chong, S.S. Screening for CGG repeat expansion in the FMR1 gene by melting curve analysis of combined 5′ and 3′ direct triplet-primed PCRs. Clin. Chem. 2012, 58, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.I.; Kerr, G.R.; Mueller, P.W. Fragile X Syndrome: Scientific Background and Screening Technologies. J. Mol. Diagn. 2015, 17, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekara, C.H.; Wijesundera, W.S.; Perera, H.N.; Chong, S.S.; Rajan-Babu, I.S. Cascade screening for Fragile X Syndrome/CGG repeat expansions in children attending special education in Sri Lanka. PLoS ONE 2015, 10, e0145537. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.X.; Loo, Y.L.; Mundhofir, F.E.; Cayami, F.K.; Faradz, S.M.; Rajan-Babu, I.S.; Chong, S.S.; Koh, Y.Y.; Guan, M. Validation of a commercially available screening tool for the rapid identification of CGG trinucleotide repeat expansions in FMR1. J. Mol. Diagn. 2015, 17, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Rajan-Babu, I.S.; Lian, M.; Tran, H.A.; Dang, T.T.; Huong Le, T.M.; Thanh, N.M.; Lee, C.G.; Chong, S.S. Defining the performance parameters of a rapid screening tool for FMR1 CGG-Repeat expansions based on direct Triplet-Primed PCR and melt curve analysis. J. Mol. Diagn. 2016, 18, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.H.; Ankathil, R.; Salmi, A.R.; Sudhikaran, W.; Limprasert, P.; Zilfalil, B.A. A new method for FMR1 gene methylation screening by multiplex methylation-specific real-time polymerase chain reaction. Genet. Test. Mol. Biomark. 2011, 15, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Methylation analysis in newborn screening for fragile X syndrome—Reply. JAMA Neurol. 2014, 71, 800–801. [Google Scholar] [CrossRef] [PubMed]
- Teo, C.R.; Rajan-Babu, I.S.; Law, H.Y.; Lee, C.G.; Chong, S.S. Methylation-specific triplet-primed PCR and melting curve analysis as a rapid screening tool for identifying actionable FMR1 genotypes. Clin. Chem. 2013, 59, 1668–1670. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Mohkamsing, S.; de Vries, B.; Devys, D.; van den Ouweland, A.; Mandel, J.L.; Galjaard, H.; Oostra, B. Rapid antibody test for fragile X syndrome. Lancet 1995, 345, 1147–1148. [Google Scholar] [CrossRef]
- Willemsen, R.; Smits, A.; Mohkamsing, S.; van Beerendonk, H.; de Haan, A.; de Vries, B.; van den Ouweland, A.; Sistermans, E.; Galjaard, H.; Oostra, B.A. Rapid antibody test for diagnosing fragile X syndrome: A validation of the technique. Hum. Genet. 1997, 99, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Anar, B.; De Diego Otero, Y.; de Vries, B.B.; Hilhorst-Hofstee, Y.; Smits, A.; van Looveren, E.; Willems, P.J.; Galjaard, H.; Oostra, B.A. Noninvasive test for fragile X syndrome, using hair root analysis. Am. J. Hum. Genet. 1999, 65, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Smits, A.; Severijnen, L.A.; Jansen, M.; Jacobs, A.; De Bruyn, E.; Oostra, B. Predictive testing for cognitive functioning in female carriers of the fragile X syndrome using hair root analysis. J. Med. Genet. 2003, 40, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Oostra, B.A.; Willemsen, R. Diagnostic tests for fragile X syndrome. Expert Rev. Mol. Diagn. 2001, 1, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.; Oosterwijk, J.C.; Los, F.J.; Galjaard, H.; Oostra, B.A. Prenatal diagnosis of fragile X syndrome. Lancet 1996, 348, 967–968. [Google Scholar] [CrossRef]
- Willemsen, R.; Los, F.; Mohkamsing, S.; van den Ouweland, A.; Deelen, W.; Galjaard, H.; Oostra, B. Rapid antibody test for prenatal diagnosis of fragile X syndrome on amniotic fluid cells: A new appraisal. J. Med. Genet. 1997, 34, 250–251. [Google Scholar] [CrossRef] [PubMed]
- Lambiris, N.; Peters, H.; Bollmann, R.; Leschik, G.; Leisti, J.; Salonen, R.; Cobet, G.; Oostra, B.A.; Willemsen, R. Rapid FMR1-protein analysis of fetal blood: An enhancement of prenatal diagnostics. Hum. Genet. 1999, 105, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Lessard, M.; Chouiali, A.; Drouin, R.; Sebire, G.; Corbin, F. Quantitative measurement of FMRP in blood platelets as a new screening test for fragile X syndrome. Clin. Genet. 2012, 82, 472–477. [Google Scholar] [CrossRef] [PubMed]
- LaFauci, G.; Adayev, T.; Kascsak, R.; Kascsak, R.; Nolin, S.; Mehta, P.; Brown, W.T.; Dobkin, C. Fragile X screening by quantification of FMRP in dried blood spots by a Luminex immunoassay. J. Mol. Diagn. 2013, 15, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Adayev, T.; LaFauci, G.; Dobkin, C.; Caggana, M.; Wiley, V.; Field, M.; Wotton, T.; Kascsak, R.; Nolin, S.L.; Glicksman, A.; et al. Fragile X protein in newborn dried blood spots. BMC Med. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.C.; Coffee, B.; Benke, P.J.; Berry-Kravis, E.; Gilbert, F.; Oostra, B.; Halley, D.; Zwick, M.E.; Cutler, D.J.; Warren, S.T. Array-based FMR1 sequencing and deletion analysis in patients with a fragile X syndrome-like phenotype. PLoS ONE 2010, 5, e9476. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajan-Babu, I.-S.; Chong, S.S. Molecular Correlates and Recent Advancements in the Diagnosis and Screening of FMR1-Related Disorders. Genes 2016, 7, 87. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7100087
Rajan-Babu I-S, Chong SS. Molecular Correlates and Recent Advancements in the Diagnosis and Screening of FMR1-Related Disorders. Genes. 2016; 7(10):87. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7100087
Chicago/Turabian StyleRajan-Babu, Indhu-Shree, and Samuel S. Chong. 2016. "Molecular Correlates and Recent Advancements in the Diagnosis and Screening of FMR1-Related Disorders" Genes 7, no. 10: 87. https://0-doi-org.brum.beds.ac.uk/10.3390/genes7100087