
The Role of the Transcriptional Response to DNA Replication Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. DNA Replication
2. DNA Replication Stress
2.1. DNA Characteristics
2.2. Obstructions to Replication Fork Progression
2.3. Replication and Transcription Collisions
2.4. Loss of Regulation of DNA Replication
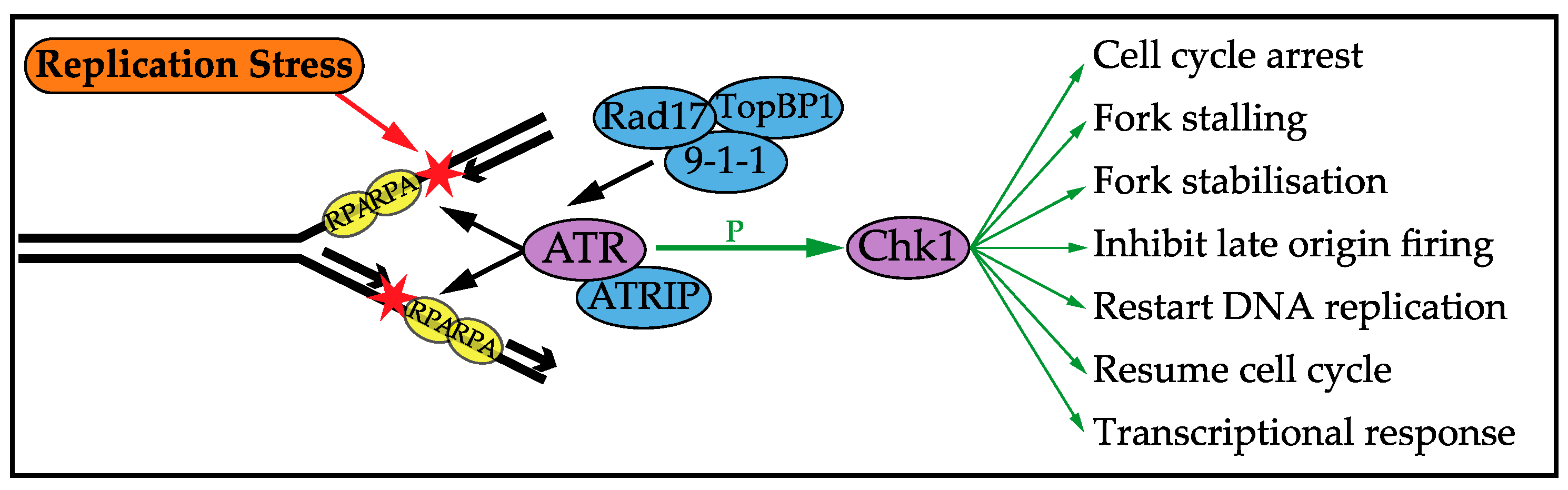
3. DNA Replication Stress Checkpoint Response
3.1. Cell Cycle Arrest
3.2. Stalling and Stabilising Replication Forks
3.3. Control of Origin Firing
3.4. Replication Restart
4. The Transcriptional Response to DNA Replication Stress
4.1. Role of the Replication Stress Transcriptional Response
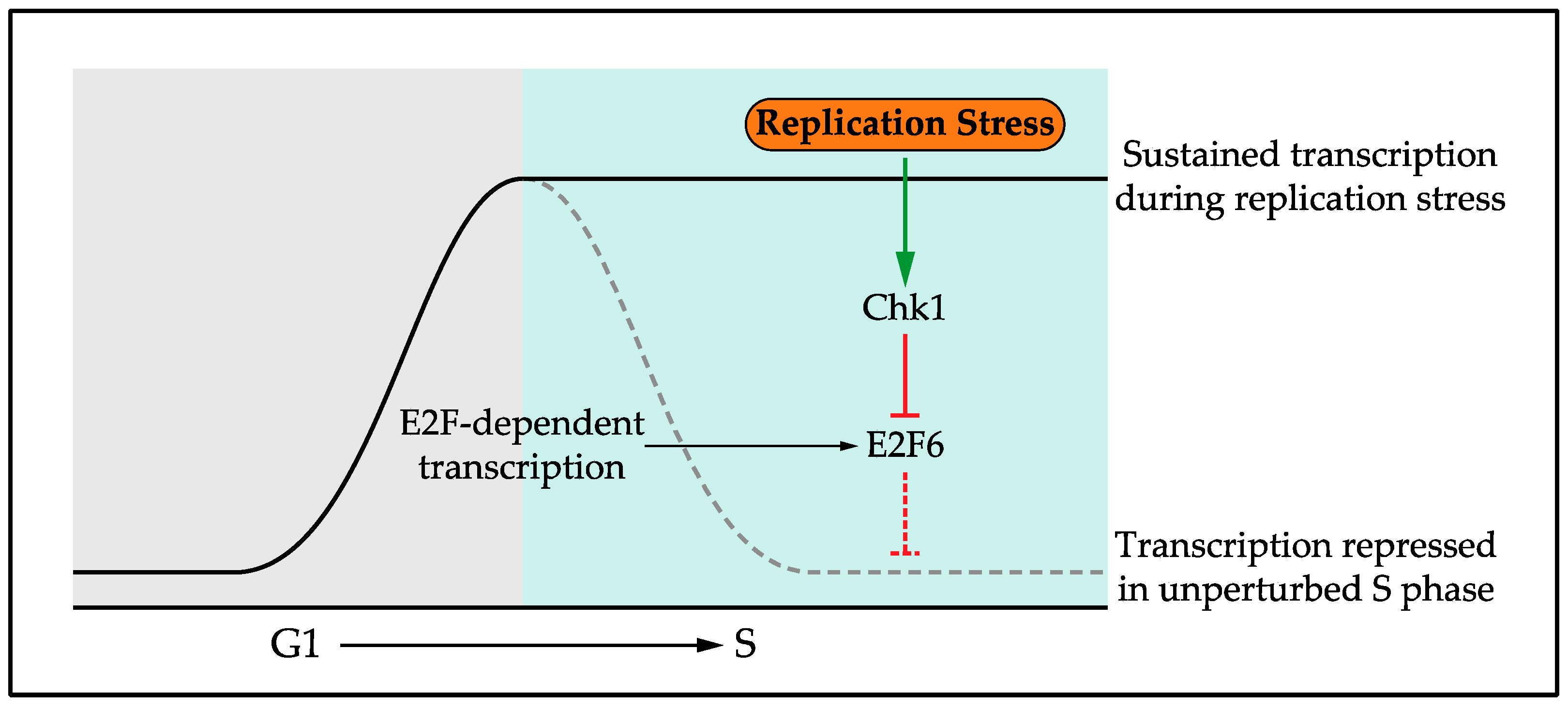
5. Regulation of the Transcriptional Response
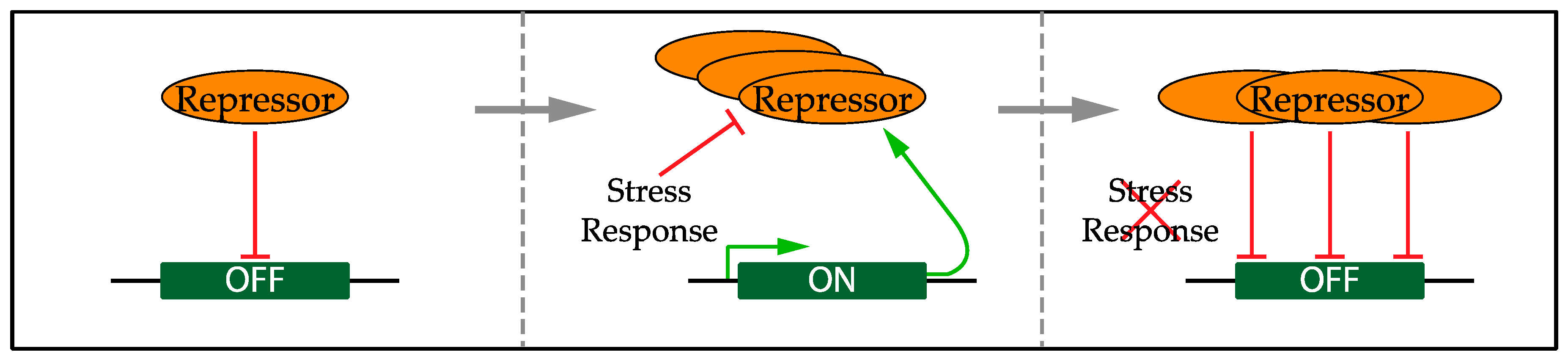
5.1. Confining the Transcriptional Response to Replication Stress
5.2. DNA Replication Restart
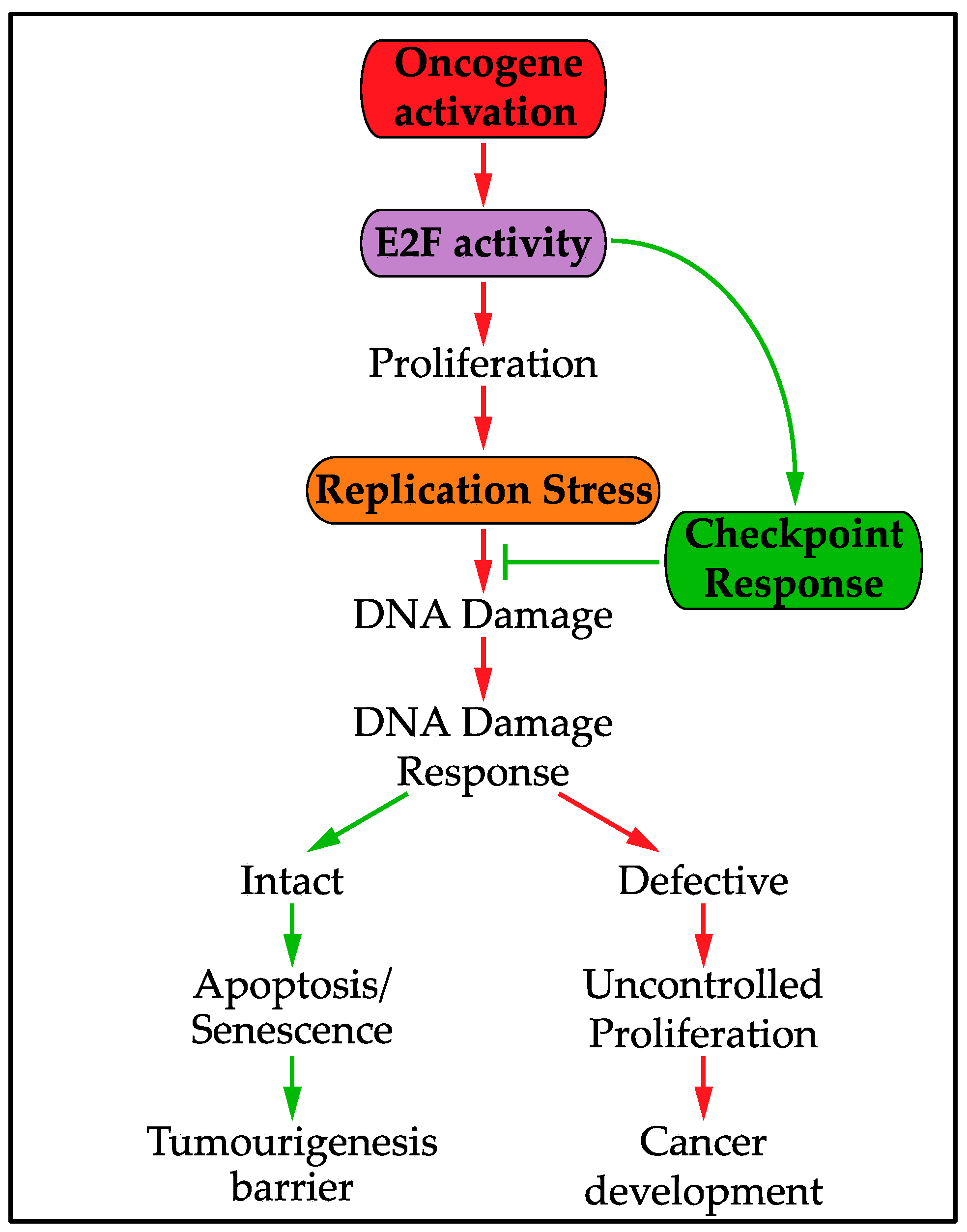
6. The Replication Stress Transcriptional Response and Oncogenic Activity
7. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fragkos, M.; Ganier, O.; Coulombe, P.; Méchali, M. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015, 16, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Deegan, T.D.; Janska, A.; Early, A.; Diffley, J.F.X. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 2015, 519, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Woo, R.A.; Poon, R.Y.C. Cyclin-dependent kinases and S phase control in mammalian cells. Cell Cycle 2003, 2, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.G.; Park, C.-H.; Burke, T.W.; Leone, G.; DeGregori, J.; Engel, A.; Nevins, J.R. Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Rialland, M.; Sola, F.; Santocanale, C. Essential role of human CDT1 in DNA replication and chromatin licensing. J. Cell Sci. 2002, 115, 1435–1440. [Google Scholar] [PubMed]
- Cayrou, C.; Coulombe, P.; Vigneron, A.; Stanojcic, S.; Ganier, O.; Peiffer, I.; Rivals, E.; Puy, A.; Laurent-Chabalier, S.; Desprat, R.; et al. Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011, 21, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Yekezare, M.; Gómez-González, B.; Diffley, J.F.X. Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally. J. Cell Sci. 2013, 126, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Santocanale, C.; Sharma, K.; Diffley, J.F.X. Activation of dormant origins of DNA replication in budding yeast. Genes Dev. 1999, 13, 2360–2364. [Google Scholar] [CrossRef] [PubMed]
- Larasati; Duncker, B. Mechanisms Governing DDK Regulation of the Initiation of DNA Replication. Genes 2016. [Google Scholar] [CrossRef] [PubMed]
- Labib, K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 2010, 24, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.P.; Jones, J.M.; Bruck, I.; Kaplan, D.L. Origin DNA Melting-An Essential Process with Divergent Mechanisms. Genes 2017. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.U.E.; Liu, Y.; Wu, X.; Shell, S.M. Functions of Human Replication Protein A (RPA): From DNA Replication to DNA Damage and Stress Responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.Q.; Co, C.; Li, J.J. Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature 2001, 411, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of Eukaryotic DNA Replication by Geminin Binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, Q.; Liao, R.; Sun, P.; Wu, X. The SCFSkp2 ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J. Biol. Chem. 2003, 278, 30854–30858. [Google Scholar] [CrossRef] [PubMed]
- Méndez, J.; Zou-Yang, X.H.; Kim, S.-Y.; Hidaka, M.; Tansey, W.P.; Stillman, B. Human Origin Recognition Complex Large Subunit Is Degraded by Ubiquitin-Mediated Proteolysis after Initiation of DNA Replication. Mol. Cell 2002, 9, 481–491. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K. a Causes and consequences of replication stress. Nat. Cell Biol. 2013, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hills, S.A.; Diffley, J.F.X. DNA Replication and Oncogene-Induced Replicative Stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Lecona, E.; Fernández-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Vassiliou, L.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R., Jr.; Kastrinakis, N.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.-R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Vasquez, K. Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes 2017. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, E.V.; Mirkin, S.M. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef] [PubMed]
- Viguera, E.; Canceill, D.; Ehrlich, S.D. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 2001, 20, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.G.; Glover, T.W. Chromosome Fragile Sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Szilard, R.K.; Jacques, P.E.; Laramée, L.; Cheng, B.; Galicia, S.; Bataille, A.R.; Yeung, M.; Mendez, M.; Bergeron, M.; Robert, F.; et al. Systematic identification of fragile sites via genome-wide location analysis of γ-H2AX. Nat. Struct. Mol. Biol. 2011, 17, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Rozenzhak, S.; Mejía-Ramírez, E.; Williams, J.S.; Schaffer, L.; Hammond, J.A.; Head, S.R.; Russell, P. Rad3ATR decorates critical chromosomal domains with γH2A to protect genome integrity during S-phase in fission yeast. PLoS Genet. 2010, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Keszthelyi, A.; Minchell, N.; Baxter, J. The Causes and Consequences of Topological Stress during DNA Replication. Genes 2016. [Google Scholar] [CrossRef] [PubMed]
- Ivessa, A.S.; Lenzmeier, B.A.; Bessler, J.B.; Goudsouzian, L.K.; Schnakenberg, S.L.; Zakian, V.A. The Saccharomyces cerevisiae Helicase Rrm3p Facilitates Replication Past Nonhistone Protein-DNA Complexes. Mol. Cell 2003, 12, 1525–1536. [Google Scholar] [CrossRef]
- Lambert, S.; Carr, A.M. Impediments to replication fork movement: Stabilisation, reactivation and genome instability. Chromosoma 2013, 122, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, S.; Méndez, J. DNA replication stress: From molecular mechanisms to human disease. Chromosoma 2016. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene 2014, 34, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Resnitzky, D.; Gossen, M.; Bujard, H.; Reed, S.I. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol. Cell. Biol. 1994, 14, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Araki, H. Multiple regulatory mechanisms to inhibit untimely initiation of DNA replication are important for stable genome maintenance. PLoS Genet. 2011, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Liontos, M.; Koutsami, M.; Sideridou, M.; Evangelou, K.; Kletsas, D.; Levy, B.; Kotsinas, A.; Nahum, O.; Zoumpourlis, V.; Kouloukoussa, M.; et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007, 67, 10899–10909. [Google Scholar] [CrossRef] [PubMed]
- Green, B.M.; Finn, K.J.; Li, J.J. Loss of DNA replication control is a potent inducer of gene amplification. Science 2010, 329, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbe, L.; Coquelle, A.; Pasero, P. Does interference between replication and transcription contribute to genomic instability in cancer cells? Cell Cycle 2010, 9, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Jossen, R.; Bermejo, R. The DNA damage checkpoint response to replication stress: A Game of Forks. Front. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F.X. DNA replication as a target of the DNA damage checkpoint. DNA Repair (Amst.) 2009, 8, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.S.; Syljuåsen, R.G. Safeguarding genome integrity: The checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012, 40, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Barnes, R.; Eckert, K. Maintenance of Genome Integrity: How Mammalian Cells Orchestrate Genome Duplication by Coordinating Replicative and Specialized DNA Polymerases. Genes 2017. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The checkpoint response to replication stress. DNA Repair (Amst.) 2009, 8, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Feng, W. Mec1/ATR, the Program Manager of Nucleic Acids Inc. Genes 2016. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Carignon, S.; Técher, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise Activation of the ATR Signaling Pathway upon Increasing Replication Stress Impacts Fragile Site Integrity. PLoS Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- McGowan, C.H.; Russell, P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993, 12, 75–85. [Google Scholar] [PubMed]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef]
- Unsal-Kaçmaz, K.; Chastain, P.D.; Qu, P.-P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Cotta-Ramusino, C.; Pellicioli, A.; Liberi, G.; Plevani, P.; Muzi-Falconi, M.; Newlon, C.S.; Foiani, M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 2001, 412, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Calzada, A.; Hodgson, B.; Kanemaki, M.; Bueno, A.; Labib, K. Molecular anatomy and regulation of a stable replisome eukaryotic DNA at a paused replication fork. Genes Dev. 2005, 19, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Lossaint, G.; Larroque, M.; Ribeyre, C.; Bec, N.; Larroque, C.; Décaillet, C.; Gari, K.; Constantinou, A. FANCD2 Binds MCM Proteins and Controls Replisome Function upon Activation of S Phase Checkpoint Signaling. Mol. Cell 2013, 51, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Masai, H.; Bartek, J. Regulation and roles of Cdc7 kinase under replication stress. Cell Cycle 2014, 13, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Watanabe, K.; Mistrik, M.; Vesela, E.; Protivankova, I.; Mailand, N.; Lee, M.; Masai, H.; Lukas, J.; Bartek, J. ATR–Chk1–APC/C Cdh1-dependent stabilization of Cdc7–ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes Dev. 2013, 27, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, I.; Koepp, D. Recovery from the DNA Replication Checkpoint. Genes 2016. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.Y.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res. 2008, 18, 8–16. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, N.; Wong, R.P.; Ulrich, H.D. Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front. Genet. 2016, 7, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the maestro of the replication fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, R.A.M.; Kalashnikova, T.I.; Aslanian, A.; Wohlschlegel, J.; Chahwan, C.; Yates, J.R.; Russell, P.; Wittenberg, C. DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc. Natl. Acad. Sci. USA 2008, 105, 11230–11235. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Patel, P.K.; Rosebrock, A.; Oliva, A.; Leatherwood, J.; Rhind, N. The DNA Replication Checkpoint Directly Regulates MBF-Dependent G1/S Transcription. Mol. Cell. Biol. 2008, 28, 5977–5985. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.; Li, J.; Eshaghi, M.; Peng, X.; Karuturi, R.K.M.; Liu, J. Modulation of Cell Cycle–specific Gene Expressions at the Onset of S Phase Arrest Contributes to the Robust DNA Replication Checkpoint Response in Fission Yeast. Mol. Biol. Cell 2007, 18, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Caetano, C.; Klier, S.; de Bruin, R.A.M. Phosphorylation of the MBF repressor Yox1p by the DNA replication checkpoint keeps the G1/S cell-cycle transcriptional program active. PLoS ONE 2011, 6, e17211. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.; Gómez-Escoda, B.; Hidalgo, E.; Ayté, J. G1/S transcription and the DNA synthesis checkpoint: Common regulatory mechanisms. Cell Cycle 2011, 10, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Escoda, B.; Ivanova, T.; Calvo, I.A.; Alves-Rodrigues, I.; Hidalgo, E.; Ayte, J. Yox1 links MBF-dependent transcription to completion of DNA synthesis. EMBO Rep 2011, 12, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.; Alves-Rodrigues, I.; Gómez-Escoda, B.; Dutta, C.; DeCaprio, J.A.; Rhind, N.; Hidalgo, E.; Ayté, J. The DNA damage and the DNA replication checkpoints converge at the MBF transcription factor. Mol. Biol. Cell 2013, 24, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007, 26, 2083–2093. [Google Scholar] [CrossRef]
- Travesa, A.; Kuo, D.; de Bruin, R.A.M.; Kalashnikova, T.I.; Guaderrama, M.; Thai, K.; Aslanian, A.; Smolka, M.B.; Yates, J.R.; Ideker, T.; et al. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012, 31, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Bastos de Oliveira, F.M.; Harris, M.R.; Brazauskas, P.; de Bruin, R.A.; Smolka, M.B. Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 2012, 31, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Klier, S.; McGowan, C.; Wittenberg, C.; de Bruin, R.A.M. Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell-cycle transcription. Curr. Biol. 2013, 23, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, R.A.; Kalashnikova, T.I.; Chahwan, C.; McDonald, W.H.; Wohlschlegel, J.; Yates, J.; Russell, P.; Wittenberg, C. Constraining G1-specific transcription to late G1 phase: The MBF-associated corepressor Nrm1 acts via negative feedback. Mol. Cell 2006, 23, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Tercero, A.; Longhese, M.P.; Diffley, J.F.X. A Central Role for DNA Replication Forks in Checkpoint Activation and Response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Bertoli, C.; Herlihy, A.E.; Pennycook, B.R.; Kriston-Vizi, J.; de Bruin, R.A.M. Sustained E2F-Dependent Transcription Is a Key Mechanism to Prevent Replication-Stress-Induced DNA Damage. Cell Rep. 2016, 15, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Huang, E.; Zuzan, H.; Spang, R.; Leone, G.; West, M.; Nevins, J.R. Role for E2F in Control of Both DNA Replication and Mitotic Functions as Revealed from DNA Microarray Analysis. Mol. Cell. Biol. 2001, 21, 4684–4699. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Gorgoulis, V.G. Involvement of E2F transcription factor family in cancer. Eur. J. Cancer 2005, 41, 2403–2414. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.; La Thangue, N.B. E2F and cell cycle control: A double-edged sword. Arch. Biochem. Biophys. 2003, 412, 157–169. [Google Scholar] [CrossRef]
- De Nadal, E.; Ammerer, G.; Posas, F. Controlling gene expression in response to stress. Nat. Rev. Genet. 2011, 12, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Contreras, A.J.; Gutierrez-Martinez, P.; Specks, J.; Rodrigo-Perez, S.; Fernandez-Capetillo, O. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J. Exp. Med. 2012, 209, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Contreras, A.J.; Specks, J.; Barlow, J.H.; Ambrogio, C.; Desler, C.; Vikingsson, S.; Rodrigo-Perez, S.; Green, H.; Rasmussen, L.J.; Murga, M.; et al. Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev. 2015, 29, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin E Deregulation Promotes Loss of Specific Genomic Regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhou, Z.; Elledge, S.J. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998, 94, 595–605. [Google Scholar] [CrossRef]
- Lubelsky, Y.; Reuven, N.; Shaul, Y. Autorepression of Rfx1 Gene Expression: Functional Conservation from Yeast to Humans in Response to DNA Replication Arrest. Mol. Cell. Biol. 2005, 25, 10665–10673. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, P.; Hofer, A.; Thelander, L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem. 2006, 281, 7834–7841. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, H. SOS response as an adaptive response to DNA damage in prokaryotes. In Stress-Inducible Cellular Responses; Feige, U., Yahara, I., Morimoto, R.I., Polla, B.S., Eds.; Birkhäuser Basel: Basel, Switzerland, 1996; pp. 221–235. [Google Scholar]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Caetano, C.; Limbo, O.; Farmer, S.; Klier, S.; Dovey, C.; Russell, P.; de Bruin, R.A.M. Tolerance of Deregulated G1/S Transcription Depends on Critical G1/S Regulon Genes to Prevent Catastrophic Genome Instability. Cell Rep. 2014, 9, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Otterness, D.M.; Chiang, G.G.; Xie, W.; Liu, Y.-C.; Mercurio, F.; Abraham, R.T. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 2005, 19, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Dorrello, N.V.; Guardavaccaro, D.; Venere, M.; Halazonetis, T.; Sherman, N.E.; Pagano, M. SCFβTrCP-Mediated Degradation of Claspin Regulates Recovery from the DNA Replication Checkpoint Response. Mol. Cell 2006, 23, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Bartek, J.; Lukas, J. Destruction of Claspin by SCFβTrCP Restrains Chk1 Activation and Facilitates Recovery from Genotoxic Stress. Mol. Cell 2006, 23, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nannenga, B.; Donehower, L.A. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005, 19, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Szyjka, S.J.; Aparicio, J.G.; Viggiani, C.J.; Knott, S.; Xu, W.; Tavaré, S.; Aparicio, O.M. Rad53 regulates replication fork restart after DNA damage in Saccharomyces cerevisiae. Genes Dev. 2008, 22, 1906–1920. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Herlihy, A.E. The DNA Replication Stress Checkpoint Transcriptional Response and Its Role in Replication Stress Tolerance. Ph.D. Thesis, University College London, London, UK, 2017. [Google Scholar]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herlihy, A.E.; De Bruin, R.A.M. The Role of the Transcriptional Response to DNA Replication Stress. Genes 2017, 8, 92. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8030092
Herlihy AE, De Bruin RAM. The Role of the Transcriptional Response to DNA Replication Stress. Genes. 2017; 8(3):92. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8030092
Chicago/Turabian StyleHerlihy, Anna E., and Robertus A.M. De Bruin. 2017. "The Role of the Transcriptional Response to DNA Replication Stress" Genes 8, no. 3: 92. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8030092