Autosomal Recessive NRL Mutations in Patients with Enhanced S-Cone Syndrome

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Analysis
2.2. Clinical Evaluation
3. Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Robson, A.G.; Holder, G.E. Pathognomonic (diagnostic) ergs. A review and update. Retina 2013, 33, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Marmor, M.F.; Kemp, C.M.; Knighton, R.W. SWS (blue) cone hypersensitivity in a newly identified retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1990, 31, 827–838. [Google Scholar]
- Marmor, M.F.; Jacobson, S.G.; Foerster, M.H.; Kellner, U.; Weleber, R.G. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am. J. Ophthalmol. 1990, 110, 124–134. [Google Scholar] [CrossRef]
- Audo, I.; Michaelides, M.; Robson, A.G.; Hawlina, M.; Vaclavik, V.; Sandbach, J.M.; Neveu, M.M.; Hogg, C.R.; Hunt, D.M.; Moore, A.T.; et al. Phenotypic variation in enhanced S-cone syndrome. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2082–2093. [Google Scholar] [CrossRef] [PubMed]
- Schorderet, D.F.; Escher, P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), goldmann-favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum. Mutat. 2009, 30, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Khanna, H.; Oh, E.C.; Hicks, D.; Mitton, K.P.; Swaroop, A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 2004, 13, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.C.; Khan, N.; Novelli, E.; Khanna, H.; Strettoi, E.; Swaroop, A. Transformation of cone precursors to functional rod photoreceptors by bZIP transcription factor NRL. Proc. Natl. Acad. Sci. USA 2007, 104, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.H.; Ahmad, O.; Ahmad, F.; Liu, J.; Chen, S. The photoreceptor-specific nuclear receptor Nr2e3 interacts with Crx and exerts opposing effects on the transcription of rod versus cone genes. Hum. Mol. Genet. 2005, 14, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Kanda, A.; Friedman, J.S.; Nishiguchi, K.M.; Swaroop, A. Retinopathy mutations in the bZIP protein NRL alter phosphorylation and transcriptional activity. Hum. Mutat. 2007, 28, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Farjo, Q.; Jackson, A.; Pieke-Dahl, S.; Scott, K.; Kimberling, W.J.; Sieving, P.A.; Richards, J.E.; Swaroop, A. Human bZIP transcription factor gene NRL: Structure, genomic sequence, and fine linkage mapping at 14q11.2 and negative mutation analysis in patients with retinal degeneration. Genomics 1997, 45, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, A.; Xu, J.Z.; Pawar, H.; Jackson, A.; Skolnick, C.; Agarwal, N. A conserved retina-specific gene encodes a basic motif/leucine zipper domain. Proc. Natl. Acad. Sci. USA 1992, 89, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.C.; Cheng, H.; Hao, H.; Jia, L.; Khan, N.W.; Swaroop, A. Rod differentiation factor NRL activates the expression of nuclear receptor Nr2e3 to suppress the development of cone photoreceptors. Brain Res. 2008, 1236, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Mears, A.J.; Kondo, M.; Swain, P.K.; Takada, Y.; Bush, R.A.; Saunders, T.L.; Sieving, P.A.; Swaroop, A. NRL is required for rod photoreceptor development. Nat. Genet. 2001, 29, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Bessant, D.A.; Holder, G.E.; Fitzke, F.W.; Payne, A.M.; Bhattacharya, S.S.; Bird, A.C. Phenotype of retinitis pigmentosa associated with the Ser50Thr mutation in the NRL gene. Arch. Ophthalmol. 2003, 121, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, X.; Chen, L.J.; Chiang, S.W.; Tam, P.O.; Lai, T.Y.; Chan, C.K.; Wang, N.; Lam, D.S.; Pang, C.P. Association of NR2E3 but not NRL mutations with retinitis pigmentosa in the Chinese population. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Friedman, J.S.; Sandberg, M.A.; Swaroop, A.; Berson, E.L.; Dryja, T.P. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc. Natl. Acad. Sci. USA 2004, 101, 17819–17824. [Google Scholar] [CrossRef] [PubMed]
- Newman, H.; Blumen, S.C.; Braverman, I.; Hanna, R.; Tiosano, B.; Perlman, I.; Ben-Yosef, T. Homozygosity for a recessive loss-of-function mutation of the NRL gene is associated with a variant of enhanced S-cone syndrome. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; van den Born, L.I.; Klevering, B.J.; de Castro-Miro, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2227–2239. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Beryozkin, A.; Shevah, E.; Kimchi, A.; Mizrahi-Meissonnier, L.; Khateb, S.; Ratnapriya, R.; Lazar, C.H.; Blumenfeld, A.; Ben-Yosef, T.; Hemo, Y.; et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci. Rep. 2015, 5, 13187. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; van Genderen, M.M.; van Schooneveld, M.J.; Visser, L.; Riemslag, F.C.; Keunen, J.E.; Bakker, B.; Zonneveld, M.N.; den Hollander, A.I.; Cremers, F.P.; et al. A homozygous frameshift mutation in LRAT causes retinitis punctata albescens. Ophthalmology 2012, 119, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. ISCEV standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. Adv. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Reddick, A.C.; Schwartz, S.B.; Ferguson, J.S.; Aleman, T.S.; Kellner, U.; Jurklies, B.; Schuster, A.; Zrenner, E.; Wissinger, B.; et al. Mutation analysis of NR2E3 and NRL genes in enhanced S cone syndrome. Hum. Mutat. 2004, 24, 439. [Google Scholar] [CrossRef] [PubMed]
- Acar, C.; Mears, A.J.; Yashar, B.M.; Maheshwary, A.S.; Andreasson, S.; Baldi, A.; Sieving, P.A.; Iannaccone, A.; Musarella, M.A.; Jacobson, S.G.; et al. Mutation screening of patients with leber congenital amaurosis or the enhanced S-cone syndrome reveals a lack of sequence variations in the NRL gene. Mol. Vis. 2003, 9, 14–17. [Google Scholar] [PubMed]
- Yzer, S.; Barbazetto, I.; Allikmets, R.; van Schooneveld, M.J.; Bergen, A.; Tsang, S.H.; Jacobson, S.G.; Yannuzzi, L.A. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol. 2013, 131, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Ripamonti, C.; Aboshiha, J.; Henning, G.B.; Sergouniotis, P.I.; Michaelides, M.; Moore, A.T.; Webster, A.R.; Stockman, A. Vision in observers with enhanced S-cone syndrome: An excess of S-cones but connected mainly to conventional S-cone pathways. Investig. Ophthalmol. Vis. Sci. 2014, 55, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Arno, G.; Sergouniotis, P.I.; Tiffin, P.; Borman, A.D.; Chandra, A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; Moore, A.T. Clinical and molecular characterization of enhanced S-cone syndrome in children. JAMA Ophthalmol. 2014, 132, 1341–1349. [Google Scholar] [CrossRef] [PubMed]



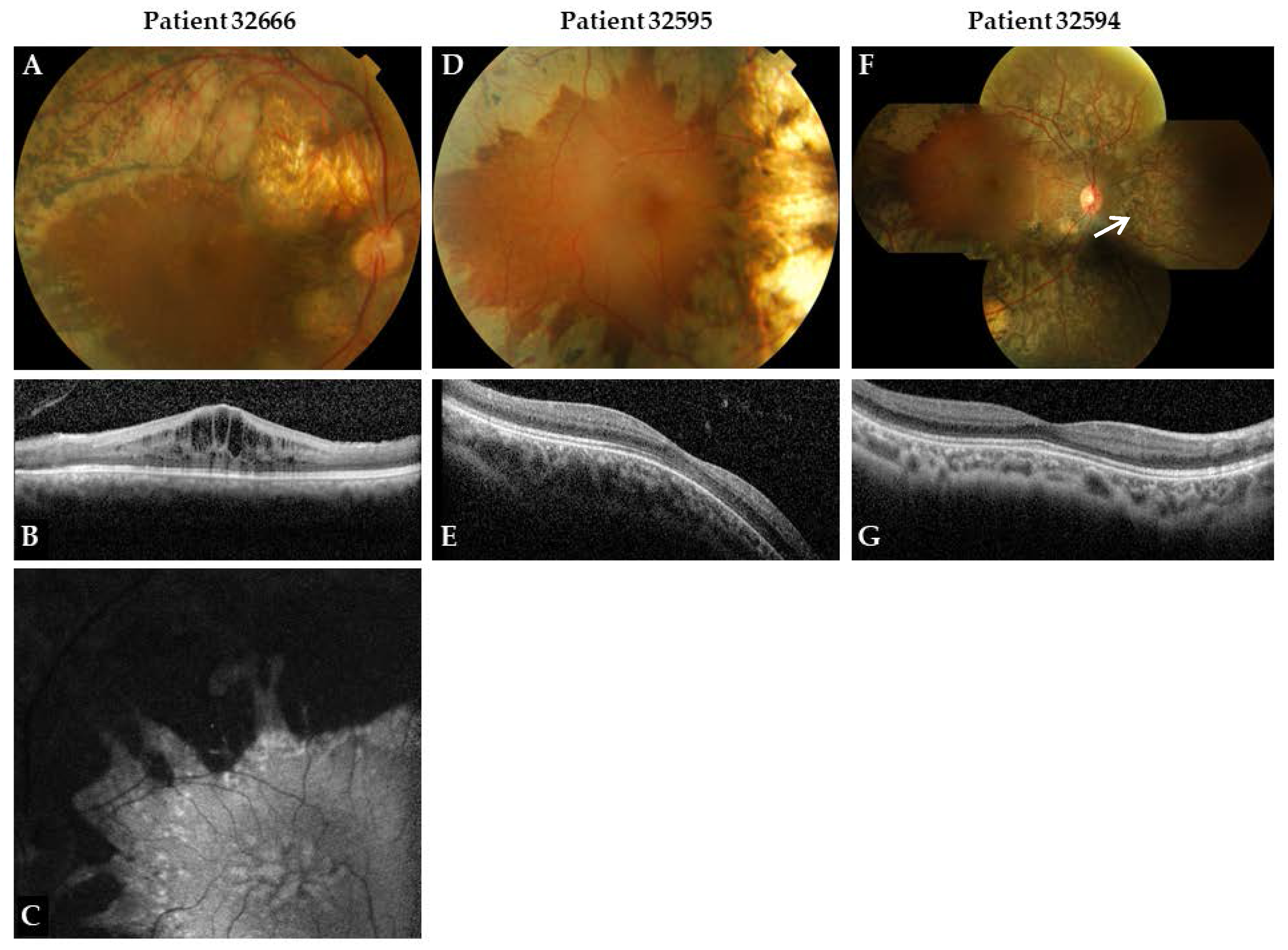{kind=link}
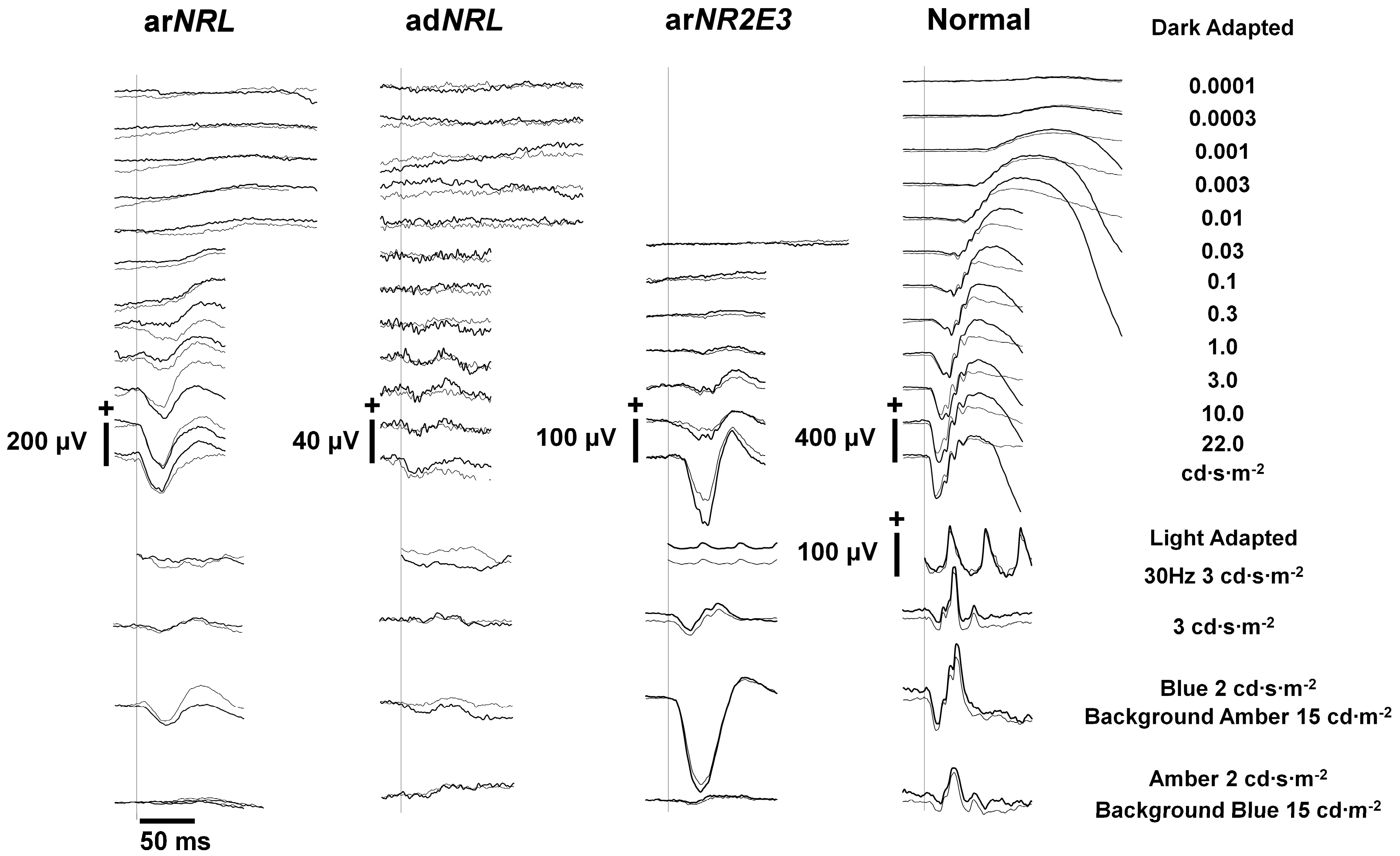{kind=link}
| Proband | Gender | Mutation (cDNA) | Mutation (Protein) | Age at Diagnosis (y) | Age at Last Examination (y) | BCVA RE, LE Refr. Error (SE) | Goldmann Perimetry | Ocular Features |
|---|---|---|---|---|---|---|---|---|
| 32666 | M | c.508C>A c.508C>A | p.Arg170Ser p.Arg170Ser | 10 | 29 | 20/125, 20/125 +4.75/+6.50 | Moderately constricted, midperipheral and central sensitivity loss | Divergent strabismus |
| 32595 | M | c.508C>A c.654del | p.Arg170Ser p.Cys219Valfs*4 | 2 | 13 | 20/40, 20/40 +3.0/+3.0 | Midperipheral and central sensitivity loss | Convergent strabismus, nystagmus |
| 32594 | M | c.508C>A c.654del | p.Arg170Ser p.Cys219Valfs*4 | 2 | 11 | 20/32, 20/63 +1.5/+3.25 | Moderately constricted, midperipheral and central sensitivity loss | Convergent strabismus, nystagmus |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Littink, K.W.; Stappers, P.T.Y.; Riemslag, F.C.C.; Talsma, H.E.; Van Genderen, M.M.; Cremers, F.P.M.; Collin, R.W.J.; Van den Born, L.I. Autosomal Recessive NRL Mutations in Patients with Enhanced S-Cone Syndrome. Genes 2018, 9, 68. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020068
Littink KW, Stappers PTY, Riemslag FCC, Talsma HE, Van Genderen MM, Cremers FPM, Collin RWJ, Van den Born LI. Autosomal Recessive NRL Mutations in Patients with Enhanced S-Cone Syndrome. Genes. 2018; 9(2):68. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020068
Chicago/Turabian StyleLittink, Karin W., Patricia T. Y. Stappers, Frans C. C. Riemslag, Herman E. Talsma, Maria M. Van Genderen, Frans P. M. Cremers, Rob W. J. Collin, and L. Ingeborgh Van den Born. 2018. "Autosomal Recessive NRL Mutations in Patients with Enhanced S-Cone Syndrome" Genes 9, no. 2: 68. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9020068