Dissecting Tissue-Specific Transcriptomic Responses from Leaf and Roots under Salt Stress in Petunia hybrida Mitchell

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Treatments
2.2. Tissue Sample and RNA Isolation
2.3. Library Preparation and Deep Sequencing
2.4. Processing of Illumina RNA-Seq Reads
2.5. Sequence Alignment to the Petunia Genome
2.6. Table Counts
2.7. Gene Expression and Differential Gene Expression
2.8. Bar Plots, Venn Diagrams and Heat Maps
2.9. Sequencing Data
2.10. Phylogenetic Analysis
2.11. Truncating the Petunia Trehalose 6 Phosphate Synthase PhTPS1 (⊗PhTPS1)
2.12. The pDB20 Yeast Vector
2.13. Cloning ⊗PhTPS1 into the pDB20 Yeast Expression Vector
2.14. Yeast Strains and Functional TPS1 Gene Analysis
3. Results and Discussion
3.1. Transcriptomic Relationship between the Samples
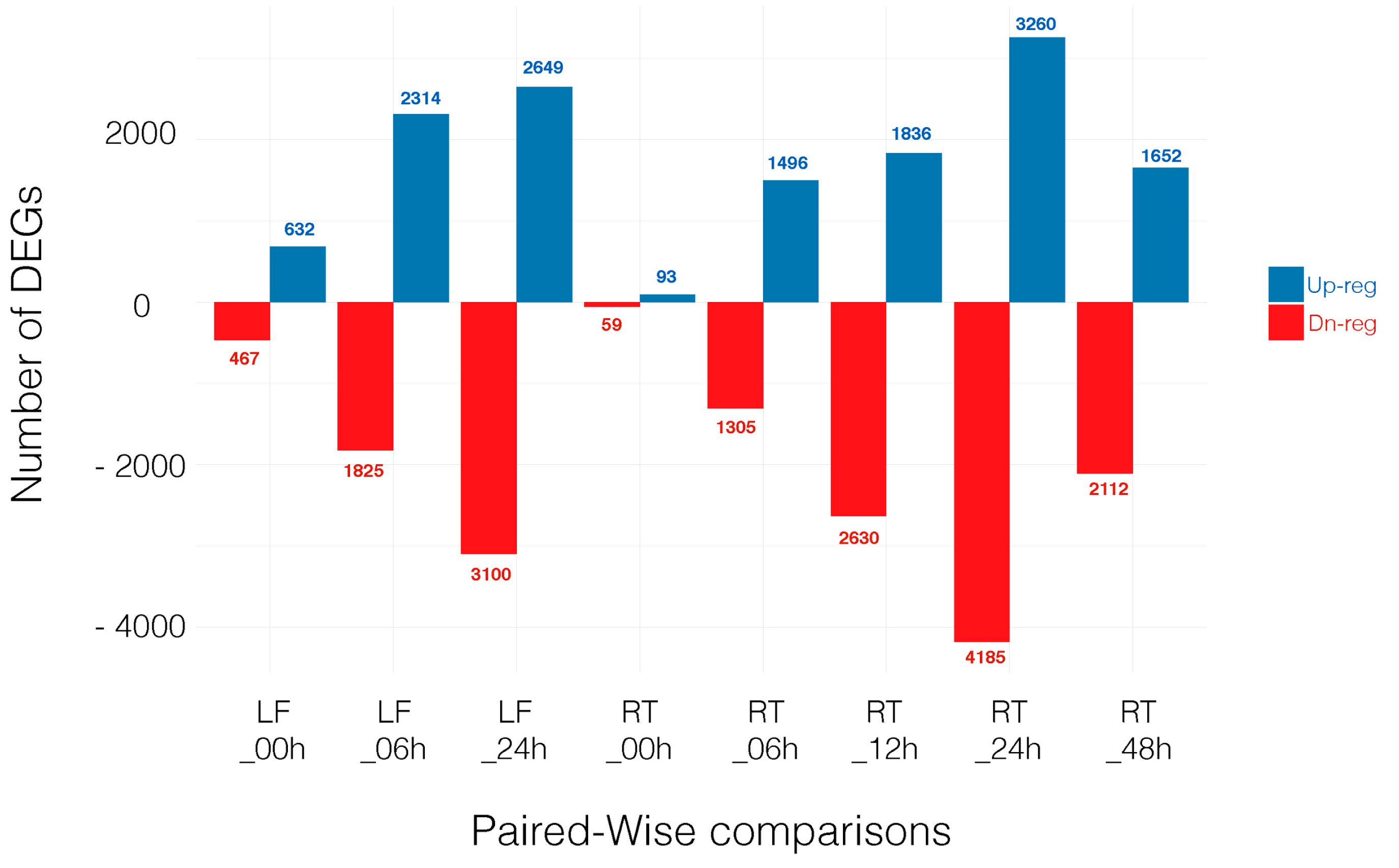
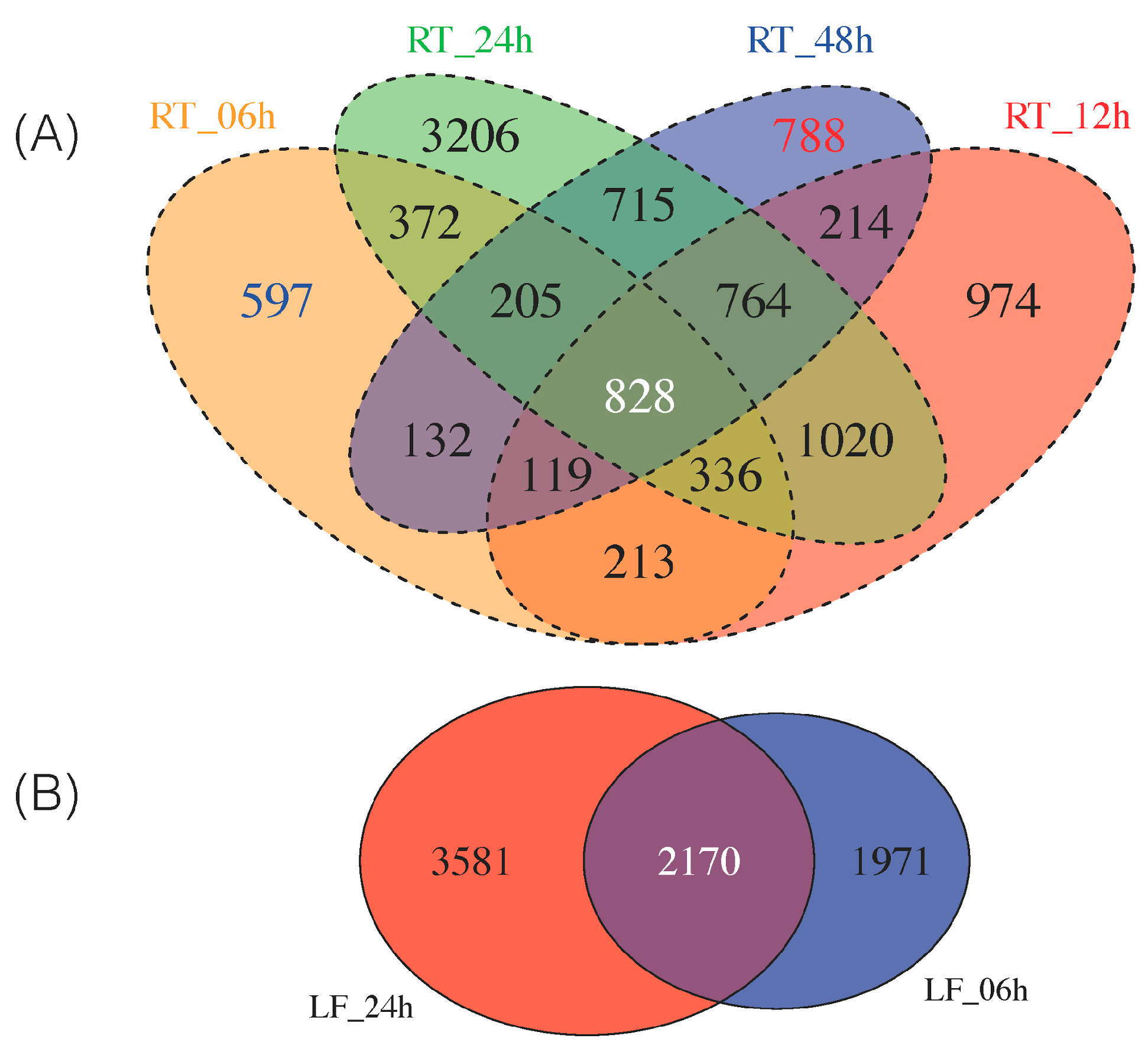
3.2. Differentially Expressed Genes across Time Point and Tissues
3.3. Expanding from Previous Transcriptomic Studies
3.4. Candidate Genes to Enhance Salt Stress
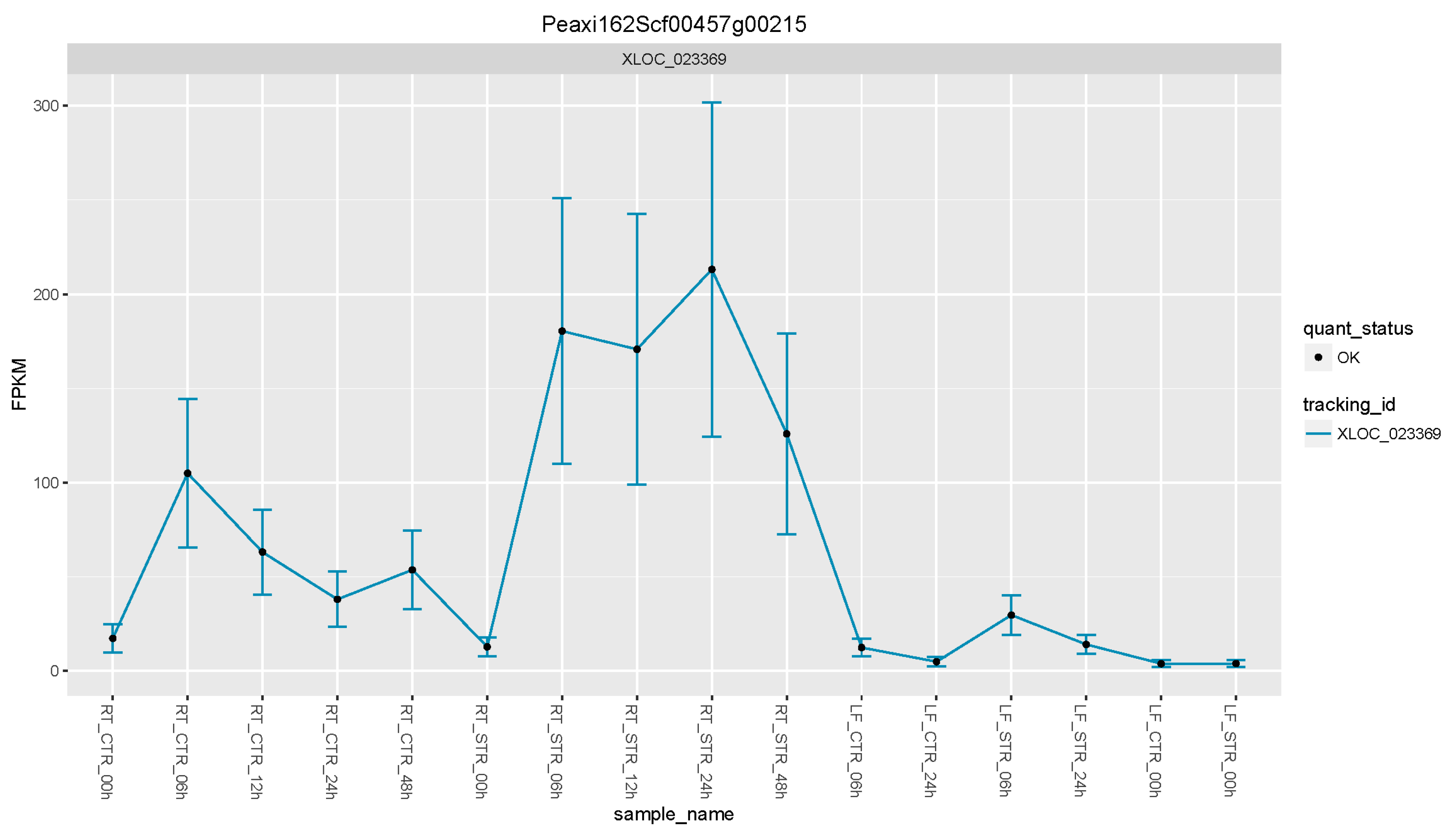
3.5. Petunia Trehalose-6-Phosphate Synthase 1 Gene, a Candidate Gene to Enhance Salt Tolerance in Solanaceae Plants
3.6. The Petunia Trehalose-6-Phosphate Synthase 1 Gene Rescue Mutant Yeast Phenotype
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mansour, M.M.F.; Salama, K.H.A. Cellular basis of salinity tolerance in plants. Environ. Exp. Bot. 2004, 52, 113–122. [Google Scholar] [CrossRef]
- Zhu, J.-K. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef]
- Munns, R. Genes and salt tolerance: Bringing them together: Tansley review. New Phytol. 2005, 167, 645–663. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.; Foolad, M.R. Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ. Exp. Bot. 2007, 59, 206–216. [Google Scholar] [CrossRef]
- Møller, I.S.; Gilliham, M.; Jha, D.; Mayo, G.M.; Roy, S.J.; Coates, J.C.; Haseloff, J.; Tester, M. Shoot Na+ Exclusion and increased salinity tolerance engineered by cell type-specific alteration of Na+ transport in Arabidopsis. Plant Cell Online 2009, 21, 2163–2178. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Ding, L.; Zhu, J.K. SOS1, a genetic locus essential for salt tolerance and potassium acquisition. Plant Cell Online 1996, 8, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Diehl, A.; Wu, F.; Vrebalov, J.; Giovannoni, J.; Siepel, A.; Tanksley, S.D. Sequencing and comparative analysis of a conserved syntenic segment in the Solanaceae. Genetics 2008, 180, 391–408. [Google Scholar] [CrossRef] [PubMed]
- USDA National Agricultural Statistics Service. Census of Agriculture, United States Summary and State Data; USDA National Agricultural Statistics Service: Washington, DC, USA, 2009; Volume 1, p. 739.
- USDA National Agricultural Statistics Service. Floriculture Crops Summary; USDA National Agricultural Statistics Service: Washington, DC, USA, 2011; p. 59.
- Broderick, S.R.; Wijeratne, S.; Wijeratn, A.J.; Chapin, L.J.; Meulia, T.; Jones, M.L. RNA-sequencing reveals early, dynamic transcriptome changes in the corollas of pollinated petunias. BMC Plant Biol. 2014, 14, 307. [Google Scholar] [CrossRef] [PubMed]
- Villarino, G.H.; Mattson, N.S. assessing tolerance to sodium chloride salinity in fourteen floriculture species. HortTechnology 2011, 21, 539–545. [Google Scholar]
- Ren, Z.-H.; Gao, J.-P.; Li, L.-G.; Cai, X.-L.; Huang, W.; Chao, D.-Y.; Zhu, M.-Z.; Wang, Z.-Y.; Luan, S.; Lin, H.-X. A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat. Genet. 2005, 37, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Rensink, W.; Hart, A.; Liu, J.; Ouyang, S.; Zismann, V.; Buell, C.R. Analyzing the potato abiotic stress transcriptome using expressed sequence tags. Genome 2005, 48, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.H.; Lippold, F.; Redestig, H.; Hannah, M.A.; Erban, A.; Krämer, U.; Kopka, J.; Udvardi, M.K. Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus: Systems analysis of Lotus japonicus under salt stress. Plant J. 2007, 53, 973–987. [Google Scholar] [CrossRef] [PubMed]
- Villarino, G.H.; Bombarely, A.; Giovannoni, J.J.; Scanlon, M.J.; Mattson, N.S. Transcriptomic analysis of Petunia hybrida in response to salt stress using high throughput RNA sequencing. PLoS ONE 2014, 9, e94651. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Yang, S.S.; Miller, S.S.; Bucciarelli, B.; Liu, J.; Rydeen, A.; Bozsoki, Z.; Uhde-Stone, C.; Tu, Z.J.; Allan, D.; et al. An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol. 2013, 161, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Cui, P.; Wang, Z.; Zhang, S.; Ali, S.; Xiong, L. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genom. 2014, 15, 431. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.H.; Oliver, B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Vinocur, B.; Altman, A. Recent advances in engineering plant tolerance to abiotic stress: Achievements and limitations. Curr. Opin. Biotechnol. 2005, 16, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Zentella, R.; Mascorro-Gallardo, J.O.; Van Dijck, P.; Folch-Mallol, J.; Bonini, B.; Van Vaeck, C.; Gaxiola, R.; Covarrubias, A.A.; Nieto-Sotelo, J.; Thevelein, J.M.; et al. A Selaginella lepidophylla Trehalose-6-Phosphate synthase complements growth and stress-tolerance defects in a yeast tps1 mutant. Plant Physiol. 1999, 119, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, M.A.; Santos, E.; Flores, C.; Martínez-Zapater, J.M.; Salinas, J.; Gancedo, C. Isolation and molecular characterization of the Arabidopsis TPS1 gene, encoding trehalose-6-phosphate synthase. Plant J. 1998, 13, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.Z.; Hanson, M.R.; Skvirsky, R.C.; Ausubel, F.M. Anther culture of Petunia: Genotypes with high frequency of callus, root, or plantlet formation. Z. Für Pflanzenphysiol. 1980, 100, 131–145. [Google Scholar] [CrossRef]
- Zhong, S.; Joung, J.-G.; Zheng, Y.; Chen, Y.-R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-Throughput Illumina strand-specific RNA sequencing library preparation. Cold Spring Harbor Protoc. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Bombarely, A.; Moser, M.; Amrad, A.; Bao, M.; Bapaume, L.; Barry, C.S.; Bliek, M.; Boersma, M.R.; Borghi, L.; Bruggmann, R.; et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2016, 2, 16074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Bioconductor—edgeR. Available online: http://bioconductor.org/packages/release/bioc/html/edgeR.html (accessed on 1 March 2017).
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T Cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Nookaew, I.; Papini, M.; Pornputtapong, N.; Scalcinati, G.; Fagerberg, L.; Uhlén, M.; Nielsen, J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: A case study in Saccharomyces cerevisiae. Nucleic Acids Res. 2012, 40, 10084–10097. [Google Scholar] [CrossRef] [PubMed]
- DeWald, D.B.; Torabinejad, J.; Jones, C.A.; Shope, J.C.; Cangelosi, A.R.; Thompson, J.E.; Prestwich, G.D.; Hama, H. Rapid accumulation of phosphatidylinositol 4,5-bisphosphate and inositol 1,4,5-trisphosphate correlates with calcium mobilization in salt-stressed Arabidopsis. Plant Physiol. 2001, 126, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Pical, C.; Westergren, T.; Dove, S.K.; Larsson, C.; Sommarin, M. Salinity and hyperosmotic stress induce rapid increases in phosphatidylinositol 4,5-bisphosphate, diacylglycerol pyrophosphate, and phosphatidylcholine in Arabidopsis thaliana cells. J. Biol. Chem. 1999, 274, 38232–38240. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; Rui, Y.E.; Hui, M.A.; Xu, Z.H.; Xue, H.W. DNA chip-based expression profile analysis indicates involvement of the phosphatidylinositol signaling pathway in multiple plant responses to hormone and abiotic treatments. Cell Res. 2004, 14, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Staxén, I.; Pical, C.; Montgomery, L.T.; Gray, J.E.; Hetherington, A.M.; McAinsh, M.R. Abscisic acid induces oscillations in guard-cell cytosolic free calcium that involve phosphoinositide-specific phospholipase C. Proc. Natl. Acad. Sci. USA 1999, 96, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.G.; Toyota, M.; Kim, S.H.; Hilleary, R.; Gilroy, S. Salt stress-induced Ca2+ waves are associated with rapid, long-distance root-to-shoot signaling in plants. Proc. Natl. Acad. Sci. USA 2014, 111, 6497–6502. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Berry, A.W.; Harpham, N.V.J.; Moshkov, I.E.; Novikova, G.V.; Kulaeva, O.N.; Hall, M.A. (Eds.) Plant hormone signal perception and transduction. In Proceedings of the International Symposium on Plant Hormone Signal Perception and Transduction, Moscow, Russia, 4–10 September 1994; Kluwer Academic Publishers: Dordrecht, The Netherlands; Boston, MA, USA, 1996. [Google Scholar]
- Apse, M.P.; Aharon, G.S.; Snedden, W.A.; Blumwald, E. Salt tolerance conferred by overexpression of a Vacuolar Na+/H+ antiport in Arabidopsis. Science 1999, 285, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, M.; Kamboj, A.; Hove, R.M.; Loveridge, S.; El-Osta, A.; Bhave, M. Analysis of the barley leaf transcriptome under salinity stress using mRNA-Seq. Acta Physiol. Plant. 2013, 35, 1915–1924. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Villarino, G.H.; Hu, Q.; Manrique, S.; Flores-Vergara, M.; Sehra, B.; Robles, L.; Brumos, J.; Stepanova, A.N.; Colombo, L.; Sundberg, E.; et al. Transcriptomic signature of the SHATTERPROOF2 Expression domain reveals the meristematic nature of Arabidopsis gynoecial medial domain. Plant Physiol. 2016, 171, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Avonce, N.; Leyman, B.; Mascorro-Gallardo, J.O.; Van Dijck, P.; Thevelein, J.M.; Iturriaga, G. The Arabidopsis trehalose-6-P synthase AtTPS1 gene is a regulator of glucose, abscisic acid, and stress signaling. Plant Physiol 2004, 136, 3649–3659. [Google Scholar] [CrossRef] [PubMed]
- Thevelein, J.M.; Hohmann, S. Trehalose synthase: Guard to the gate of glycolysis in yeast? Trends Biochem. Sci. 1995, 20, 3–10. [Google Scholar] [CrossRef]
- Paul, M.J. Trehalose 6-phosphate: A signal of sucrose status. Biochem. J. 2008, 412, e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Schluepmann, H. Trehalose mediated growth inhibition of Arabidopsis seedlings is due to trehalose-6-phosphate accumulation. Plant Physiol. 2004, 135, 879–890. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Short Name | Annotation | Cufflinks | edgeR | Arabidopsis Homolog | 3IPR | 4GO | ||||||||
| RT_CTR_06h | RT_STR_06h | Fold Change | p-Value | p-Value | Fold Change | 1LR | p-Value | 2FDR | ||||||
| 6h-Up Reg | Peaxi162Scf00500g00067 | Late embryogenesis abundant protein, putative | 0.23 | 10.82 | 46.54 | 2.60E-03 | 6.61E-03 | 84.23 | 62.39 | 2.82E-15 | 2.11E-13 | AT3G53040.1 | IPR004238 | |
| 6h-Up Reg | Peaxi162Scf00845g00031 | Zinc finger CCCH domain-containing protein 2 | 0.36 | 8.43 | 23.11 | 2.20E-02 | 4.21E-02 | 122.75 | 114.94 | 8.09E-27 | 3.64E-24 | sp|Q9ZWA1|C3H2_ARATH | IPR000571 | GO:0046872 |
| 6h-Up Reg | Peaxi162Scf00921g00256 | Protein NRT1/PTR FAMILY 6.4 | 0.29 | 6.13 | 20.91 | 7.90E-03 | 1.74E-02 | 107.55 | 43.79 | 3.66E-11 | 1.25E-09 | sp|Q9LVE0|PTR33_ARATH | IPR000109,IPR016196 | GO:0016020,GO:0006810,GO:0005215 |
| 6h-Up Reg | Peaxi162Scf00366g00816 | Mannan endo-1,4-β-mannosidase 7 | 1.52 | 14.22 | 9.38 | 4.50E-04 | 1.39E-03 | 4.17 | 20.00 | 7.73E-06 | 7.88E-05 | sp|Q9FJZ3|MAN7_ARATH | IPR017853 | GO:0004553,GO:0005975,GO:0003824 |
| 6h-Up Reg | Peaxi162Scf00166g00824 | Cyclic nucleotide-gated channel 14 | 0.40 | 3.68 | 9.31 | 4.00E-03 | 9.62E-03 | 5.88 | 22.42 | 2.19E-06 | 2.61E-05 | AT2G24610.1 | IPR014710 | |
| 6h-Up Reg | Peaxi162Scf00002g00037 | Myb domain protein 42 | 0.73 | 6.32 | 8.65 | 2.24E-02 | 4.28E-02 | 6.34 | 21.16 | 4.23E-06 | 4.65E-05 | AT4G12350.1 | IPR009057 | GO:0003677,GO:0003682 |
| 6h-Up Reg | Peaxi162Scf00104g00017 | ERD (early-responsive to dehydration stress) family protein | 1.23 | 9.87 | 8.00 | 5.00E-05 | 1.85E-04 | 89.09 | 48.40 | 3.48E-12 | 1.44E-10 | AT4G02900.1 | IPR027815,IPR003864 | GO:0016020 |
| 6h-Up Reg | Peaxi162Scf00171g00525 | glutamate decarboxylase | 0.35 | 2.74 | 7.89 | 2.29E-02 | 4.37E-02 | 19.10 | 31.81 | 1.70E-08 | 3.48E-07 | AT5G17330.1 | IPR015424,IPR002129 | GO:0019752,GO:0016831,GO:0006536,GO:0004351,GO:0003824,GO:0030170 |
| 6h-Up Reg | Peaxi162Scf00102g00178 | Unknown protein | 12.73 | 95.62 | 7.51 | 5.00E-05 | 1.85E-04 | 3.01 | 12.15 | 4.91E-04 | 2.81E-03 | |||
| 6h-Up Reg | Peaxi162Scf00101g00031 | Protein of unknown function, DUF584 | 2.50 | 14.97 | 5.98 | 3.50E-03 | 8.56E-03 | 7.44 | 25.98 | 3.45E-07 | 5.26E-06 | AT5G60680.1 | IPR007608 | |
| 6h-Dn Reg | Peaxi162Scf00332g10028 | Plant protein of unknown function (DUF247) | 3.54 | 0.33 | 10.60 | 8.00E-03 | 1.76E-02 | 92.06 | 53.75 | 2.27E-13 | 1.19E-11 | AT3G02645.1 | IPR004158 | |
| 6h-Dn Reg | Peaxi162Scf00551g00011 | ZCF37, putative [Theobroma cacao] | 6.97 | 0.69 | 10.17 | 1.13E-02 | 2.36E-02 | 19.27 | 13.25 | 2.72E-04 | 1.70E-03 | ref|XP_007029922.1| | ||
| 6h-Dn Reg | Peaxi162Scf00030g00127 | EXORDIUM like 2 | 4.80 | 0.49 | 9.88 | 3.95E-03 | 9.52E-03 | 7.93 | 15.93 | 6.58E-05 | 5.07E-04 | AT5G64260.1 | IPR006766 | |
| 6h-Dn Reg | Peaxi162Scf00380g00817 | basic helix-loop-helix (bHLH) DNA-binding superfamily protein | 4.34 | 0.56 | 7.78 | 9.25E-03 | 1.99E-02 | 7.82 | 9.98 | 1.58E-03 | 7.44E-03 | AT4G37850.1 | IPR011598 | GO:0046983 |
| 6h-Dn Reg | Peaxi162Scf00074g00355 | R2R3-MYB transcription factor [Prunus avium] | 6.33 | 0.82 | 7.71 | 2.25E-02 | 4.30E-02 | 14.13 | 15.54 | 8.06E-05 | 6.02E-04 | gb|ADY15314.1| | IPR009057 | GO:0003677,GO:0003682 |
| 6h-Dn Reg | Peaxi162Scf01372g00039 | Protein of unknown function (DUF1442) | 3.53 | 0.49 | 7.24 | 6.55E-03 | 1.48E-02 | 18.97 | 13.35 | 2.58E-04 | 1.63E-03 | AT5G62280.1 | IPR009902 | |
| 6h-Dn Reg | Peaxi162Scf00980g00018 | Basic helix-loop-helix (bHLH) DNA-binding superfamily protein | 2.94 | 0.67 | 4.40 | 2.28E-02 | 4.35E-02 | 3.50 | 15.69 | 7.45E-05 | 5.63E-04 | AT5G48560.1 | IPR011598 | GO:0046983 |
| 6h-Dn Reg | Peaxi162Scf00058g00179 | Unknown protein | 5.74 | 1.37 | 4.19 | 2.68E-02 | 5.00E-02 | 3.70 | 14.11 | 1.73E-04 | 1.15E-03 | |||
| 6h-Dn Reg | Peaxi162Scf00258g00114 | Unknown protein | 16.26 | 4.63 | 3.51 | 1.80E-03 | 4.79E-03 | 2.56 | 10.35 | 1.30E-03 | 6.30E-03 | |||
| 6h-Dn Reg | Peaxi162Scf00616g00625 | BTB/POZ domain-containing protein | 2.64 | 0.85 | 3.10 | 2.21E-02 | 4.23E-02 | 2.77 | 13.73 | 2.11E-04 | 1.37E-03 | AT5G60050.1 | IPR011333 | |
| Gene Short Name | Annotation | Cufflinks | edgeR | Arabidopsis Homolog | IPR | GO | ||||||||
| RT_CTR_48h | RT_STR_48h | Fold Change | p-Value | p-Value | Fold Change | LR | p-Value | FDR | ||||||
| 48h-Up Reg | Peaxi162Scf00672g00810 | GDSL esterase/lipase | 3.56 | 63.04 | 17.70 | 5.00E-05 | 1.85E-04 | 20.26 | 63.67 | 1.47E-15 | 1.16E-13 | sp|Q9FHW9|GDL90_ARATH | IPR001087 | GO:0016787,GO:0016788,GO:0006629 |
| 48h-Up Reg | Peaxi162Scf00106g01710 | Cytochrome P450, family 96, subfamily A, polypeptide 10 | 0.29 | 4.71 | 16.00 | 2.65E-02 | 4.94E-02 | 64.53 | 34.40 | 4.48E-09 | 1.21E-07 | AT4G39490.1 | IPR001128 | GO:0020037,GO:0016705,GO:0005506,GO:0055114 |
| 48h-Up Reg | Peaxi162Scf00516g00330 | Palmitoyl-acyl carrier protein thioesterase, chloroplastic | 0.37 | 5.59 | 15.22 | 2.65E-02 | 4.95E-02 | 17.64 | 23.08 | 1.55E-06 | 2.43E-05 | sp|Q9SJE2|FATB_ARATH | IPR002864 | GO:0016790,GO:0006633 |
| 48h-Up Reg | Peaxi162Scf00069g01724 | Protein phosphatase 2C family protein | 1.42 | 20.73 | 14.58 | 5.00E-05 | 1.85E-04 | 18.73 | 61.31 | 4.87E-15 | 3.56E-13 | AT3G15260.1 | IPR001932,IPR015655 | GO:0003824 |
| 48h-Up Reg | Peaxi162Scf00059g01920 | CASP-like protein | 10.01 | 139.24 | 13.91 | 5.00E-05 | 1.85E-04 | 16.09 | 59.37 | 1.30E-14 | 8.92E-13 | sp|A7PJ32|CSPL2_VITVI | IPR006702 | |
| 48h-Up Reg | Peaxi162Scf00037g00185 | Unknown protein | 0.91 | 12.48 | 13.72 | 2.50E-04 | 8.18E-04 | 18.31 | 29.13 | 6.77E-08 | 1.42E-06 | |||
| 48h-Up Reg | Peaxi162Scf00074g01735 | WRKY DNA-binding protein 24 | 1.39 | 19.09 | 13.71 | 5.85E-03 | 1.34E-02 | 13.10 | 19.77 | 8.76E-06 | 1.12E-04 | AT5G41570.1 | IPR003657 | GO:0006355,GO:0043565,GO:0003700 |
| 48h-Up Reg | Peaxi162Scf01058g00011 | Cytochrome P450, family 86, subfamily A, polypeptide 1 | 5.21 | 60.55 | 11.62 | 5.00E-05 | 1.85E-04 | 14.35 | 25.94 | 3.52E-07 | 6.36E-06 | AT5G58860.1 | IPR001128 | GO:0020037,GO:0016705,GO:0005506,GO:0055114 |
| 48h-Up Reg | Peaxi162Scf00083g01919 | Aldehyde dehydrogenase family 3 member F1 | 0.44 | 4.92 | 11.07 | 1.90E-03 | 5.02E-03 | 5.87 | 6.71 | 9.61E-03 | 4.03E-02 | sp|Q70E96|AL3F1_ARATH | IPR016161,IPR012394 | GO:0016620,GO:0006081,GO:0008152,GO:0055114,GO:0016491,GO:0004030 |
| 48h-Up Reg | Peaxi162Scf00901g00418 | Metal ion binding protein, putative [Ricinus communis] | 0.66 | 6.95 | 10.56 | 2.64E-02 | 4.92E-02 | 112.76 | 17.10 | 3.54E-05 | 3.81E-04 | ref|XP_002526528.1| | IPR006121 | GO:0046872,GO:0030001 |
| 48h-Dn Reg | Peaxi162Scf00047g02040 | FASCICLIN-like arabinogalactan 2 | 45.62 | 0.62 | 73.43 | 1.65E-03 | 4.43E-03 | 160.00 | 85.31 | 2.56E-20 | 4.49E-18 | AT4G12730.1 | IPR008700,IPR000782 | |
| 48h-Dn Reg | Peaxi162Scf00559g00019 | 2-Oxoglutarate (2OG) and FeII-dependent oxygenase superfamily | 61.64 | 1.00 | 61.82 | 5.00E-05 | 1.85E-04 | 86.97 | 75.19 | 4.27E-18 | 5.00E-16 | AT3G12900.1 | IPR002283,IPR027443,IPR026992 | GO:0016706,GO:0005506,GO:0055114,GO:0016491 |
| 48h-Dn Reg | Peaxi162Scf72209g00001 | Unknown protein | 13.41 | 0.41 | 33.04 | 2.52E-02 | 4.73E-02 | 63.68 | 28.31 | 1.03E-07 | 2.09E-06 | |||
| 48h-Dn Reg | Peaxi162Scf00195g01222 | Unknown protein | 110.22 | 3.73 | 29.57 | 6.50E-03 | 1.47E-02 | 237.25 | 25.02 | 5.69E-07 | 9.79E-06 | |||
| 48h-Dn Reg | Peaxi162Scf01049g00041 | Unknown protein | 5.14 | 0.23 | 22.65 | 1.01E-02 | 2.14E-02 | 73.82 | 32.17 | 1.41E-08 | 3.47E-07 | |||
| 48h-Dn Reg | Peaxi162Scf00117g00052 | UDP-glycosyltransferase superfamily protein | 4.60 | 0.21 | 22.09 | 2.65E-03 | 6.71E-03 | 25.50 | 26.80 | 2.26E-07 | 4.21E-06 | AT4G15480.1 | IPR002213 | GO:0008152,GO:0016758 |
| 48h-Dn Reg | Peaxi162Scf00206g00311 | Expansin B3 | 17.99 | 0.89 | 20.20 | 5.00E-04 | 1.53E-03 | 17.81 | 38.66 | 5.05E-10 | 1.63E-08 | AT4G28250.1 | IPR007118 | GO:0019953,GO:0005576 |
| 48h-Dn Reg | Peaxi162Scf00665g00142 | Xyloglucan endotransglucosylase/hydrolase 26 | 14.12 | 0.81 | 17.39 | 1.05E-03 | 2.97E-03 | 25.99 | 11.02 | 9.03E-04 | 6.04E-03 | AT4G28850.1 | IPR016455,IPR008264,IPR008985 | GO:0048046,GO:0016762,GO:0004553,GO:0005618,GO:0006073,GO:0005975 |
| 48h-Dn Reg | Peaxi162Scf00025g00283 | Transducin/WD40 repeat-like superfamily protein | 4.57 | 0.26 | 17.30 | 2.65E-03 | 6.71E-03 | 25.81 | 44.36 | 2.73E-11 | 1.11E-09 | AT5G23730.1 | IPR015943 | GO:0005515 |
| 48h-Dn Reg | Peaxi162Scf00326g00712 | Peroxidase superfamily protein | 25.15 | 1.52 | 16.59 | 5.00E-05 | 1.85E-04 | 17.04 | 18.30 | 1.89E-05 | 2.20E-04 | AT1G30870.1 | IPR010255 | GO:0006979,GO:0020037,GO:0004601,GO:0055114 |
| Annotation | Leaf 6 h | Leaf 24 h | Root 6 h | Root 12 h | Root 24 h | Root 48 h | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cufflinks | edgeR | Cufflinks | edgeR | Cufflinks | edgeR | Cufflinks | edgeR | Cufflinks | edgeR | Cufflinks | edgeR | |||||||||||||
| FC | q-Value | FC | FDR | FC | q-Value | FC | FDR | FC | q-Value | FC | FDR | FC | q-Value | FC | FDR | FC | q-Value | FC | FDR | FC | q-Value | FC | FDR | |
| BEL1-like homeodomain 6 | 1.7 | 3.80E-03 | 1.6 | 3.31E-03 | 1.7 | 4.27E-03 | 1.6 | 7.24E-03 | 1.5 | 1.75E-02 | 1.8 | 3.07E-04 | 1.7 | 2.72E-03 | 1.8 | 3.07E-04 | 1.8 | 3.54E-04 | 1.8 | 2.08E-04 | 1.7 | 4.08E-03 | 2.0 | 9.05E-05 |
| Cold-regulated 413-plasma membrane 2 | 5.3 | 1.75E-04 | 5.4 | 2.83E-11 | 4.0 | 1.75E-04 | 3.6 | 6.01E-07 | 2.8 | 1.85E-04 | 4.2 | 3.05E-08 | 3.2 | 1.85E-04 | 4.2 | 3.05E-08 | 1.8 | 1.25E-03 | 2.4 | 4.87E-04 | 2.7 | 1.85E-04 | 3.2 | 2.29E-05 |
| Basic helix-loop-helix (bHLH) DNA-binding | 2.0 | 1.75E-04 | 1.8 | 1.86E-03 | 1.6 | 4.50E-03 | 1.6 | 1.02E-02 | 3.2 | 1.85E-04 | 1.8 | 1.21E-03 | 1.6 | 1.10E-02 | 1.8 | 1.21E-03 | 3.1 | 1.85E-04 | 1.8 | 7.63E-04 | 2.2 | 1.85E-04 | 2.8 | 8.79E-07 |
| Protein NRT1/PTR FAMILY 1.2 | 2.2 | 1.75E-04 | 2.2 | 2.22E-03 | 3.0 | 1.75E-04 | 3.3 | 3.24E-06 | 1.7 | 2.12E-02 | 2.6 | 4.27E-04 | 1.9 | 6.71E-03 | 2.6 | 4.27E-04 | 3.1 | 1.25E-03 | 4.7 | 4.12E-08 | 1.6 | 4.45E-02 | 2.0 | 2.30E-02 |
| Pleckstrin (PH) domain-containing protein | 3.9 | 1.75E-04 | 4.3 | 1.24E-12 | 30.9 | 1.75E-04 | 40.1 | 1.12E-56 | 2.4 | 1.85E-04 | 2.5 | 2.37E-05 | 1.7 | 1.94E-03 | 2.5 | 2.37E-05 | 3.1 | 1.85E-04 | 3.2 | 1.50E-08 | 2.3 | 1.85E-04 | 2.8 | 2.39E-06 |
| Unknown protein | 2.2 | 1.75E-04 | 2.1 | 4.16E-05 | 4.9 | 1.75E-04 | 4.9 | 1.16E-19 | 2.4 | 3.54E-04 | 2.3 | 5.40E-06 | 2.0 | 6.68E-04 | 2.3 | 5.40E-06 | 1.6 | 9.20E-03 | 2.0 | 4.75E-05 | 2.4 | 1.85E-04 | 2.5 | 2.85E-07 |
| NAD(P)-binding Rossmann-fold | 7.4 | 1.75E-04 | 7.0 | 5.98E-16 | 6.6 | 1.75E-04 | 7.0 | 1.44E-15 | 2.1 | 1.85E-04 | 3.1 | 5.66E-06 | 2.7 | 1.85E-04 | 3.1 | 5.66E-06 | 5.5 | 1.85E-04 | 5.7 | 7.46E-13 | 3.6 | 1.85E-04 | 3.7 | 3.95E-07 |
| Plant invertase/pectin methylesterase | 3.2 | 1.75E-04 | 2.9 | 3.51E-07 | 2.3 | 1.75E-04 | 2.6 | 6.70E-06 | 2.1 | 1.85E-04 | 1.7 | 1.41E-02 | 2.6 | 1.85E-04 | 1.7 | 1.41E-02 | 2.3 | 1.85E-04 | 1.8 | 7.46E-03 | 2.5 | 1.85E-04 | 3.3 | 1.11E-06 |
| Dehydration-responsive element-binding | 4.6 | 1.75E-04 | 4.7 | 1.89E-04 | 5.5 | 1.75E-04 | 6.1 | 1.24E-05 | 3.7 | 1.85E-04 | 9.6 | 8.42E-07 | 6.9 | 3.54E-04 | 9.6 | 8.42E-07 | 4.7 | 1.80E-03 | 8.5 | 1.73E-06 | 5.8 | 3.47E-03 | 7.4 | 1.90E-04 |
| Sec14p-like phosphatidylinositol transfer | 2.1 | 1.75E-04 | 2.0 | 1.79E-02 | 4.5 | 1.75E-04 | 4.6 | 1.46E-08 | 3.0 | 1.85E-04 | 2.3 | 1.16E-02 | 2.3 | 3.54E-04 | 2.3 | 1.16E-02 | 2.8 | 3.54E-04 | 4.1 | 9.30E-06 | 1.7 | 1.98E-02 | 2.3 | 2.03E-02 |
| Major facilitator superfamily protein | 2.0 | 1.75E-04 | 2.2 | 5.73E-04 | 2.4 | 1.75E-04 | 2.5 | 4.34E-05 | 2.3 | 1.85E-04 | 7.0 | 1.15E-16 | 4.7 | 1.85E-04 | 7.0 | 1.15E-16 | 3.9 | 1.85E-04 | 4.4 | 3.14E-10 | 6.3 | 1.85E-04 | 8.9 | 3.99E-15 |
| Trehalose phosphate synthase | 2.4 | 1.75E-04 | 2.5 | 1.62E-03 | 2.9 | 1.75E-04 | 2.6 | 9.50E-04 | 1.7 | 3.34E-03 | 3.4 | 1.70E-05 | 2.7 | 1.85E-04 | 3.4 | 1.70E-05 | 5.5 | 1.85E-04 | 5.4 | 9.51E-10 | 2.3 | 1.85E-04 | 2.7 | 7.49E-04 |
| Glycosyl hydrolase family protein | 3.4 | 1.75E-04 | 3.6 | 1.10E-07 | 4.1 | 1.75E-04 | 4.3 | 8.05E-10 | 4.1 | 1.85E-04 | 2.0 | 8.91E-03 | 2.7 | 1.85E-04 | 2.0 | 8.91E-03 | 2.4 | 1.85E-04 | 3.2 | 2.84E-06 | 2.4 | 1.85E-04 | 2.3 | 1.67E-03 |
| Heat stress transcription factor C-1 | 3.4 | 1.75E-04 | 3.4 | 3.91E-08 | 3.1 | 1.75E-04 | 3.0 | 1.54E-06 | 8.1 | 1.85E-04 | 3.6 | 2.75E-08 | 3.8 | 1.85E-04 | 3.6 | 2.75E-08 | 2.5 | 1.85E-04 | 2.7 | 1.67E-05 | 3.0 | 1.85E-04 | 3.2 | 5.09E-05 |
| Conserved hypothetical protein | 1.5 | 2.02E-02 | 1.5 | 4.44E-02 | 2.6 | 1.75E-04 | 2.4 | 8.21E-08 | 1.6 | 9.73E-03 | 3.2 | 3.42E-11 | 2.4 | 1.85E-04 | 3.2 | 3.42E-11 | 3.3 | 1.85E-04 | 3.5 | 2.86E-13 | 2.1 | 1.85E-04 | 2.7 | 1.75E-07 |
| Peroxidase superfamily protein | 2.4 | 1.75E-04 | 2.3 | 1.17E-02 | 2.2 | 1.75E-04 | 2.1 | 2.00E-02 | 2.5 | 1.85E-04 | 4.6 | 1.38E-06 | 4.2 | 1.85E-04 | 4.6 | 1.38E-06 | 6.4 | 1.85E-04 | 6.1 | 5.53E-09 | 5.7 | 1.85E-04 | 5.5 | 8.27E-08 |
| α/β-Hydrolases | 1.8 | 6.34E-04 | 1.8 | 6.61E-03 | 4.3 | 1.75E-04 | 4.3 | 1.17E-13 | 2.2 | 1.85E-04 | 2.4 | 5.69E-05 | 2.1 | 1.85E-04 | 2.4 | 5.69E-05 | 3.6 | 1.85E-04 | 3.9 | 1.38E-11 | 2.0 | 1.85E-04 | 2.1 | 7.83E-04 |
| Nucleobase-ascorbate transporter 12 | 0.6 | 4.61E-03 | 0.6 | 2.37E-02 | 0.7 | 1.91E-02 | 0.6 | 1.99E-02 | 0.5 | 6.18E-03 | 0.4 | 7.86E-05 | 0.4 | 1.85E-04 | 0.4 | 7.86E-05 | 0.3 | 1.85E-04 | 0.3 | 2.91E-08 | 0.4 | 1.85E-04 | 0.5 | 2.01E-03 |
| Glycine-rich RNA-binding protein | 0.3 | 1.75E-04 | 0.4 | 9.99E-05 | 0.6 | 2.61E-03 | 0.5 | 6.83E-03 | 0.2 | 1.85E-04 | 0.3 | 2.24E-09 | 0.2 | 1.85E-04 | 0.3 | 2.24E-09 | 0.3 | 1.85E-04 | 0.3 | 1.50E-06 | 0.4 | 1.85E-04 | 0.4 | 2.59E-05 |
| ACT domain repeat 4 | 0.6 | 2.11E-03 | 0.5 | 2.64E-03 | 0.1 | 1.75E-04 | 0.0 | 7.17E-47 | 0.3 | 1.85E-04 | 0.4 | 2.25E-05 | 0.4 | 1.85E-04 | 0.4 | 2.25E-05 | 0.6 | 1.53E-03 | 0.7 | 3.83E-02 | 0.2 | 1.85E-04 | 0.2 | 7.95E-18 |
| Homeodomain-like superfamily protein | 0.4 | 1.52E-02 | 0.3 | 5.53E-04 | 0.2 | 3.34E-04 | 0.3 | 2.22E-05 | 0.4 | 2.03E-02 | 0.2 | 1.21E-05 | 0.4 | 9.83E-03 | 0.2 | 1.21E-05 | 0.1 | 4.90E-03 | 0.2 | 1.87E-06 | 0.4 | 3.56E-02 | 0.4 | 2.24E-02 |
| Glucose-6-phosphate dehydrogenase 2 | 0.6 | 3.69E-03 | 0.6 | 8.22E-03 | 0.5 | 1.75E-04 | 0.5 | 1.82E-04 | 0.4 | 1.85E-04 | 0.4 | 5.31E-08 | 0.3 | 1.85E-04 | 0.4 | 5.31E-08 | 0.3 | 1.85E-04 | 0.4 | 1.94E-07 | 0.4 | 1.85E-04 | 0.4 | 1.74E-06 |
| ACT domain repeat 4 | 0.4 | 1.75E-04 | 0.4 | 1.03E-04 | 0.4 | 1.75E-04 | 0.4 | 8.36E-04 | 0.5 | 6.51E-03 | 0.3 | 6.15E-05 | 0.3 | 6.68E-04 | 0.3 | 6.15E-05 | 0.4 | 1.85E-04 | 0.4 | 2.38E-03 | 0.2 | 1.85E-04 | 0.2 | 5.76E-08 |
| Nodulin MtN21/EamA-like transporter | 0.5 | 1.22E-02 | 0.5 | 6.71E-03 | 0.2 | 1.75E-04 | 0.2 | 4.98E-13 | 0.5 | 4.43E-03 | 0.4 | 7.14E-05 | 0.3 | 1.85E-04 | 0.4 | 7.14E-05 | 0.3 | 1.85E-04 | 0.4 | 1.42E-05 | 0.4 | 1.85E-04 | 0.4 | 1.51E-04 |
| Protein COBRA | 0.3 | 1.75E-04 | 0.3 | 9.82E-08 | 0.2 | 1.75E-04 | 0.2 | 2.49E-14 | 0.3 | 8.77E-03 | 0.3 | 1.16E-04 | 0.3 | 2.84E-03 | 0.3 | 1.16E-04 | 0.3 | 1.85E-04 | 0.3 | 1.21E-05 | 0.4 | 1.53E-03 | 0.4 | 2.30E-03 |
| Ferredoxin-3, chloroplastic | 0.3 | 4.86E-04 | 0.3 | 3.74E-06 | 0.3 | 1.98E-03 | 0.4 | 1.88E-04 | 0.5 | 1.85E-04 | 0.3 | 1.15E-07 | 0.3 | 1.85E-04 | 0.3 | 1.15E-07 | 0.1 | 1.85E-04 | 0.2 | 3.99E-14 | 0.1 | 1.85E-04 | 0.1 | 2.68E-18 |
| Fatty acid desaturase 2 | 0.6 | 3.34E-04 | 0.6 | 1.44E-02 | 0.3 | 1.75E-04 | 0.3 | 1.32E-07 | 0.5 | 1.67E-03 | 0.5 | 1.15E-03 | 0.4 | 1.85E-04 | 0.5 | 1.15E-03 | 0.2 | 1.85E-04 | 0.3 | 2.39E-10 | 0.3 | 1.85E-04 | 0.3 | 7.93E-10 |
| Ferredoxin—NADP reductase, root isozyme | 0.1 | 1.75E-04 | 0.1 | 1.15E-33 | 0.5 | 1.75E-04 | 0.5 | 2.43E-03 | 0.5 | 1.85E-04 | 0.4 | 3.52E-06 | 0.4 | 1.85E-04 | 0.4 | 3.52E-06 | 0.3 | 1.85E-04 | 0.3 | 4.63E-10 | 0.4 | 1.85E-04 | 0.5 | 2.27E-03 |
| Xyloglucan endotransglucosylase/hydrolase | 0.1 | 1.75E-04 | 0.1 | 1.18E-07 | 0.2 | 1.75E-04 | 0.2 | 6.29E-05 | 0.1 | 1.85E-04 | 0.1 | 1.39E-14 | 0.0 | 1.85E-04 | 0.1 | 1.39E-14 | 0.0 | 1.85E-04 | 0.0 | 2.27E-17 | 0.1 | 1.85E-04 | 0.1 | 7.61E-11 |
| Fatty acid hydroxylase superfamily | 0.3 | 3.34E-04 | 0.2 | 2.22E-09 | 0.3 | 3.34E-04 | 0.3 | 1.58E-08 | 0.5 | 1.11E-02 | 0.3 | 1.03E-05 | 0.3 | 3.54E-04 | 0.3 | 1.03E-05 | 0.1 | 1.01E-02 | 0.1 | 1.23E-16 | 0.2 | 2.39E-02 | 0.2 | 1.33E-10 |
| 3-Oxo-5-α-steroid 4-dehydrogenase | 0.3 | 1.75E-04 | 0.2 | 3.37E-09 | 0.3 | 1.75E-04 | 0.3 | 3.63E-08 | 0.2 | 2.29E-02 | 0.1 | 3.96E-10 | 0.1 | 2.72E-03 | 0.1 | 3.96E-10 | 0.2 | 1.85E-04 | 0.1 | 1.20E-13 | 0.2 | 1.85E-04 | 0.2 | 3.52E-08 |
| Unknown protein | 0.5 | 3.34E-04 | 0.5 | 4.33E-03 | 0.3 | 1.75E-04 | 0.3 | 1.16E-10 | 0.5 | 6.68E-04 | 0.2 | 2.42E-12 | 0.2 | 1.85E-04 | 0.2 | 2.42E-12 | 0.2 | 1.85E-04 | 0.3 | 1.31E-08 | 0.1 | 1.85E-04 | 0.1 | 1.03E-22 |
| Phosphoenolpyruvate carboxylase 1 | 0.2 | 1.75E-04 | 0.2 | 2.31E-11 | 0.4 | 1.75E-04 | 0.4 | 6.10E-05 | 0.4 | 1.85E-04 | 0.3 | 5.72E-08 | 0.3 | 1.85E-04 | 0.3 | 5.72E-08 | 0.3 | 1.85E-04 | 0.3 | 2.16E-09 | 0.2 | 1.85E-04 | 0.2 | 2.81E-16 |
| Protein phosphatase 2C family protein | 0.3 | 1.75E-04 | 0.3 | 2.83E-09 | 0.3 | 1.75E-04 | 0.2 | 1.36E-12 | 0.2 | 1.85E-04 | 0.2 | 2.03E-10 | 0.2 | 1.85E-04 | 0.2 | 2.03E-10 | 0.2 | 1.85E-04 | 0.2 | 8.05E-15 | 0.3 | 1.85E-04 | 0.4 | 9.01E-05 |
| Protein NRT1/PTR FAMILY 6.1 | 0.4 | 1.75E-04 | 0.4 | 3.99E-04 | 0.3 | 1.75E-04 | 0.3 | 5.75E-05 | 0.2 | 1.94E-03 | 0.2 | 1.28E-06 | 0.2 | 4.20E-03 | 0.2 | 1.28E-06 | 0.1 | 2.46E-03 | 0.1 | 3.80E-10 | 0.2 | 4.20E-03 | 0.2 | 8.55E-07 |
| Transmembrane amino acid transporter | 0.5 | 6.34E-04 | 0.5 | 5.23E-03 | 0.6 | 1.63E-02 | 0.6 | 3.48E-02 | 0.6 | 2.67E-02 | 0.4 | 1.00E-04 | 0.3 | 1.85E-04 | 0.4 | 1.00E-04 | 0.1 | 1.85E-04 | 0.1 | 1.16E-18 | 0.3 | 1.85E-04 | 0.3 | 9.29E-06 |
| Tyrosine phosphatase family protein | 0.4 | 2.33E-02 | 0.2 | 2.37E-06 | 0.2 | 1.19E-03 | 0.2 | 1.27E-08 | 0.2 | 1.85E-04 | 0.5 | 1.22E-02 | 0.3 | 1.85E-04 | 0.5 | 1.22E-02 | 0.3 | 1.85E-04 | 0.5 | 6.36E-03 | 0.3 | 1.85E-04 | 0.4 | 1.29E-04 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villarino, G.H.; Hu, Q.; Scanlon, M.J.; Mueller, L.; Bombarely, A.; Mattson, N.S. Dissecting Tissue-Specific Transcriptomic Responses from Leaf and Roots under Salt Stress in Petunia hybrida Mitchell. Genes 2017, 8, 195. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8080195
Villarino GH, Hu Q, Scanlon MJ, Mueller L, Bombarely A, Mattson NS. Dissecting Tissue-Specific Transcriptomic Responses from Leaf and Roots under Salt Stress in Petunia hybrida Mitchell. Genes. 2017; 8(8):195. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8080195
Chicago/Turabian StyleVillarino, Gonzalo H., Qiwen Hu, Michael J. Scanlon, Lukas Mueller, Aureliano Bombarely, and Neil S. Mattson. 2017. "Dissecting Tissue-Specific Transcriptomic Responses from Leaf and Roots under Salt Stress in Petunia hybrida Mitchell" Genes 8, no. 8: 195. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8080195