Investigating the Epigenetic Discrimination of Identical Twins Using Buccal Swabs, Saliva, and Cigarette Butts in the Forensic Setting

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Sample Preparation
2.3. Genome-Wide DNA Methylation Profiling and Data Processing
2.4. Candidate Twin Differentially Methylated CpG Site Selection
2.5. MethyLight
2.6. Quantitative PCR Data Analysis
3. Results
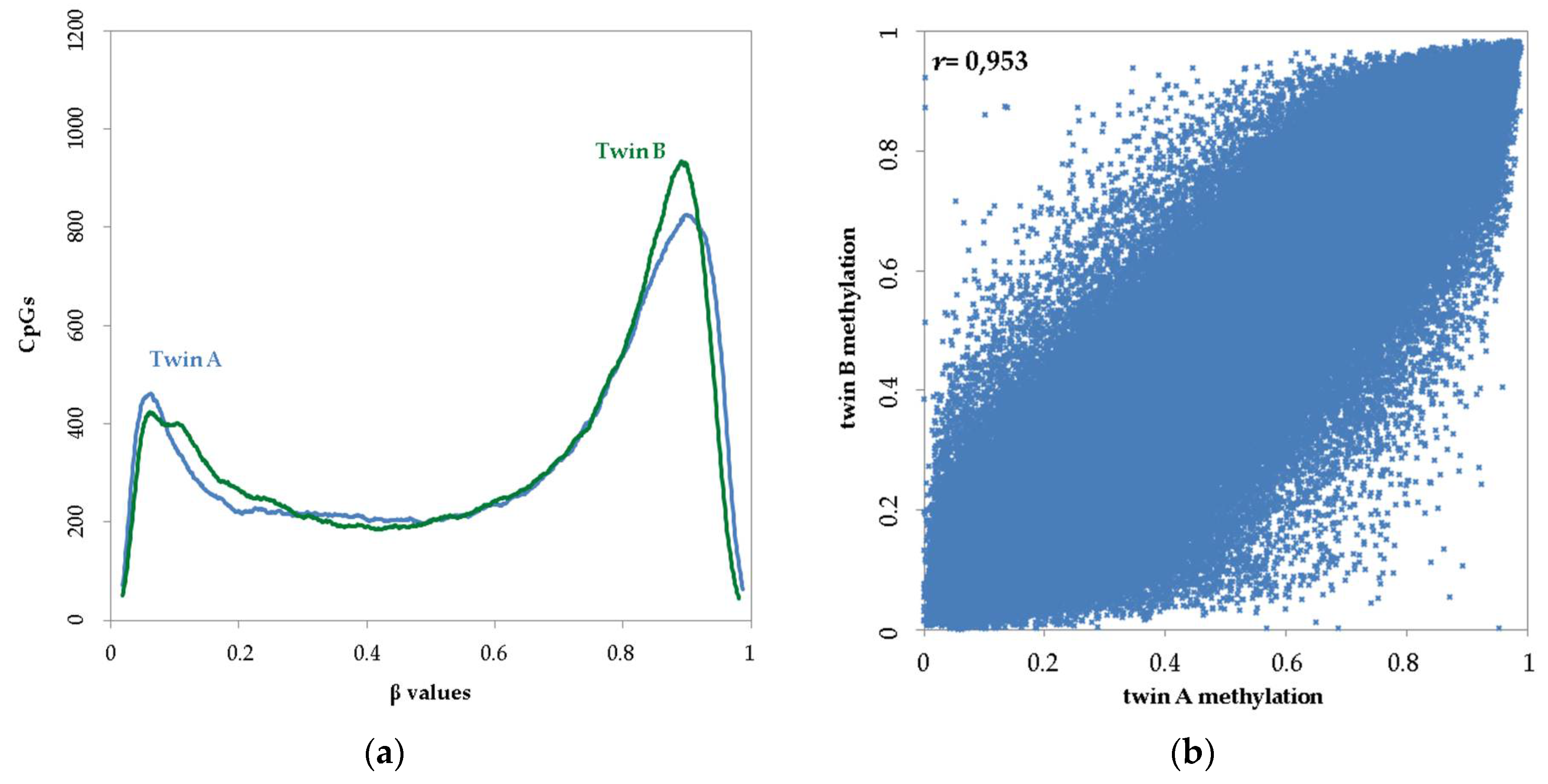
3.1. Distribution and Twin–Twin Correlation of Genome-Wide Methylation Array Data in Reference-Type Buccal DNA
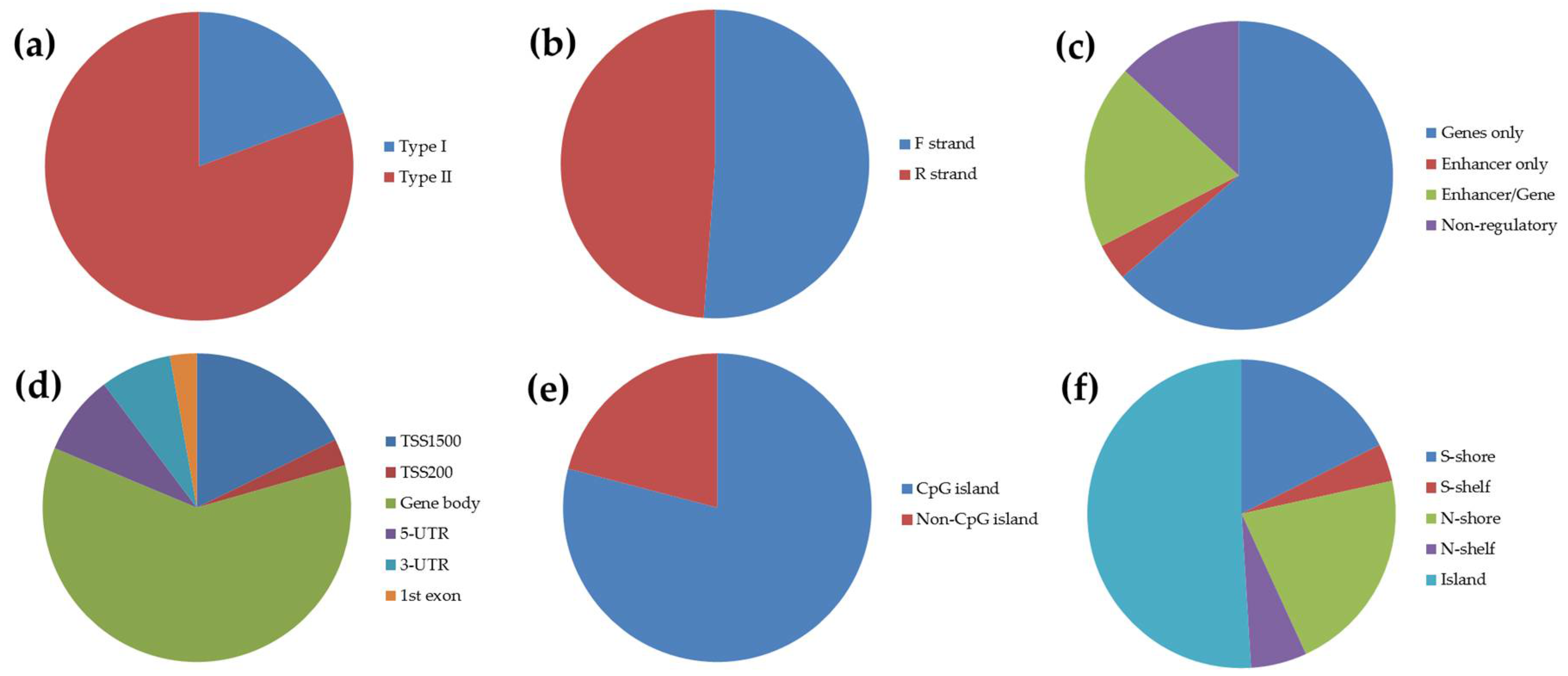
3.2. Identification of Candidate Twin Differentially Methylated CpG Sites from Microarray Screening in Reference-Type Buccal DNA
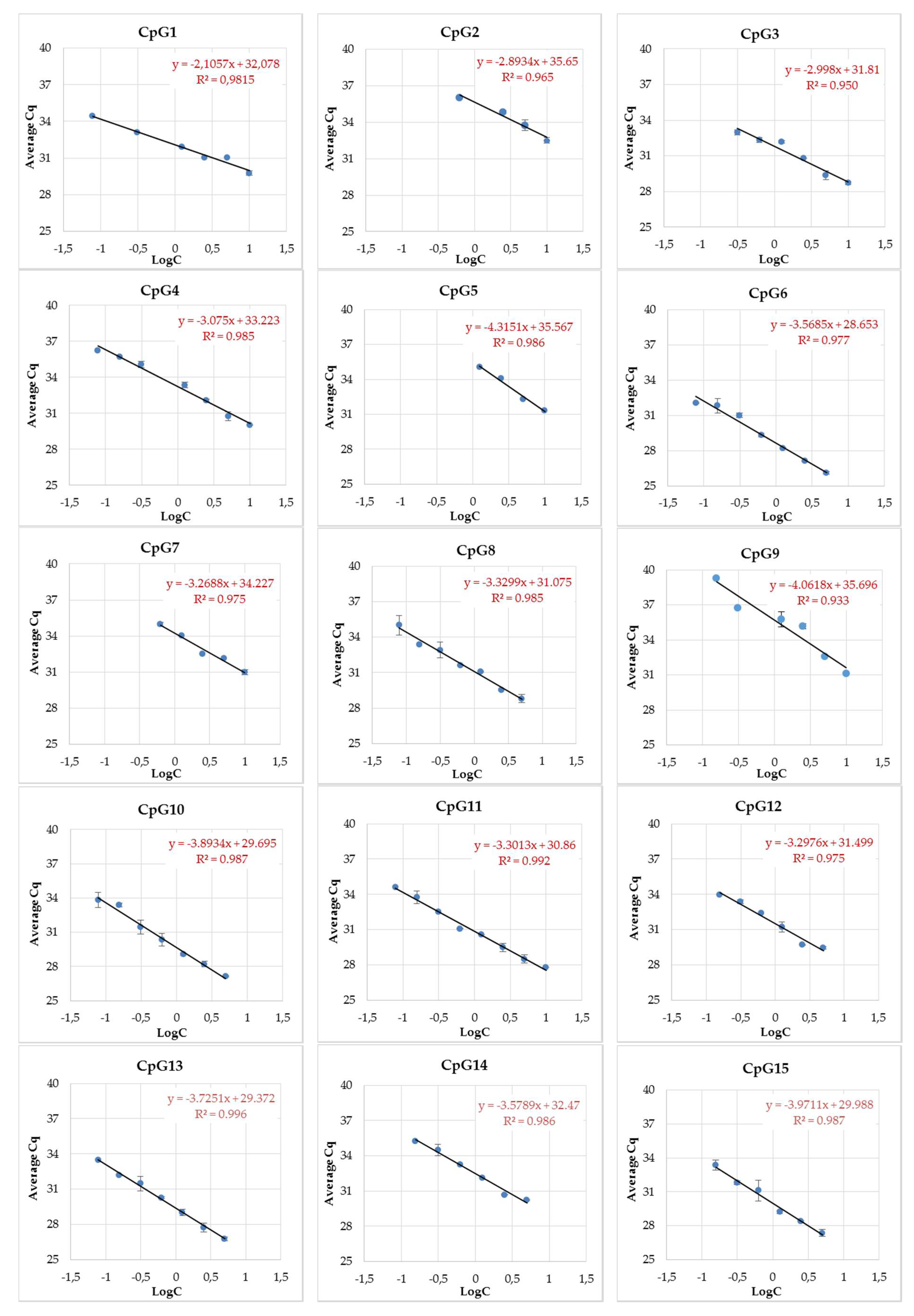
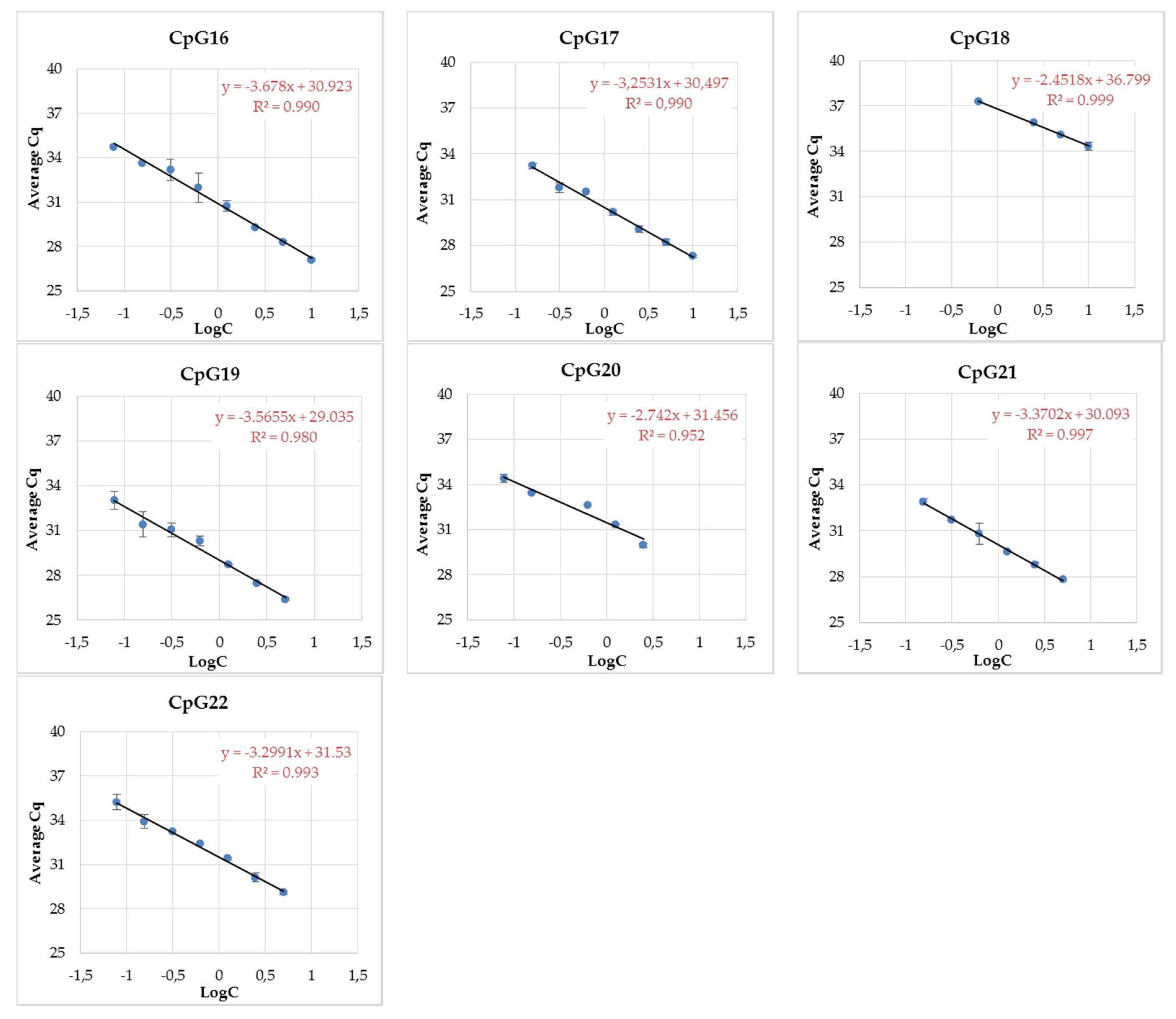
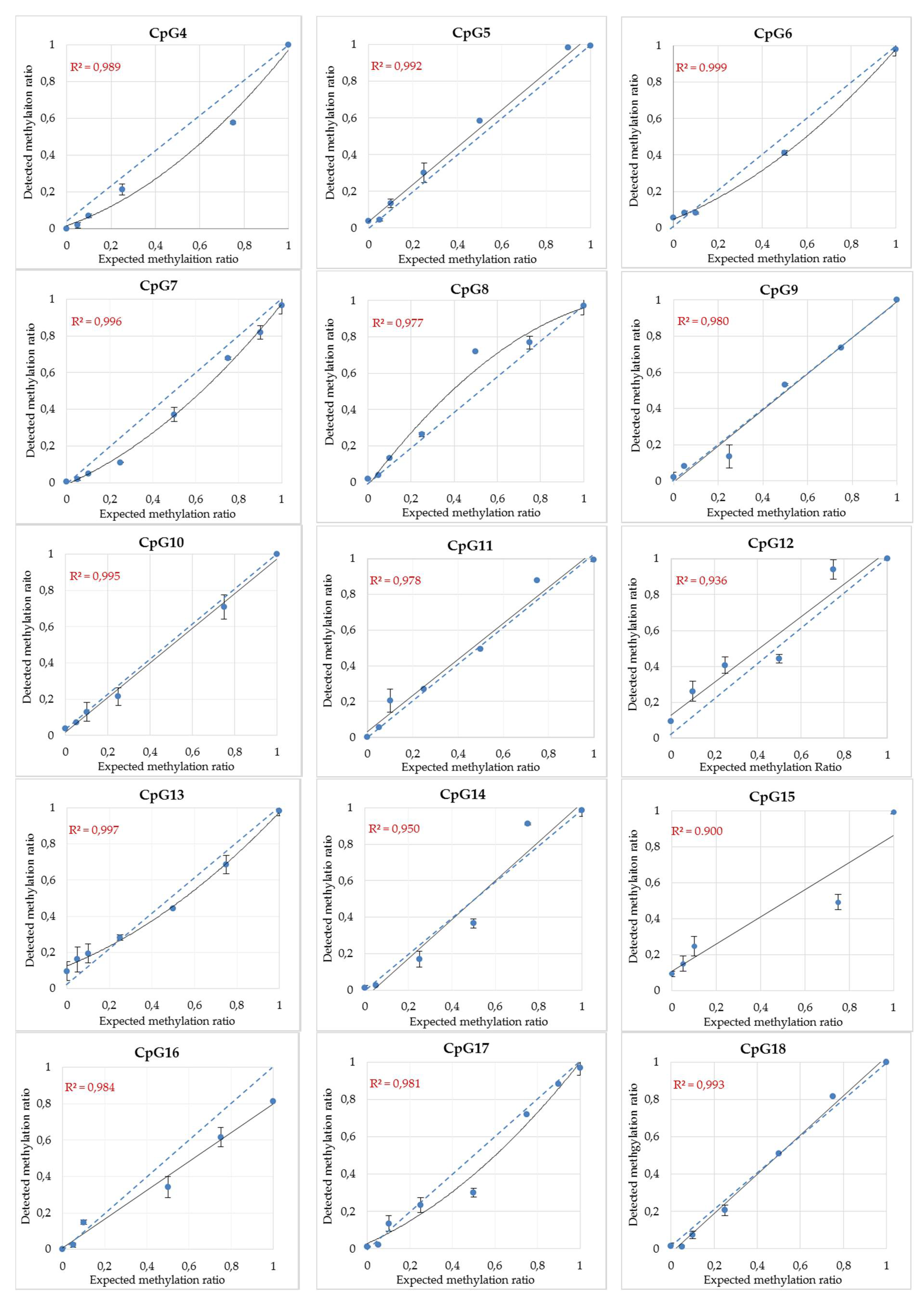
3.3. MethyLight Method Development for Top Candidate Twin Differentially Methylated CpG Sites and Performance Assessment
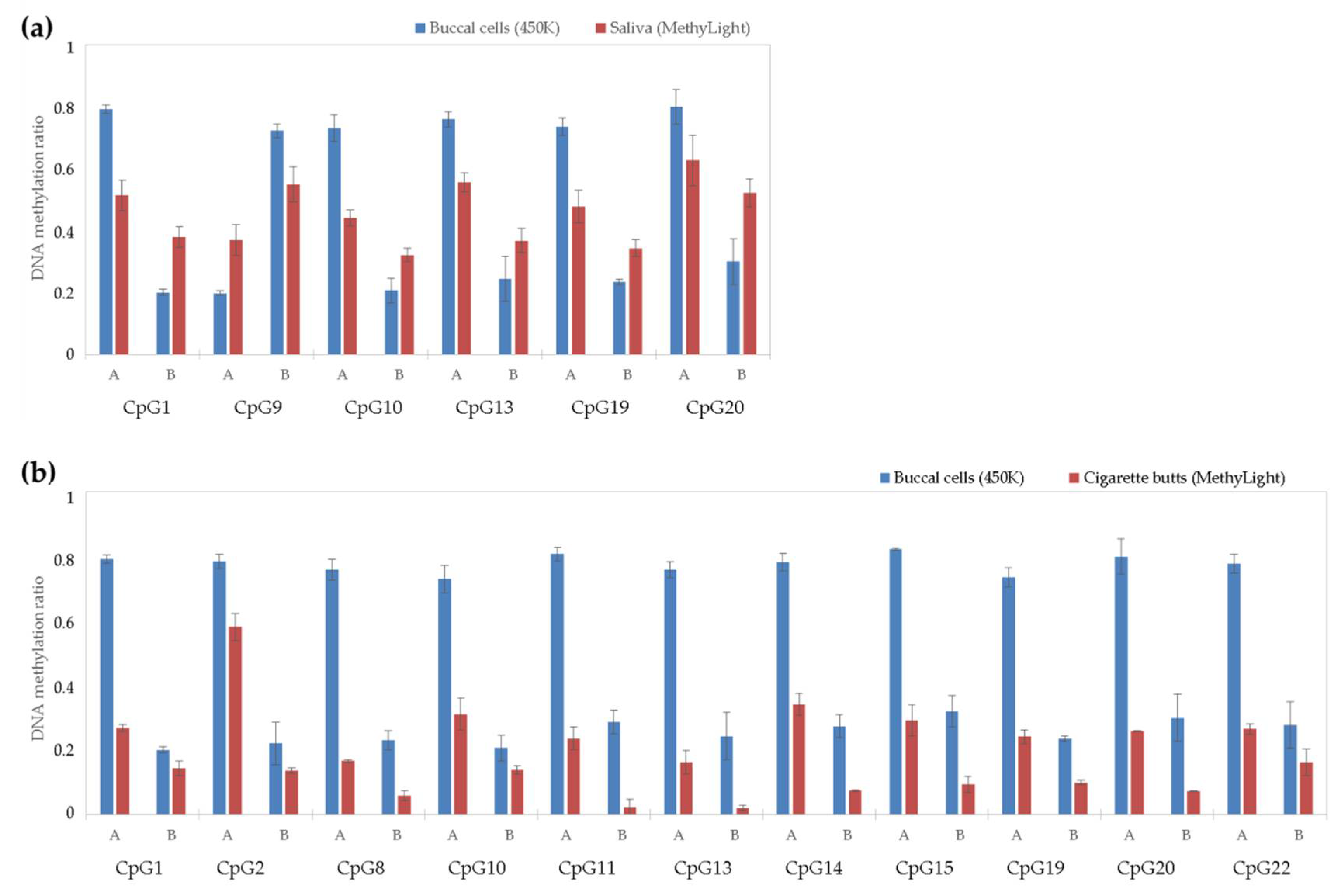
3.4. Saliva DNA Analysis of 22 Top Twin Differentially Methylated CpG Sites Selected from Microarray Data in Reference-Type Buccal DNA
3.5. Cigarette Butt DNA Analysis of 22 Top tDMSs Selected from Microarray Data in Reference-Type Buccal DNA
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | CpG | Primer Sequence (5′-3′) 1 | Primer Length (bp) | Ta (°C) | Primer Concentration (µM) | PCR Length (bp) | |
|---|---|---|---|---|---|---|---|
| CpG1 | cg01115923 | F | TTTTTTTGTTTATAGTGGGAG | 21 | 60 | 2/1 | 86 |
| R | ACTACTAATCCCAAAACTAAAAA | 23 | |||||
| Pm | CCRTACCRAAAACTATAACG 2 | 20 | |||||
| CpG2 | cg23449764 | F | TGTGTGATAGTGAGAAGTATAAA | 23 | 60 | 2/1 | 250 |
| R | AACTTCTAAATCCAATACCA | 20 | |||||
| Pm | CGAAATAAACATATATAATAACACG | 25 | |||||
| CpG3 | cg14525379 | F | GGGTTTATAGATTGTTTTTAGGT | 23 | 60 | 2/1 | 249 |
| R | AAATCTCCTTTACCCTTTTACTT | 23 | |||||
| Pm | AACAAATATAATAAACTTAAATACCG | 26 | |||||
| CpG4 | cg26857315 | F | TTTAGGAGGGAAGTATAGGAA | 21 | 58 | 1.2/0.6 | 286 |
| R | CCCAAAATACTAAAAAACCAAA | 22 | |||||
| Pm | AAATAAATAAACGCGTCCCG | 20 | |||||
| CpG5 | cg18812079 | F | GTATTTAGAGGAGTAGATA | 19 | 58 | 1.2/0.6 | 143 |
| R | TACACCTAAAAAAAATCCCA | 20 | |||||
| Pm | CTAAAAACCCAATCCTACCG | 20 | |||||
| CpG6 | cg07033292 | F | TGTTGATAGTTGTATAGTAG | 20 | 57 | 0.8/0.4 | 120 |
| R | ATAACTAAAAAACCCAACC | 19 | |||||
| Pm | ACCRTAATTAAAACCAAAACCG | 22 | |||||
| CpG7 | cg17434062 | F | TTTTTTGAGGTAGTGTA | 17 | 59 | 1.2/0.6 | 167 |
| R | RCTTCCCCAAAATAAAAATAATC | 23 | |||||
| Pm | AAATTCCAATATCTAAAATACCCG | 24 | |||||
| CpG8 | cg17854471 | F | GTTGTGTTAGTTATTTATTTTTGGG | 25 | 57 | 0.8/0.4 | 224 |
| R | TAAAACCAACCTCATTCTT | 19 | |||||
| Pm | AAAAATTAACACTATACTATCAACG | 25 | |||||
| CpG9 | cg13038544 | F | GGGGGAATTAGGTATTATTTTTA | 23 | 57 | 1.2/0.6 | 248 |
| R | CAAATATAAAAACCCTACTC | 20 | |||||
| Pm | CGTAACAAAATAAAATCCGCTCG | 23 | |||||
| CpG10 | cg23041250 | F | GGTTTTTTTTTTAGGTGT | 18 | 57 | 0.8/0.4 | 177 |
| R | AAAACCTCACCCTAACCTAA | 20 | |||||
| Pm | ACCTCRCCCTCCACRCG | 17 | |||||
| CpG11 | cg14737704 | F | AATTGTTGTGTGTTGGTGGATA | 22 | 55 | 0.8/0.4 | 220 |
| R | ACCAACAATAATAAAAAC | 18 | |||||
| Pm | AATAAAACTAATAAACTCCACG | 22 | |||||
| CpG12 | cg18562578 | F | GAGGATTTTTGTTTGGTTTTT | 21 | 59 | 0.8/0.4 | 255 |
| R | RTAACTCCCTTTCTATATAT | 20 | |||||
| Pm | CTTCTTTACCAACCACRATACG | 22 | |||||
| CpG13 | cg05415840 | F | ATTTTTGAGTTGGGGTTGATT | 21 | 55 | 0.8/0.4 | 142 |
| R | CCCTACAAAAAAAAAAACT | 19 | |||||
| Pm | ACATCTAAATAACACRAATATATACG | 26 | |||||
| CpG14 | cg20482280 | F | TTGGTTGTTTAGGAAGTGTAT | 21 | 56 | 0.8/0.4 | 262 |
| R | AATCTTCCTATAAACAAAAA | 20 | |||||
| Pm | TCTCCTCTATAACCATATAAAACACG | 26 | |||||
| CpG15 | cg15904939 | F | TTTTTAAGGTTGTGAGTTAG | 20 | 56 | 0.8/0.4 | 142 |
| R | ACTAACCCTACTAAAATA | 18 | |||||
| Pm | AATAACCTAAAATTATCCACCACG | 24 | |||||
| CpG16 | cg03353765 | F | TTAGTGGATAGGAAAGTTAA | 20 | 60 | 1.2/0.6 | 216 |
| R | TTTCAAACAACACAAAAACC | 20 | |||||
| Pm | CGACTACCAAATAAAAACTACTTACG | 26 | |||||
| CpG17 | cg03571301 | F | ATTTGTGAATAGTATTATGGGGA | 23 | 60 | 1.2/0.6 | 231 |
| R | CATTTCTCTACCAACAAAAA | 20 | |||||
| Pm | CCATCCCTATATTTACTAACG | 21 | |||||
| CpG18 | cg02961798 | F | GGTTGTTYGGTATTTTTTAGTAGT | 24 | 58 | 1.2/0.6 | 191 |
| R | ATCCTAACTTCTTCCTA | 17 | |||||
| Pm | CRAAATATACCTAAATATAAAACTCCG | 27 | |||||
| CpG19 | cg00134667 | F | AGGAGGGATTTTTTTTAAGGTA | 22 | 56 | 0.8/0.4 | 146 |
| R | AAAAATACCCAACTCTATCT | 20 | |||||
| Pm | AAATTAATACTTTCCAAATACCG | 23 | |||||
| CpG20 | cg02886509 | F | GGAATATTTGTGGGTAAATT | 20 | 58 | 0.8/0.4 | 222 |
| R | RCCACTACTACTTTATTCTCTAA | 23 | |||||
| Pm | CTCAAAAATCATCACRTCCG | 20 | |||||
| CpG21 | cg13460168 | F | TTTTTGATATTTTTGTGGGTGG | 22 | 60 | 0.8/0.4 | 187 |
| R | ATCCCCCRAATTTTATTCTTAAC | 23 | |||||
| Pm | TCCAAACTTAACAATAAATAACG | 23 | |||||
| CpG22 | cg10399269 | F | TTTTTTTATGGTTTGTTGGT | 20 | 57 | 0.8/0.4 | 196 |
| R | AACCTCTATAACCTCAAAAT | 20 | |||||
| Pm | CRAATAAATAAATATCCCCG | 20 | |||||
| Alu | F | GGTTAGGTATAGTGGTTTATATTTGTAATTTTAGTA | 36 | 60 | 2/1 | 98 | |
| R | ATTAACTAAACTAATCTTAAACTCCTAACCTCA | 33 | |||||
| Pm | CCTACCTTAACCTCCC | 16 | |||||
| Assay | Min DNA Input (ng) | Average SD | Equation for Data Normalization |
|---|---|---|---|
| CpG1 | 0.078 | 0.036 | y = 0.8664x + 0.0182 |
| CpG2 | 0.625 | 0.027 | y = 1.0412x + 0.0308 |
| CpG3 | 0.313 | 0.026 | y = 1.056x |
| CpG4 | 0.078 | 0.010 | y = 1.0325x + 0.0348 |
| CpG5 | 0.0625 | 0.023 | y = 0.9772x − 0.0302 |
| CpG6 | 0.078 | 0.020 | y = −0.5173x2 + 1.5932x − 0.0643 |
| CpG7 | 0.625 | 0.020 | y = 1.0135x + 0.0613 |
| CpG8 | 0.078 | 0.022 | y = 0.5787x2 + 0.4094x + 0.0314 |
| CpG8 | 0.156 | 0.025 | y = 0.9843x + 0.014 |
| CpG10 | 0.078 | 0.034 | y = 1.0439x − 0.0177 |
| CpG11 | 0.078 | 0.025 | y = 0.9695x − 0.0221 |
| CpG12 | 0.156 | 0.036 | y = −0.1247x2 + 1.1728x − 0.1337 |
| CpG13 | 0.078 | 0.044 | y = 1.1769x − 0.0989 |
| CpG14 | 0.156 | 0.024 | y = 0.8913x + 0.0585 |
| CpG15 | 0.156 | 0.038 | y = −1.2737x2 + 2.5978x − 0.3106 |
| CpG16 | 0.078 | 0.027 | y = 1.2423x − 0.0023 |
| CpG17 | 0.078 | 0.026 | y = 1.0117x + 0.029 |
| CpG18 | 0.625 | 0.018 | y = 0.9449x + 0.023 |
| CpG19 | 0.078 | 0.036 | y = 1.0686x − 0.0282 |
| CpG20 | 0.078 | 0.030 | y = 0.9236x + 0.0198 |
| CpG21 | 0.156 | 0.031 | y = 0.4054x2 + 0.6791x − 0.0246 |
| CpG22 | 0.078 | 0.033 | y = 0.9962x + 0.043 |








References
- Jobling, M.A.; Gill, P. Encoded evidence: DNA in forensic analysis. Nat. Rev. Genet. 2004, 5, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Kayser, M.; de Knijff, P. Improving human forensics through advances in genetics, genomics and molecular biology. Nat. Rev. Genet. 2011, 12, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Yang, Y.; Zhang, X.N.; Quan, X.L.; Wu, Y.M. Tri-allelic pattern of short tandem repeats identifies the murderer among identical twins and suggests an embryonic mutational origin. Forensic Sci. Int. Genet. 2015, 16, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, C. Despite DNA Evidence, Twins Charged in Heist Go Free. Available online: http://content.time.com/time/world/article/0,8599,1887111,00.html (accessed on 20 November 2017).
- Humphreys, A. New Brunswick Twins Identical DNA Made for Difficult Conviction in Deadly Home Invasion. Available online: http://news.nationalpost.com/news/canada/twins-convicted (accessed on 20 November 2017).
- Krawczak, M.; Cooper, D.N.; Fandrich, F.; Engel, W.; Schmidtke, J. How to distinguish genetically between an alleged father and his monozygotic twin: A thought experiment. Forensic Sci. Int. Genet. 2012, 6, e129–e130. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lehmann, J.; Schilling, E.; Gradl, G.; Richter, D.C.; Wiehler, J.; Rolf, B. Finding the needle in the haystack: Differentiating “identical” twins in paternity testing and forensics by ultra-deep next generation sequencing. Forensic Sci. Int. Genet. 2014, 9, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Inbar-Feigenberg, M.; Choufani, S.; Butcher, D.T.; Roifman, M.; Weksberg, R. Basic concepts of epigenetics. Fertil. Steril. 2013, 99, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Spector, T.D. A twin approach to unraveling epigenetics. Trends Genet. 2011, 27, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.A.; Tang, T.; Wang, S.C.; Ptak, C.; Oh, G.H.T.; Wong, A.H.C.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C.; et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Ehli, E.A.; Slieker, R.C.; Bartels, M.; Weber, Z.M.; Davies, G.E.; Slagboom, P.E.; Heijmans, B.T.; Boomsma, D.I. Epigenetic variation in monozygotic twins a genome-wide analysis of DNA methylation in buccal cells. Genes 2014, 5, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Gervin, K.; Hammero, M.; Akselsen, H.E.; Moe, R.; Nygard, H.; Brandt, I.; Gjessing, H.K.; Harris, J.R.; Undlien, D.E.; Lyle, R. Extensive variation and low heritability of DNA methylation identified in a twin study. Genome Res. 2011, 21, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Kremer, D.; Tobi, E.W.; Boomsma, D.I.; Slagboom, P.E. Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Hum. Mol. Genet. 2007, 16, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirazzini, C.; Giuliani, C.; Bacalini, M.G.; Boattini, A.; Capri, M.; Fontanesi, E.; Marasco, E.; Mantovani, V.; Pierini, M.; Pini, E.; et al. Space/Population and Time/Age in DNA methylation variability in humans: A study on IGF2/H19 locus in different Italian populations and in mono- and di-zygotic twins of different age. Aging (Albany NY) 2012, 4, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.; Loke, Y.J.; Gordon, L.; Ollikainen, M.; Cruickshank, M.N.; Saffery, R.; Craig, J.M. Longitudinal, genome-scale analysis of DNA methylation in twins from birth to 18 months of age reveals rapid epigenetic change in early life and pair-specific effects of discordance. Genome Biol. 2013, 14, R42. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Christiansen, L.; Thomassen, M.; Kruse, T.A.; Christensen, K. Twins for epigenetic studies of human aging and development. Aging Res. Rev. 2013, 12, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ribel-Madsen, R.; Fraga, M.F.; Jacobsen, S.; Bork-Jensen, J.; Lara, E.; Calvanese, V.; Fernandez, A.F.; Friedrichsen, M.; Vind, B.F.; Hojlund, K.; et al. Genome-wide analysis of DNA methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS ONE 2012, 7, e51302. [Google Scholar] [CrossRef] [PubMed]
- Malki, K.; Koritskaya, E.; Harris, F.; Bryson, K.; Herbster, M.; Tosto, M.G. Epigenetic differences in monozygotic twins discordant for major depressive disorder. Transl. Psychiatry 2016, 6, e839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, L.; van Dongen, J.; Bell, C.G.; Burri, A.; Deloukas, P.; Boomsma, D.I.; Spector, T.D.; Bell, J.T. Integrative DNA methylome analysis of pan-cancer biomarkers in cancer discordant monozygotic twin-pairs. Clin. Epigenet. 2016, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Fernandez, J.E.; Spector, T.D.; Bell, J.T. Epigenetics of discordant monozygotic twins: Implications for disease. Genome Med. 2014, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, S.; Que, T.; Li, L.; Zhao, S. Identical but not the same: The value of DNA methylation profiling in forensic discrimination within monozygotic twins. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e337–e338. [Google Scholar] [CrossRef]
- Li, C.; Zhao, S.; Zhang, N.; Zhang, S.; Hou, Y. Differences of DNA methylation profiles between monozygotic twins’ blood samples. Mol. Biol. Rep. 2013, 40, 5275–5280. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Zhu, G.; Fu, G.; Zhang, X.; Fu, L.; Li, S.; Cong, B. A Genome-Wide Scan of DNA Methylation Markers for Distinguishing Monozygotic Twins. Twin Res. Hum. Genet. 2015, 18, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fu, G.; Yan, L.; Craig, J.M.; Zhang, X.; Fu, L.; Ma, C.; Li, S.; Cong, B. LINE-1 DNA methylation: A potential forensic marker for discriminating monozygotic twins. Forensic Sci. Int. Genet. 2015, 19, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.; Evans, N.; Bexon, K.J.; van der Meer, D.J.; Williams, G.A. Differentiating between monozygotic twins through DNA methylation-specific high-resolution melt curve analysis. Anal. Biochem. 2015, 476, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhao, S.; Zhang, S.H.; Chen, J.; Lu, D.; Shen, M.; Li, C. Intra-monozygotic twin pair discordance and longitudinal variation of whole-genome scale DNA methylation in adults. PLoS ONE 2015, 10, e0135022. [Google Scholar] [CrossRef] [PubMed]
- Espada, J.; Esteller, M. DNA methylation and the functional organization of the nuclear compartment. Semin. Cell Dev. Biol. 2010, 21, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, A.; Diez Lopez, C.; Carnero-Montoro, E.; Ralf, A.; Ward, K.; Spector, T.; Bell, J.T.; Kayser, M. Epigenetic discrimination of identical twins from blood under the forensic scenario. Forensic Sci. Int. Genet. 2017, 31, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Lokk, K.; Modhukur, V.; Rahashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltsina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, Y.; Lin, N.; Lowdon, R.F.; Hong, C.; Nagarajan, R.P.; Cheng, J.B.; Li, D.; Stevens, M.; Lee, H.J.; et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013, 23, 1522–1540. [Google Scholar] [CrossRef] [PubMed]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- De Bustos, C.; Ramos, E.; Young, J.M.; Tran, R.K.; Menzel, U.; Langford, C.F.; Eichler, E.E.; Hsu, L.; Henikoff, S.; Dumanski, J.P.; et al. Tissue-specific variation in DNA methylation levels along human chromosome 1. Epigenet. Chromatin 2009, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Baron, U.; Turbachova, I.; Hellwag, A.; Eckhardt, F.; Berlin, K.; Hoffmuller, U.; Gardina, P.; Olek, S. DNA methylation analysis as a tool for cell typing. Epigenetics 2006, 1, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016, 17, 72. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Nivard, M.G.; Willemsen, G.; Hottenga, J.J.; Helmer, Q.; Dolan, C.V.; Ehli, E.A.; Davies, G.E.; van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Touleimat, N.; Tost, J. Complete pipeline for Infinium Human Methylation 450 K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalisation of 450K methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Campan, M.; Weisenberger, D.J.; Trinh, B.; Laird, P.W.; MethyLight, J.T. (Eds.) DNA Methylation—Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; Volume 507, pp. 325–337. ISBN 978-1-59745-522-0. [Google Scholar]
- Zerbino, D.; Achuthan, P.; Akanni, W.; Amode, M.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; García, G.C.; et al. Ensembl 2018. Available online: http://grch37.ensembl.org/index.html (accessed on 20 November 2017).
- Tusnady, G.E.; Simon, I.; Varadi, A.; Aranyi, T. BiSearch: Primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005, 33, e9. [Google Scholar] [CrossRef] [PubMed]
- Vallone, P.M.; Butler, J.M. AutoDimer: A screening tool for primer-dimer and hairpin structures. BioTechniques 2004, 37, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Snellenberg, S.; De Strooper, L.M.A.; Hesselink, A.T.; Meijer, C.J.L.M.; Snijders, P.J.F.; Heideman, D.A.M.; Steenbergen, R.D.M. Development of a multiplex methylation-specific PCR as candidate triage test for women with an HPV-positive cervical scrape. BMC Cancer 2012, 12, 551. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chen, H.Y.; Bai, E.Q.; Luo, Y.X.; Fu, R.J.; He, Y.S.; Jiang, J.; Wang, H.Q. Development of a multiplex MethyLight assay for the detection of multigene methylation in human colorectal cancer. Cancer Genet. Cytogenet. 2010, 202, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Olkhov-Mitsel, E.; Zdravic, D.; Kron, K.; van der Kwast, T.; Fleshner, N.; Bapat, B. Novel multiplex MethyLight protocol for detection of DNA methylation in patient tissues and bodily fluids. Sci. Rep. 2014, 4, 4432. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Heyn, H.; Moran, S.; Serra-Musach, J.; Pujana, M.A.; Bibikova, M.; Esteller, M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011, 6, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Siegmund, K.D. An evaluation of processing methods for HumanMethylation450 BeadChip data. BMC Genom. 2016, 17, 469. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, A.; Ballard, D.; Aliferi, A.; Miller, T.H.; Barron, L.P.; Syndercombe Court, D. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci. Int. Genet. 2017, 28, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, E.A.; Zavgorodnij, M.G.; Majorova, S.P.; Vorobjev, I.A.; Jandaghi, P.; Bure, I.V.; Hoheisel, J.D. Correction of PCR-bias in quantitative DNA methylation studies by means of cubic polynomial regression. Nucleic Acids Res. 2011, 39, e77. [Google Scholar] [CrossRef] [PubMed]
- Vidaki, A.; Giangasparo, F.; Syndercombe Court, D. Discovery of potential DNA methylation markers for forensic tissue identification using bisulphite pyrosequencing. Electrophoresis 2016, 37, 2767–2779. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Li, J.; Nakanishi, M.; Asada, T.; Ikesue, M.; Goto, Y.; Fukushima, Y.; Iwai, N. Establishment of the MethyLight Assay for Assessing Aging, Cigarette Smoking, and Alcohol Consumption. Biomed. Res. Int. 2015, 2015, 451981. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Prange-Krex, G.; Freiberg-Richter, J.; Bornhäuser, M.; Ehninger, G. Buccal swabs but not mouthwash samples can be used to obtain pretransplant DNA fingerprints from recipients of allogeneic bone marrow transplants. Bone Marrow Transplant. 2000, 25, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Aps, J.K.M.; van den Maagdenberg, K.; Delanghe, J.R.; Martens, L.C. Flow cytometry as a new method to quantify the cellular content of human saliva and its relation to gingivitis. Clin. Chim. Acta 2002, 321, 35–41. [Google Scholar] [CrossRef]
- Frobel, J.; Bozic, T.; Lenz, M.; Uciechowski, P.; Han, Y.; Herwartz, R.; Strathmann, K.; Isfort, S.; Panse, J.; Esser, A.; et al. Leukocyte counts based on site-specific DNA methylation analysis. bioRxiv 2017, 150110. [Google Scholar] [CrossRef]



| Normalization Method | Number of Potential tDMSs (% of Total Analyzed CpGs) | ||
|---|---|---|---|
| Threshold | > 0.495 | >0.395 | >0.295 |
| FUNNORM | 239 (0.07%) | 998 (0.29%) | 4.208 (1.22%) |
| SWAN | 66 (0.02%) | 411 (0.18%) | 2.068 (0.60%) |
| Shared by both methods | 61 (0.02%) | 363 (0.11%) | 1.751 (0.51%) |
| CpG | Assay | FUNNORM | SWAN | Average Diff | ||||
|---|---|---|---|---|---|---|---|---|
| A | B | Diff | A | B | Diff | |||
| cg01115923 | CpG1 | 0.801 | 0.207 | 0.594 | 0.783 | 0.193 | 0.590 | 0.592 |
| cg23449764 | CpG2 | 0.768 | 0.173 | 0.595 | 0.799 | 0.266 | 0.533 | 0.564 |
| cg14525379 | CpG3 | 0.769 | 0.194 | 0.575 | 0.841 | 0.295 | 0.546 | 0.561 |
| cg26857315 | CpG4 | 0.258 | 0.850 | 0.592 | 0.294 | 0.807 | 0.513 | 0.553 |
| cg18812079 | CpG5 | 0.831 | 0.310 | 0.521 | 0.833 | 0.252 | 0.581 | 0.551 |
| cg07033292 | CpG6 | 0.757 | 0.157 | 0.600 | 0.700 | 0.212 | 0.488 | 0.544 |
| cg17434062 | CpG7 | 0.815 | 0.247 | 0.568 | 0.827 | 0.314 | 0.513 | 0.541 |
| cg11777917 | (-) 1 | 0.841 | 0.245 | 0.596 | 0.749 | 0.279 | 0.470 | 0.533 |
| cg17854471 | CpG8 | 0.782 | 0.209 | 0.573 | 0.737 | 0.25 | 0.487 | 0.530 |
| cg13038544 | CpG9 | 0.193 | 0.737 | 0.544 | 0.202 | 0.707 | 0.505 | 0.525 |
| cg23041250 | CpG10 | 0.760 | 0.178 | 0.582 | 0.699 | 0.234 | 0.465 | 0.534 |
| cg12047941 | (-) 1 | 0.800 | 0.210 | 0.590 | 0.743 | 0.289 | 0.454 | 0.522 |
| cg14737704 | CpG11 | 0.823 | 0.312 | 0.511 | 0.793 | 0.26 | 0.533 | 0.522 |
| cg18562578 | CpG12 | 0.242 | 0.831 | 0.589 | 0.314 | 0.766 | 0.452 | 0.521 |
| cg05415840 | CpG13 | 0.742 | 0.192 | 0.550 | 0.777 | 0.294 | 0.483 | 0.517 |
| cg17080283 | (-) 1 | 0.719 | 0.157 | 0.562 | 0.691 | 0.224 | 0.467 | 0.515 |
| cg20482280 | CpG14 | 0.802 | 0.249 | 0.553 | 0.764 | 0.298 | 0.466 | 0.510 |
| cg15904939 | CpG15 | 0.826 | 0.353 | 0.473 | 0.821 | 0.285 | 0.536 | 0.505 |
| cg03353765 | CpG16 | 0.196 | 0.706 | 0.51 | 0.274 | 0.772 | 0.498 | 0.504 |
| cg03571301 | CpG17 | 0.762 | 0.256 | 0.506 | 0.731 | 0.232 | 0.499 | 0.503 |
| cg02961798 | CpG18 | 0.206 | 0.701 | 0.495 | 0.217 | 0.726 | 0.509 | 0.502 |
| cg00134667 | CpG19 | 0.756 | 0.228 | 0.528 | 0.715 | 0.240 | 0.475 | 0.502 |
| cg02886509 | CpG20 | 0.838 | 0.247 | 0.591 | 0.761 | 0.351 | 0.410 | 0.501 |
| cg13460168 | CpG21 | 0.207 | 0.729 | 0.522 | 0.229 | 0.708 | 0.479 | 0.501 |
| cg10399269 | CpG22 | 0.789 | 0.226 | 0.572 | 0.757 | 0.329 | 0.428 | 0.500 |
| Twin | Top CpG | Methylation Ratio | Top CpG | Methylation Ratio | ||||
|---|---|---|---|---|---|---|---|---|
| Buccal 1 | Saliva 2 | Cigarettes 2 | Buccal 1 | Saliva 2 | Cigarettes 2 | |||
| A | 1 | 0.792 | 0.513 | 0.268 | 12 | 0.278 | 0.591 | 0.428 |
| B | 0.200 | 0.378 | 0.144 | 0.799 | 0.303 | 0.016 | ||
| A | 2 | 0.784 | 0.452 | 0.581 | 13 | 0.760 | 0.557 | 0.162 |
| B | 0.220 | 0.371 | 0.135 | 0.243 | 0.367 | 0.020 | ||
| A | 3 | 0.805 | 0.193 | 0.171 | 14 | 0.783 | 0.330 | 0.341 |
| B | 0.245 | 0.115 | 0.091 | 0.274 | 0.477 | 0.074 | ||
| A | 4 | 0.276 | 0.078 | 0.092 | 15 | 0.824 | 0.357 | 0.292 |
| B | 0.829 | 0.107 | 0.077 | 0.319 | 0.605 | 0.093 | ||
| A | 5 | 0.832 | 0.195 | 0.151 | 16 | 0.235 | 0.513 | 0.219 |
| B | 0.281 | 0.254 | 0.185 | 0.739 | 0.559 | 0.119 | ||
| A | 6 | 0.729 | 0.727 | 0.405 | 17 | 0.747 | 0.164 | 0.086 |
| B | 0.185 | 0.640 | 0.331 | 0.244 | 0.137 | 0.047 | ||
| A | 7 | 0.821 | 0.244 | 0.234 | 18 | 0.212 | 0.968 | 0.408 |
| B | 0.281 | 0.216 | 0.205 | 0.714 | 0.413 | 0.490 | ||
| A | 8 | 0.760 | 0.260 | 0.165 | 19 | 0.736 | 0.477 | 0.241 |
| B | 0.230 | 0.327 | 0.057 | 0.234 | 0.343 | 0.099 | ||
| A | 9 | 0.198 | 0.368 | 0.259 | 20 | 0.800 | 0.627 | 0.259 |
| B | 0.722 | 0.549 | 0.075 | 0.299 | 0.523 | 0.072 | ||
| A | 10 | 0.730 | 0.440 | 0.311 | 21 | 0.218 | 0.452 | 0.243 |
| B | 0.206 | 0.320 | 0.138 | 0.719 | 0.249 | 0.090 | ||
| A | 11 | 0.808 | 0.274 | 0.235 | 22 | 0.778 | 0.429 | 0.265 |
| B | 0.286 | 0.530 | 0.023 | 0.278 | 0.477 | 0.162 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vidaki, A.; Kalamara, V.; Carnero-Montoro, E.; Spector, T.D.; Bell, J.T.; Kayser, M. Investigating the Epigenetic Discrimination of Identical Twins Using Buccal Swabs, Saliva, and Cigarette Butts in the Forensic Setting. Genes 2018, 9, 252. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9050252
Vidaki A, Kalamara V, Carnero-Montoro E, Spector TD, Bell JT, Kayser M. Investigating the Epigenetic Discrimination of Identical Twins Using Buccal Swabs, Saliva, and Cigarette Butts in the Forensic Setting. Genes. 2018; 9(5):252. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9050252
Chicago/Turabian StyleVidaki, Athina, Vivian Kalamara, Elena Carnero-Montoro, Timothy D. Spector, Jordana T. Bell, and Manfred Kayser. 2018. "Investigating the Epigenetic Discrimination of Identical Twins Using Buccal Swabs, Saliva, and Cigarette Butts in the Forensic Setting" Genes 9, no. 5: 252. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9050252