Zebrafish in Translational Cancer Research: Insight into Leukemia, Melanoma, Glioma and Endocrine Tumor Biology



Abstract
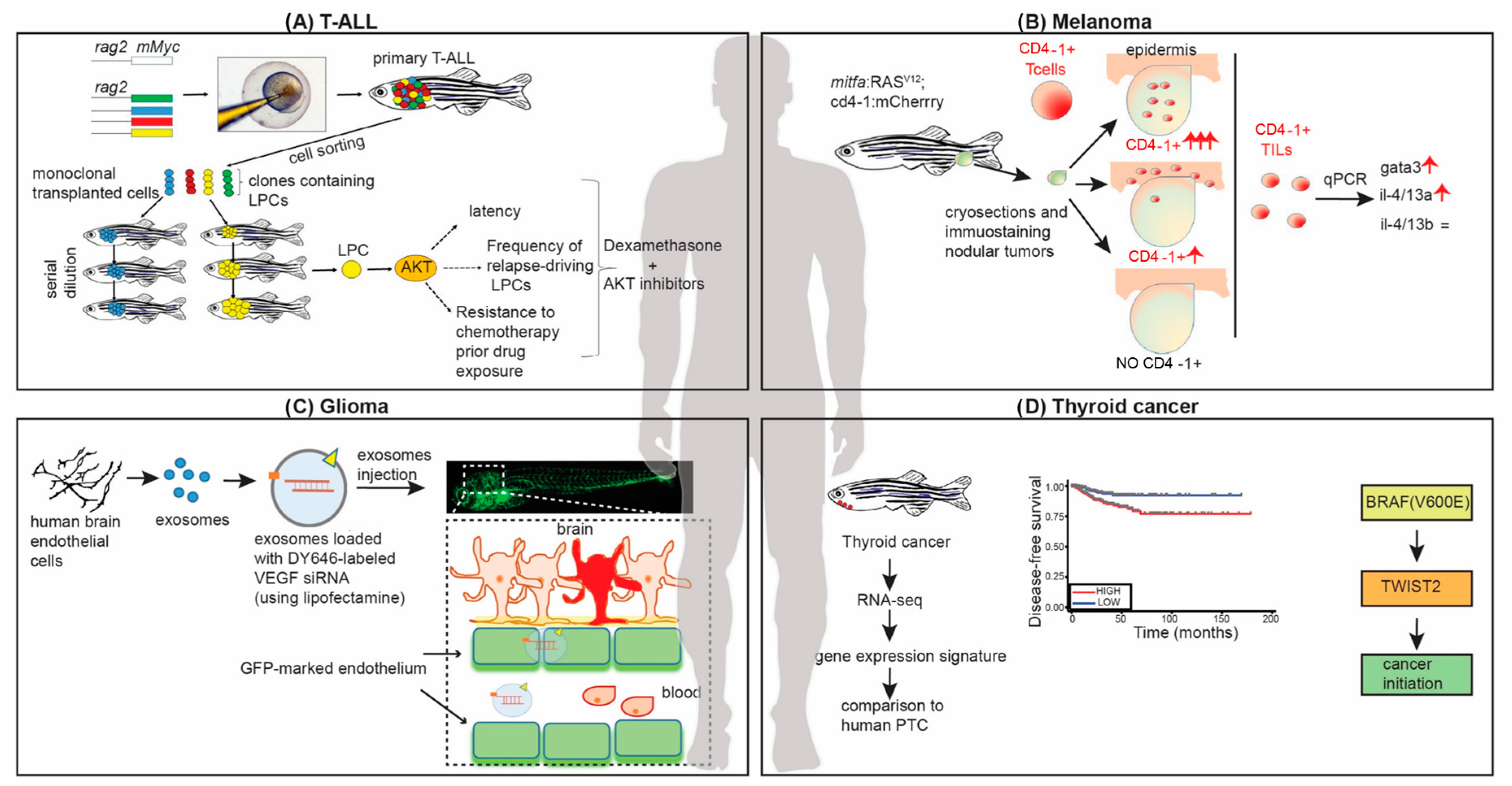
:1. Introduction
2. Zebrafish as a Tool to Study Human Leukemia
2.1. Zebrafish Hematopoietic Program
2.2. Human Leukemias
2.3. Zebrafish Transgenic Models of T-Cell Acute Lymphoblastic Leukemia (T-ALL)
2.4. T-ALL Transplantation in Zebrafish
2.5. Chemical Screen to Find New Pathways and Therapies in T-ALL
2.6. Acute Myeloid Leukemia (AML) and Myelo-Erythroid Proliferative Disorder
2.7. Chemical Screen in Zebrafish AML Models
2.8. Summary and Translational Impact of Zebrafish Models in Leukemia Research
3. Zebrafish as a Model for Studying Human Melanoma
3.1. Zebrafish Melanoma Models
3.1.1. BRAFV600E/p53
3.1.2. N-RASQ61K
3.1.3. H-RASG12V
3.2. Chemical Screen to Identify Anti Melanoma Drugs
3.3. Xenograft Models
3.4. Perspectives on the Use of Zebrafish Melanoma Models for Tumor Heterogeneity and Cancer Immunotherapy
4. Human Glioma and Relevant Target Pathways
4.1. Zebrafish Glioma Models
4.2. Human Glioma Transplantation in Zebrafish Embryos
4.3. Glioma Zebrafish Models and Therapeutic Insights
4.4. Translational Impact of Zebrafish Models in Glioma Research
5. Studying Endocrine Tumors Using Zebrafish
5.1. Pituitary Tumor
5.2. Papillary Thyroid Carcinoma
5.3. Neuroendocrine Tumors
5.4. Summary and Translational Impact of Zebrafish Models in Endocrine Tumors Research
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Huang, J.; Ye, J. A fresh look at zebrafish from the perspective of cancer research. J. Exp. Clin. Cancer Res. 2015, 34, 80. [Google Scholar] [CrossRef] [PubMed]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the big one: The potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. Dis. Model. Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.S.; Redfield, S.E.; Zon, L.I. Chemical screening in zebrafish for novel biological and therapeutic discovery. Methods Cell Biol. 2017, 138, 651–679. [Google Scholar] [PubMed]
- Zhu, S.; Thomas Look, A. Neuroblastoma and Its Zebrafish Model. Adv. Exp. Med. Biol. 2016, 916, 451–478. [Google Scholar] [PubMed]
- Hwang, K.L.; Goessling, W. Baiting for Cancer: Using the Zebrafish as a Model in Liver and Pancreatic Cancer. Adv. Exp. Med. Biol. 2016, 916, 391–410. [Google Scholar] [PubMed]
- Jing, L.; Zon, L.I. Zebrafish as a model for normal and malignant hematopoiesis. Dis. Model. Mech. 2011, 4, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kwan, W.; North, T.E. Netting Novel Regulators of Hematopoiesis and Hematologic Malignancies in Zebrafish. Curr. Top. Dev. Biol. 2017, 124, 125–160. [Google Scholar] [PubMed]
- Paik, E.J.; Zon, L.I. Hematopoietic development in the zebrafish. Int. J. Dev. Biol. 2010, 54, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Stachura, D.L.; Traver, D. Cellular dissection of zebrafish hematopoiesis. Methods Cell Biol. 2011, 101, 75–110. [Google Scholar] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.L.; Akkapeddi, P.; Alcobia, I.; Almeida, A.R.; Cardoso, B.A.; Fragoso, R.; Serafim, T.L.; Barata, J.T. From the outside, from within: Biological and therapeutic relevance of signal transduction in T-cell acute lymphoblastic leukemia. Cell. Signal. 2017, 38, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Progr. 2012, 2012, 389–396. [Google Scholar]
- Bhatnagar, B.; Garzon, R. The use of molecular genetics to refine prognosis in acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2014, 9, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Mullighan, C.G. Genomics in acute lymphoblastic leukaemia: Insights and treatment implications. Nat. Rev. Clin. Oncol. 2015, 12, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Qi, X.; Chen, J.; Chen, W.; Qiu, G.; Wu, Z.; Wu, N. CRISPR/Cas9 in zebrafish: An efficient combination for human genetic diseases modeling. Hum. Genet. 2017, 136, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R.; Ferrando, A.A. How I treat T-cell acute lymphoblastic leukemia in adults. Blood 2015, 126, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.J.; Weinberg, V.; Ellison, R.R.; Henderson, E.S.; Terebelo, H.; Rafla, S.; Cuttner, J.; Silver, R.T.; Carey, R.W.; Levy, R.N. Efficacy of daunorubicin in the therapy of adult acute lymphocytic leukemia: A prospective randomized trial by cancer and leukemia group B. Blood 1984, 64, 267–274. [Google Scholar] [PubMed]
- Scavino, H.F.; George, J.N.; Sears, D.A. Remission induction in adult acute lymphocytic leukemia. Use of vincristine and prednisone alone. Cancer 1976, 38, 672–677. [Google Scholar] [CrossRef]
- Langenau, D.M.; Traver, D.; Ferrando, A.A.; Kutok, J.L.; Aster, J.C.; Kanki, J.P.; Lin, S.; Prochownik, E.; Trede, N.S.; Zon, L.I.; et al. Myc-induced T cell leukemia in transgenic zebrafish. Science 2003, 299, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.-R.J.; Munson, K.M.; Elagib, K.E.; Goldfarb, A.N.; Sweetser, D.A.; Peterson, R.T. Discovering chemical modifiers of oncogene-regulated hematopoietic differentiation. Nat. Chem. Biol. 2009, 5, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Langenau, D.M.; Feng, H.; Berghmans, S.; Kanki, J.P.; Kutok, J.L.; Look, A.T. Cre/lox-regulated transgenic zebrafish model with conditional myc-induced T cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2005, 102, 6068–6073. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Langenau, D.M.; Madge, J.A.; Quinkertz, A.; Gutierrez, A.; Neuberg, D.S.; Kanki, J.P.; Look, A.T. Heat-shock induction of T-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br. J. Haematol. 2007, 138, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Grebliunaite, R.; Feng, H.; Kozakewich, E.; Zhu, S.; Guo, F.; Payne, E.; Mansour, M.; Dahlberg, S.E.; Neuberg, D.S.; et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Suresh, S.; Irvine, A.E. The NOTCH signaling pathway in normal and malignant blood cell production. J. Cell Commun. Signal. 2015, 9, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jette, C.; Kanki, J.P.; Aster, J.C.; Look, A.T.; Griffin, J.D. NOTCH1-induced T-cell leukemia in transgenic zebrafish. Leukemia 2007, 21, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.H.; Raimondi, A.R.; Salthouse, C.D.; Ignatius, M.S.; Blackburn, J.S.; Mizgirev, I.V.; Storer, N.Y.; de Jong, J.L.O.; Chen, A.T.; Zhou, Y.; et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood 2010, 115, 3296–3303. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, J.S.; Liu, S.; Raiser, D.M.; Martinez, S.A.; Feng, H.; Meeker, N.D.; Gentry, J.; Neuberg, D.; Look, A.T.; Ramaswamy, S.; Bernards, A.; et al. Notch signaling expands a pre-malignant pool of T-cell acute lymphoblastic leukemia clones without affecting leukemia-propagating cell frequency. Leukemia 2012, 26, 2069–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, J.S.; Liu, S.; Wilder, J.L.; Dobrinski, K.P.; Lobbardi, R.; Moore, F.E.; Martinez, S.A.; Chen, E.Y.; Lee, C.; Langenau, D.M. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell 2014, 25, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Abdelfattah, N.S.; Blackburn, J.S.; Moore, J.C.; Martinez, S.A.; Moore, F.E.; Lobbardi, R.; Tenente, I.M.; Ignatius, M.S.; et al. Optimized cell transplantation using adult rag2 mutant zebrafish. Nat. Methods 2014, 11, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Elnour, I.B.; Ahmed, S.; Halim, K.; Nirmala, V. Omenn’s Syndrome: A rare primary immunodeficiency disorder. Sultan Qaboos Univ. Med. J. 2007, 7, 133–138. [Google Scholar] [PubMed]
- Moore, J.C.; Tang, Q.; Yordán, N.T.; Moore, F.E.; Garcia, E.G.; Lobbardi, R.; Ramakrishnan, A.; Marvin, D.L.; Anselmo, A.; Sadreyev, R.I.; et al. Single-cell imaging of normal and malignant cell engraftment into optically clear prkdc-null SCID zebrafish. J. Exp. Med. 2016, 213, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- North, T.E.; Goessling, W.; Walkley, C.R.; Lengerke, C.; Kopani, K.R.; Lord, A.M.; Weber, G.J.; Bowman, T.V.; Jang, I.-H.; Grosser, T.; et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007, 447, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; Allen, R.S.; Guan, X.; Jin, P.; Uchida, N.; Dovey, M.; Harris, J.M.; Metzger, M.E.; Bonifacino, A.C.; Stroncek, D.; et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell 2011, 8, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Cutler, C.; Multani, P.; Robbins, D.; Kim, H.T.; Le, T.; Hoggatt, J.; Pelus, L.M.; Desponts, C.; Chen, Y.-B.; Rezner, B.; et al. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood 2013, 122, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Ridges, S.; Heaton, W.L.; Joshi, D.; Choi, H.; Eiring, A.; Batchelor, L.; Choudhry, P.; Manos, E.J.; Sofla, H.; Sanati, A.; et al. Zebrafish screen identifies novel compound with selective toxicity against leukemia. Blood 2012, 119, 5621–5631. [Google Scholar] [CrossRef] [PubMed]
- Langenau, D.M.; Ferrando, A.A.; Traver, D.; Kutok, J.L.; Hezel, J.-P.D.; Kanki, J.P.; Zon, L.I.; Look, A.T.; Trede, N.S. In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc. Natl. Acad. Sci. USA 2004, 101, 7369–7374. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Pan, L.; Groen, R.W.J.; Baleydier, F.; Kentsis, A.; Marineau, J.; Grebliunaite, R.; Kozakewich, E.; Reed, C.; Pflumio, F.; et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Investig. 2014, 124, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Pruvot, B.; Jacquel, A.; Droin, N.; Auberger, P.; Bouscary, D.; Tamburini, J.; Muller, M.; Fontenay, M.; Chluba, J.; Solary, E. Leukemic cell xenograft in zebrafish embryo for investigating drug efficacy. Haematologica 2011, 96, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: Biology and clinical significance. Br. J. Haematol. 1999, 106, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Troke, P.J.F.; Kindle, K.B.; Collins, H.M.; Heery, D.M. MOZ fusion proteins in acute myeloid leukaemia. Biochem. Soc. Symp. 2006, 23–39. [Google Scholar] [CrossRef]
- Borrow, J.; Shearman, A.M.; Stanton, V.P., Jr.; Becher, R.; Collins, T.; Williams, A.J.; Dubé, I.; Katz, F.; Kwong, Y.L.; Morris, C.; et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat. Genet. 1996, 12, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.G.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [PubMed]
- Dombret, H. Gene mutation and AML pathogenesis. Blood 2011, 118, 5366–5367. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, J.; Paggetti, J.; Martin, L.; Hammann, A.; Solary, E.; Bastie, J.-N.; Delva, L. MOZ/TIF2-induced acute myeloid leukaemia in transgenic fish. Br. J. Haematol. 2008, 143, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.-R.J.; Munson, K.M.; Chao, Y.L.; Peterson, Q.P.; Macrae, C.A.; Peterson, R.T. AML1-ETO reprograms hematopoietic cell fate by downregulating scl expression. Development 2008, 135, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Forrester, A.M.; Grabher, C.; McBride, E.R.; Boyd, E.R.; Vigerstad, M.H.; Edgar, A.; Kai, F.-B.; Da’as, S.I.; Payne, E.; Look, A.T.; et al. NUP98-HOXA9-transgenic zebrafish develop a myeloproliferative neoplasm and provide new insight into mechanisms of myeloid leukaemogenesis. Br. J. Haematol. 2011, 155, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Alghisi, E.; Distel, M.; Malagola, M.; Anelli, V.; Santoriello, C.; Herwig, L.; Krudewig, A.; Henkel, C.V.; Russo, D.; Mione, M.C. Targeting oncogene expression to endothelial cells induces proliferation of the myelo-erythroid lineage by repressing the Notch pathway. Leukemia 2013, 27, 2229–2241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, J.; Wheat, J.; Chen, X.; Jin, S.; Sadrzadeh, H.; Fathi, A.T.; Peterson, R.T.; Kung, A.L.; Sweetser, D.A.; et al. AML1-ETO mediates hematopoietic self-renewal and leukemogenesis through a COX/β-catenin signaling pathway. Blood 2013, 121, 4906–4916. [Google Scholar] [CrossRef] [PubMed]
- Dee, C.T.; Nagaraju, R.T.; Athanasiadis, E.I.; Gray, C.; Fernandez Del Ama, L.; Johnston, S.A.; Secombes, C.J.; Cvejic, A.; Hurlstone, A.F.L. CD4-Transgenic Zebrafish Reveal Tissue-Resident Th2- and Regulatory T Cell-like Populations and Diverse Mononuclear Phagocytes. J. Immunol. 2016, 197, 3520–3530. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Fogarty, B.; LaForge, B.; Aziz, S.; Pham, T.; Lai, L.; Bai, S. Delivery of small interfering RNA to inhibit vascular endothelial growth factor in zebrafish using natural brain endothelia cell-secreted exosome nanovesicles for the treatment of brain cancer. AAPS J. 2017, 19, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Villefranc, J.A.; Chhangawala, S.; Martinez-McFaline, R.; Riva, E.; Nguyen, A.; Verma, A.; Bareja, R.; Chen, Z.; Scognamiglio, T.; et al. Oncogenic BRAF disrupts thyroid morphogenesis and function via twist expression. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Schartl, M.; Larue, L.; Goda, M.; Bosenberg, M.W.; Hashimoto, H.; Kelsh, R.N. What is a vertebrate pigment cell? Pigment Cell Melanoma Res. 2016, 29, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadendorf, D.; Hauschild, A. Melanoma in 2013: Melanoma—The run of success continues. Nat. Rev. Clin. Oncol. 2014, 11, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Omholt, K.; Platz, A.; Kanter, L.; Ringborg, U.; Hansson, J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin. Cancer Res. 2003, 9, 6483–6488. [Google Scholar] [PubMed]
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. 2014, 9, 239–271. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.M.; et al. BRAF Mutations Are Sufficient to Promote Nevi Formation and Cooperate with p53 in the Genesis of Melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.M.; Baker, A.C.; Beroukhim, R.; Winckler, W.; Feng, W.; Marmion, J.M.; Laine, E.; Greulich, H.; Tseng, H.; Gates, C.; et al. Modeling genomic diversity and tumor dependency in malignant melanoma. Cancer Res. 2008, 68, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Macgregor, S.; Montgomery, G.W.; Liu, J.Z.; Zhao, Z.Z.; Henders, A.K.; Stark, M.; Schmid, H.; Holland, E.A.; Duffy, D.L.; Zhang, M.; et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat. Genet. 2011, 43, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Dovey, M.; White, R.M.; Zon, L.I. Oncogenic NRAS cooperates with p53 loss to generate melanoma in zebrafish. Zebrafish 2009, 6, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, C.; Jones, M.; Walker, P.; Kamarashev, J.; Kelly, A.; Hurlstone, A.F.L. Dissecting the roles of Raf- and PI3K-signalling pathways in melanoma formation and progression in a zebrafish model. Dis. Model. Mech. 2009, 2, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoriello, C.; Gennaro, E.; Anelli, V.; Distel, M.; Kelly, A.; Köster, R.W.; Hurlstone, A.; Mione, M. Kita driven expression of oncogenic HRAS leads to early onset and highly penetrant melanoma in zebrafish. PLoS ONE 2010, 5, e15170. [Google Scholar] [CrossRef] [PubMed]
- Colanesi, S.; Taylor, K.L.; Temperley, N.D.; Lundegaard, P.R.; Liu, D.; North, T.E.; Ishizaki, H.; Kelsh, R.N.; Patton, E.E. Small molecule screening identifies targetable zebrafish pigmentation pathways. Pigment Cell Melanoma Res. 2012, 25, 131–143. [Google Scholar] [CrossRef] [PubMed]
- White, R.M.; Cech, J.; Ratanasirintrawoot, S.; Lin, C.Y.; Rahl, P.B.; Burke, C.J.; Langdon, E.; Tomlinson, M.L.; Mosher, J.; Kaufman, C.; et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 2011, 471, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, C.A.; Rennekamp, A.J.; Peterson, R.T. Chemical Screening in Zebrafish. Methods Mol. Biol. 2016, 1451, 3–16. [Google Scholar] [PubMed]
- McLean, J.E.; Neidhardt, E.A.; Grossman, T.H.; Hedstrom, L. Multiple inhibitor analysis of the brequinar and leflunomide binding sites on human dihydroorotate dehydrogenase. Biochemistry 2001, 40, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, E.; Fazio, M.; Zon, L.I. From fish bowl to bedside: The power of zebrafish to unravel melanoma pathogenesis and discover new therapeutics. Pigment Cell Melanoma Res. 2017, 30, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Del Ama, L.; Jones, M.; Walker, P.; Chapman, A.; Braun, J.A.; Mohr, J.; Hurlstone, A.F.L. Reprofiling using a zebrafish melanoma model reveals drugs cooperating with targeted therapeutics. Oncotarget 2016, 7, 40348–40361. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.M.J.; Seftor, E.A.; Bonde, G.; Cornell, R.A.; Hendrix, M.J.C. The fate of human malignant melanoma cells transplanted into zebrafish embryos: Assessment of migration and cell division in the absence of tumor formation. Dev. Dyn. 2005, 233, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, S.; Presta, M. The zebrafish/tumor xenograft angiogenesis assay. Nat. Protoc. 2007, 2, 2918–2923. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; White, R.M.; Zon, L.I. Transplantation in zebrafish. Methods Cell Biol. 2011, 105, 403–417. [Google Scholar] [PubMed]
- Topczewska, J.M.; Postovit, L.-M.; Margaryan, N.V.; Sam, A.; Hess, A.R.; Wheaton, W.W.; Nickoloff, B.J.; Topczewski, J.; Hendrix, M.J.C. Embryonic and tumorigenic pathways converge via Nodal signaling: Role in melanoma aggressiveness. Nat. Med. 2006, 12, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Haldi, M.; Ton, C.; Seng, W.L.; McGrath, P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis 2006, 9, 139–151. [Google Scholar] [CrossRef] [PubMed]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Van Der Ent, W.; Burrello, C.; Teunisse, A.F.A.S.; Ksander, B.R.; Van Der Velden, P.A.; Jager, M.J.; Jochemsen, A.G.; Snaar-Jagalska, B.E. Modeling of human uveal melanoma in zebrafish xenograft embryos. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6612–6622. [Google Scholar] [CrossRef] [PubMed]
- Kansler, E.R.; Verma, A.; Langdon, E.M.; Simon-Vermot, T.; Yin, A.; Lee, W.; Attiyeh, M.; Elemento, O.; White, R.M. Melanoma genome evolution across species. BMC Genom. 2017, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, S.; Ratnakumar, K.; Langdon, E.M.; Kansler, E.R.; Kim, I.S.; Campbell, N.R.; Perry, E.B.; McMahon, A.J.; Kaufman, C.K.; van Rooijen, E.; et al. A Quantitative System for Studying Metastasis Using Transparent Zebrafish. Cancer Res. 2015, 75, 4272–4282. [Google Scholar] [CrossRef] [PubMed]
- Niezgoda, A.; Niezgoda, P.; Czajkowski, R. Novel Approaches to Treatment of Advanced Melanoma: A Review on Targeted Therapy and Immunotherapy. Biomed Res. Int. 2015, 2015, 851387. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Minichillo, S.; Brandes, A.A. Pharmacotherapy of Glioblastoma: Established Treatments and Emerging Concepts. CNS Drugs 2017, 31, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stavrovskaya, A.A.; Shushanov, S.S.; Rybalkina, E.Y. Problems of Glioblastoma Multiforme Drug Resistance. Biochemistry 2016, 81, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W. Moving the needle: Optimizing classification for glioma. Sci. Transl. Med. 2016, 8, 350fs14. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. TCGA Research Network The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Vengoechea, J.; Zheng, S.; Sloan, A.E.; Chen, Y.; Brat, D.J.; O’Neill, B.P.; de Groot, J.; Yust-Katz, S.; Yung, W.-K.A.; et al. Molecular subtypes of glioblastoma are relevant to lower grade glioma. PLoS ONE 2014, 9, e91216. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.-Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Speidel, D. The role of DNA damage responses in p53 biology. Arch. Toxicol. 2015, 89, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.M.; Mehdipour, P. Exploration of Involved Key Genes and Signaling Diversity in Brain Tumors. Cell. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Lamfers, M.L.M.; Dirven, C.M.F.; Leenstra, S. Genetic biomarkers of drug response for small-molecule therapeutics targeting the RTK/Ras/PI3K, p53 or Rb pathway in glioblastoma. CNS Oncol. 2016, 5, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, J.-S.; Jean, W.; Waldman, T. Conspirators in a capital crime: Co-deletion of p18INK4c and p16INK4a/p14ARF/p15INK4b in glioblastoma multiforme. Cancer Res. 2008, 68, 8657–8660. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Padmanabhan, A.; de Groh, E.D.; Lee, J.-S.; Haidar, S.; Dahlberg, S.; Guo, F.; He, S.; Wolman, M.A.; Granato, M.; et al. Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis. Model. Mech. 2012, 5, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.H.; Leem, G.L.; Jung, D.E.; Kim, M.H.; Kim, E.Y.; Kim, S.H.; Park, H.-C.; Park, S.W. Glioma is formed by active Akt1 alone and promoted by active Rac1 in transgenic zebrafish. Neuro Oncol. 2013, 15, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Chen, W.; Spitsbergen, J.M.; Lu, J.; Vogel, P.; Peters, J.L.; Wang, Y.-D.; Orr, B.A.; Wu, J.; Henson, H.E.; et al. Activation of Sonic hedgehog signaling in neural progenitor cells promotes glioma development in the zebrafish optic pathway. Oncogenesis 2014, 3, e96. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Chen, W.; Orr, B.A.; Spitsbergen, J.M.; Jia, S.; Eden, C.J.; Henson, H.E.; Taylor, M.R. Oncogenic KRAS promotes malignant brain tumors in zebrafish. Mol. Cancer 2015, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, M.; Gourain, V.; Reischl, M.; Affaticati, P.; Jenett, A.; Joly, J.-S.; Benelli, M.; Demichelis, F.; Poliani, P.L.; Sieger, D.; et al. A novel brain tumour model in zebrafish reveals the role of YAP activation in MAPK- and PI3K-induced malignant growth. Dis. Model. Mech. 2017, 10, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Mione, M.C.; Trede, N.S. The zebrafish as a model for cancer. Dis. Model. Mech. 2010, 3, 517–523. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Rose, K.; Zon, L. Zebrafish cancer: The state of the art and the path forward. Nat. Rev. Cancer 2013, 13, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Diekmann, H.; Goldsmith, P. Functional characterisation of the maturation of the blood-brain barrier in larval zebrafish. PLoS ONE 2013, 8, e77548. [Google Scholar] [CrossRef] [PubMed]
- Eden, C.J.; Ju, B.; Murugesan, M.; Phoenix, T.N.; Nimmervoll, B.; Tong, Y.; Ellison, D.W.; Finkelstein, D.; Wright, K.; Boulos, N.; et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene 2015, 34, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Tobia, C.; Gariano, G.; De Sena, G.; Presta, M. Zebrafish embryo as a tool to study tumor/endothelial cell cross-talk. Biochim. Biophys. Acta 2013, 1832, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Lally, B.E.; Geiger, G.A.; Kridel, S.; Arcury-Quandt, A.E.; Robbins, M.E.; Kock, N.D.; Wheeler, K.; Peddi, P.; Georgakilas, A.; Kao, G.D.; et al. Identification and biological evaluation of a novel and potent small molecule radiation sensitizer via an unbiased screen of a chemical library. Cancer Res. 2007, 67, 8791–8799. [Google Scholar] [CrossRef] [PubMed]
- Geiger, G.A.; Fu, W.; Kao, G.D. Temozolomide-mediated radiosensitization of human glioma cells in a zebrafish embryonic system. Cancer Res. 2008, 68, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Cui, W.; Gu, A.; Xu, C.; Yu, S.-C.; Li, T.-T.; Cui, Y.-H.; Zhang, X.; Bian, X.-W. A novel zebrafish xenotransplantation model for study of glioma stem cell invasion. PLoS ONE 2013, 8, e61801. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; La Du, J.; Tanguay, R.L.; Greenwood, J.A. Calpain 2 is required for the invasion of glioblastoma cells in the zebrafish brain microenvironment. J. Neurosci. Res. 2012, 90, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Persano, L.; Pistollato, F.; Moro, E.; Frasson, C.; Porazzi, P.; Della Puppa, A.; Bresolin, S.; Battilana, G.; Indraccolo, S.; et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013, 4, e500. [Google Scholar] [CrossRef] [PubMed]
- Welker, A.M.; Jaros, B.D.; Puduvalli, V.K.; Imitola, J.; Kaur, B.; Beattie, C.E. Correction: Standardized orthotopic xenografts in zebrafish reveal glioma cell-line-specific characteristics and tumor cell heterogeneity. Dis. Model. Mech. 2016, 9, 1063–1065. [Google Scholar] [CrossRef] [PubMed]
- Welker, A.M.; Jaros, B.D.; An, M.; Beattie, C.E. Changes in tumor cell heterogeneity after chemotherapy treatment in a xenograft model of glioblastoma. Neuroscience 2017, 356, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, L.; Astell, K.R.; Velikova, G.; Sieger, D. A Zebrafish live imaging model reveals differential responses of microglia toward glioblastoma cells in vivo. Zebrafish 2016, 13, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.; Larsson, S.; Danielsson, A.; Elbæk, K.J.; Kettunen, P.; Tisell, M.; Sabel, M.; Lannering, B.; Nordborg, C.; Schepke, E.; et al. Stem cell cultures derived from pediatric brain tumors accurately model the originating tumors. Oncotarget 2017, 8, 18626–18639. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Lah Turnšek, T. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 2017, 8, 25482–25499. [Google Scholar] [CrossRef] [PubMed]
- Vittori, M.; Breznik, B.; Gredar, T.; Hrovat, K.; Bizjak Mali, L.; Lah, T.T. Imaging of human glioblastoma cells and their interactions with mesenchymal stem cells in the zebrafish (Danio rerio) embryonic brain. Radiol. Oncol. 2016, 50, 159–167. [Google Scholar] [PubMed]
- Lawson, N.D.; Weinstein, B.M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cui, W.; Yu, S.; Xu, C.; Chen, G.; Gu, A.; Li, T.; Cui, Y.; Zhang, X.; Bian, X. A synthetic dl-nordihydroguaiaretic acid (Nordy), inhibits angiogenesis, invasion and proliferation of glioma stem cells within a zebrafish xenotransplantation model. PLoS ONE 2014, 9, e85759. [Google Scholar] [CrossRef] [PubMed]
- Hernan, R.; Fasheh, R.; Calabrese, C.; Frank, A.J.; Maclean, K.H.; Allard, D.; Barraclough, R.; Gilbertson, R.J. ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res. 2003, 63, 140–148. [Google Scholar] [PubMed]
- Modzelewska, K.; Boer, E.F.; Mosbruger, T.L.; Picard, D.; Anderson, D.; Miles, R.R.; Kroll, M.; Oslund, W.; Pysher, T.J.; Schiffman, J.D.; et al. MEK inhibitors reverse growth of embryonal brain tumors derived from oligoneural precursor cells. Cell Rep. 2016, 17, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Lister, J.A.; Robertson, C.P.; Lepage, T.; Johnson, S.L.; Raible, D.W. nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development 1999, 126, 3757–3767. [Google Scholar] [PubMed]
- Schwartzbaum, J.A.; Fisher, J.L.; Aldape, K.D.; Wrensch, M. Epidemiology and molecular pathology of glioma. Nat. Clin. Pract. Neurol. 2006, 2, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Löhr, H.; Hammerschmidt, M. Zebrafish in endocrine systems: Recent advances and implications for human disease. Annu. Rev. Physiol. 2011, 73, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-A.; Jiang, H.; Ben-Shlomo, A.; Wawrowsky, K.; Fan, X.-M.; Lin, S.; Melmed, S. Targeting zebrafish and murine pituitary corticotroph tumors with a cyclin-dependent kinase (CDK) inhibitor. Proc. Natl. Acad. Sci. USA 2011, 108, 8414–8419. [Google Scholar] [CrossRef] [PubMed]
- Cirello, V.; Gaudenzi, G.; Grassi, E.S.; Colombo, C.; Vicentini, L.; Ferrero, S.; Persani, L.; Vitale, G.; Fugazzola, L. Tumor and normal thyroid spheroids: From tissues to zebrafish. Minerva Endocrinol. 2017. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-derived xenograft in zebrafish embryos: A new platform for translational research in neuroendocrine tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Tumor Type | Fish Line | Compounds Screened | Read out | Hits | Major Findings | Clinical Trials | References |
|---|---|---|---|---|---|---|---|
| Leukemia | wild type AB strain | 2480 | in situ hybridization for runx1 and cmyb | PGE2 | PGE2 is a regulator of HSCs number | Phase I | [38] |
| Tg(lck:lck-EGFP) | 26400 | T-cells (GFP+) | Lenaldekar (LDK) | LDK delays mitosis and inhibits PI3K/AKT/mTOR pathway | [41] | ||
| Tg(rag2:EGFP-mMyc) | 4880 | T-cells (GFP+) | Perphenazine (PPZ) | PPZ activates the tumor suppressor protein phosphatase 2A (PP2A) | [43] | ||
| Melanoma | AB and nacre mutant | 1280 | changes in pigment cell phenotype | More than 50 | More than 50 compound affect pigmentation, migration and differentiation | [74] | |
| Tg(mitfa:BRAFV600E); p53zdf1/zdf1 | 2000 | in situ hybridization for crestin | NSC210627 and Leflunomide | Crestin is aberrantly expressed in tumor cells, leflunomide and NSC210627 suppress crestin expression | Phase I/II | [75] | |
| Tg(mitfa:HRASV12; mitfa:GFP) | 640 | melanin quantification through absorbance reading at 340 nm | Rapamycin, disulfiram, tanshinone | Rapamycin, disulfiram and tanshinone cooperate with MEKi to suppress growth of melanocytes | [79] | ||
| Endocrine tumors | Tg:Pomc-Pttg;Pomc-eGFP | CDK inhibitors | pituitary cells (GFP+) | Roscovitine | Roscovitine reverse corticotrophs expansion in embryos | Phase II | [139] |
| Tumor Type | Transplanted Cells | Recipient Fish | Drug Treatments | Major Findings | References |
|---|---|---|---|---|---|
| Leukemia | zf T-ALL (GFP+) | wild type AB strain irradiated | - | Leukemic cells infiltrate region adjacent to the olfactory bulb | [24] |
| zf T-ALL (GFP+) | CG1 strain | - | T-ALLs can be initiated from a single cell; T-ALLs exhibit wide differences in tumor-iniating potential | [32] | |
| zf T-ALL (GFP+) | CG1 strain | - | Notch signaling expands a population of pre-malignant thymocytes in T-ALL | [33] | |
| zf T-ALL (several fluorescent clonal markers) | CG1 strain | MK2206+ dexamethasone | AKT pathway increases the number of LPC; MK2206+dexamethasone kill T-ALL cells | [34] | |
| zf T-ALL (several fluorescent clonal markers) | prkdc mutant | - | Clonal dominance emerges as a consequence of neutral stochastic drifts | [37] | |
| hu leukemic cell lines (K562, K562-R, Jurkat, NB4) | wild type AB strain | anti-leukemic drugs | Imatinib and Oxaphorines decrease the leukemic burden in xenografted animals | [44] | |
| Melanoma | hu metastatic melanoma cells (GFP+) | wild type AB strain | - | Metastatic melanoma cell lines are able to retain their dedifferentiated state | [80] |
| hu metastatic melanoma cells (C8161) (GFP+) | wild type AB strain | - | Nodal expression is involved in the regulation of melanoma progression | [83] | |
| hu metastatic melanoma cells (WM-266-4) (CM-DiI+) | wild type AB strain | - | Transplanted cells proliferate, migrate and stimulate angiogenesis | [84] | |
| zf melanoma cells | casper irradiated fish | - | Tranplanted cells can be tracked in adult transparent casper fish | [85] | |
| hu primary and metastatic uveal melanoma cell lines | Tg(fli1a:EGFP) | Quinostat, MLN-4924, dasatinib | Quinostat and MLN-4924 decrease migration and proliferation of primary and metastic UM cells | [86] | |
| Glioma | mo primary brain tumor cells (RFP+) | Tg(fli1a:EGFP) | 5-FU, Erlotinib | 5-FU and Erlotinib combination is able to inhibit tumor proliferation | [119] |
| hu U251 (RFP+) | Tg(fli1a:EGFP) | NS-123 | NS-123 synergies the effects of ionizing radiation on inhibition of tumor growth | [121] | |
| hu U251 (RFP+) | Tg(fli1a:EGFP) | Temozolomide | Experiments done to test the efficacy of combined temozolomide and radiation | [122] | |
| hu CD133+ U87 (RFP+) | Tg(fli1a:EGFP) | AG-L-66085 | U87 GSCs have enhanced expression MMP-9, which regulates glioma invasion | [123] | |
| hu U87 | Tg(fli1a:EGFP) | - | Calpain 2 expression is required for glioblastoma cell invasion | [124] | |
| hu primary GBM cells | Tg(7xTCF-Xla.Siam:GFP) | - | WNT pathway activation promotes neuronal differentiation | [125] | |
| hu GBM9, hu X12 (GFP+) | wild type ABLF | Temozolomide, bortezomib | Standardization of orthotopic xenograft methods; chemotherapy changes tumor cell heteregeneity | [126,127] | |
| hu U251, U87 (mCherry+) | Tg(mpeg1:EGFP);irf8-/- | BLZ945 | Microglia promotes human glioblastoma cell growth | [128] | |
| hu primary pediatric brain tumor cells (RFP+) | wild type AB strain | - | Stem cells derived from pediatric brain tumor are tumorigenic in zebrafish | [129] | |
| hu MSC (DiO), hu U373 (eEGFP), hu U87 (dsRed+) | wild type AB strain | - | MSCs contribuite to GBM tumoral heterogeneity and invasion | [130] | |
| hu GSCs U87 (RFP+) | Tg(fli1a:EGFP) | Nordy, Axitinib, Suntinib, Vatalani | Nordy suppresses the proliferation of GSCs | [133] | |
| hu U87MG (Cell Brite® DiD) | Tg(fli1a:EGFP) | - | anti-VEGF siRNAs cross the BBB and inhibit VEGF expression in a xenotransplanted brain tumor | [56] | |
| zf primary brain tumor cells | nacre, p53zdf1/zdf1 | AZD6244, U0126 | MEK inhibitors decrease brain tumor growth | [135] | |
| Endocrine tumors | hu PTC spheroids | Tg(fli1a:EGFP) | - | Spheroids are able to activate neoangiogenesis | [140] |
| hu neuroendocrine tumors | Tg(fli1a:EGFP) | - | Neuroendocrine tumors are pro-angiogenic and invasive | [141] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idilli, A.I.; Precazzini, F.; Mione, M.C.; Anelli, V. Zebrafish in Translational Cancer Research: Insight into Leukemia, Melanoma, Glioma and Endocrine Tumor Biology. Genes 2017, 8, 236. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8090236
Idilli AI, Precazzini F, Mione MC, Anelli V. Zebrafish in Translational Cancer Research: Insight into Leukemia, Melanoma, Glioma and Endocrine Tumor Biology. Genes. 2017; 8(9):236. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8090236
Chicago/Turabian StyleIdilli, Aurora Irene, Francesca Precazzini, Maria Caterina Mione, and Viviana Anelli. 2017. "Zebrafish in Translational Cancer Research: Insight into Leukemia, Melanoma, Glioma and Endocrine Tumor Biology" Genes 8, no. 9: 236. https://0-doi-org.brum.beds.ac.uk/10.3390/genes8090236