Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Genotypes and Illumina Sequences Used in the Analyses
2.2. Long Terminal Repeats-Retrotransposon Redundancy Estimation
2.3. Retrotransposon Distribution along the Sunflower (HanXRQInbred Line) Genome
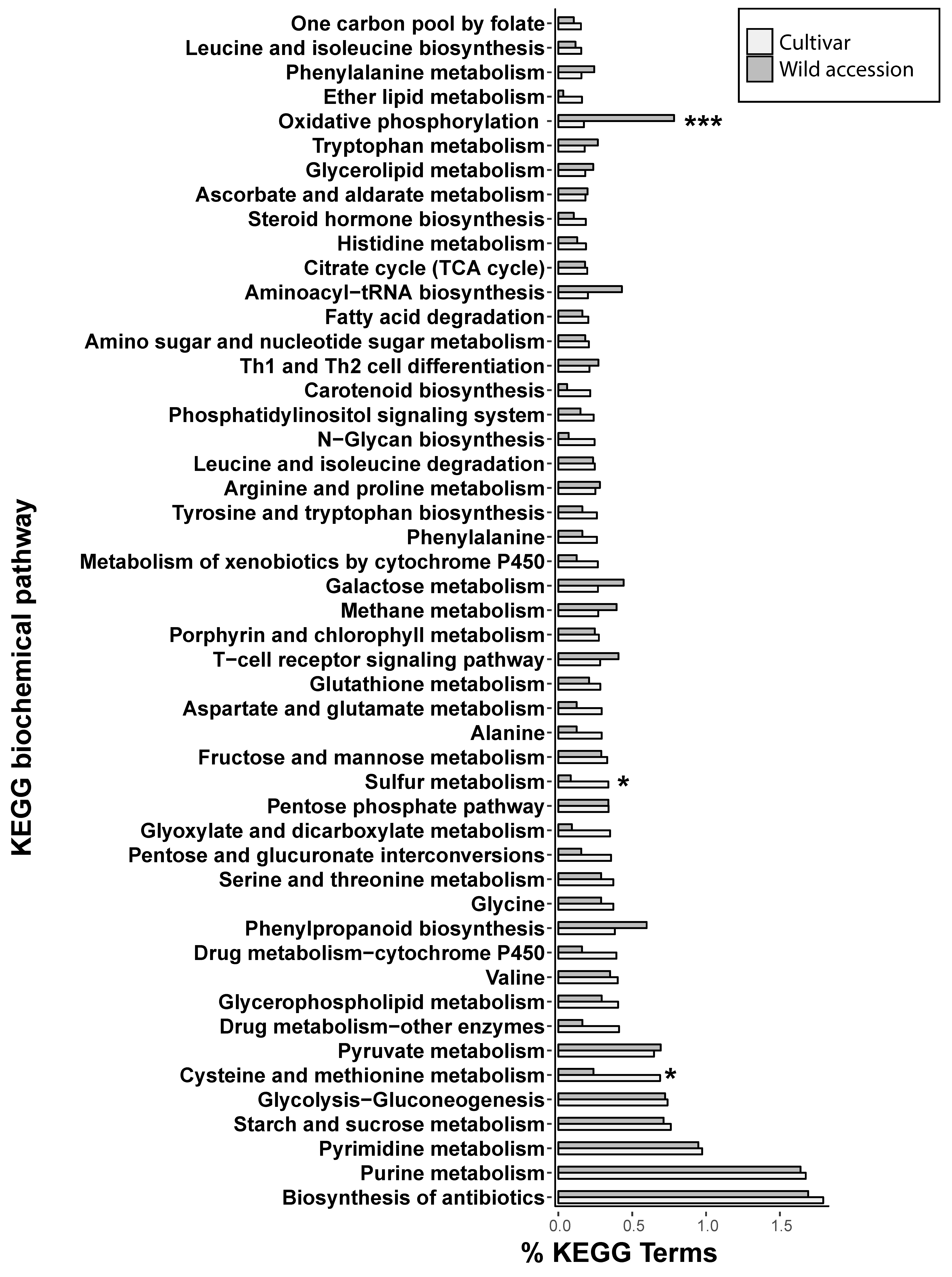
2.4. Analysis of Proximity of Long Terminal Repeats-Retrotransposons to Genes
3. Results
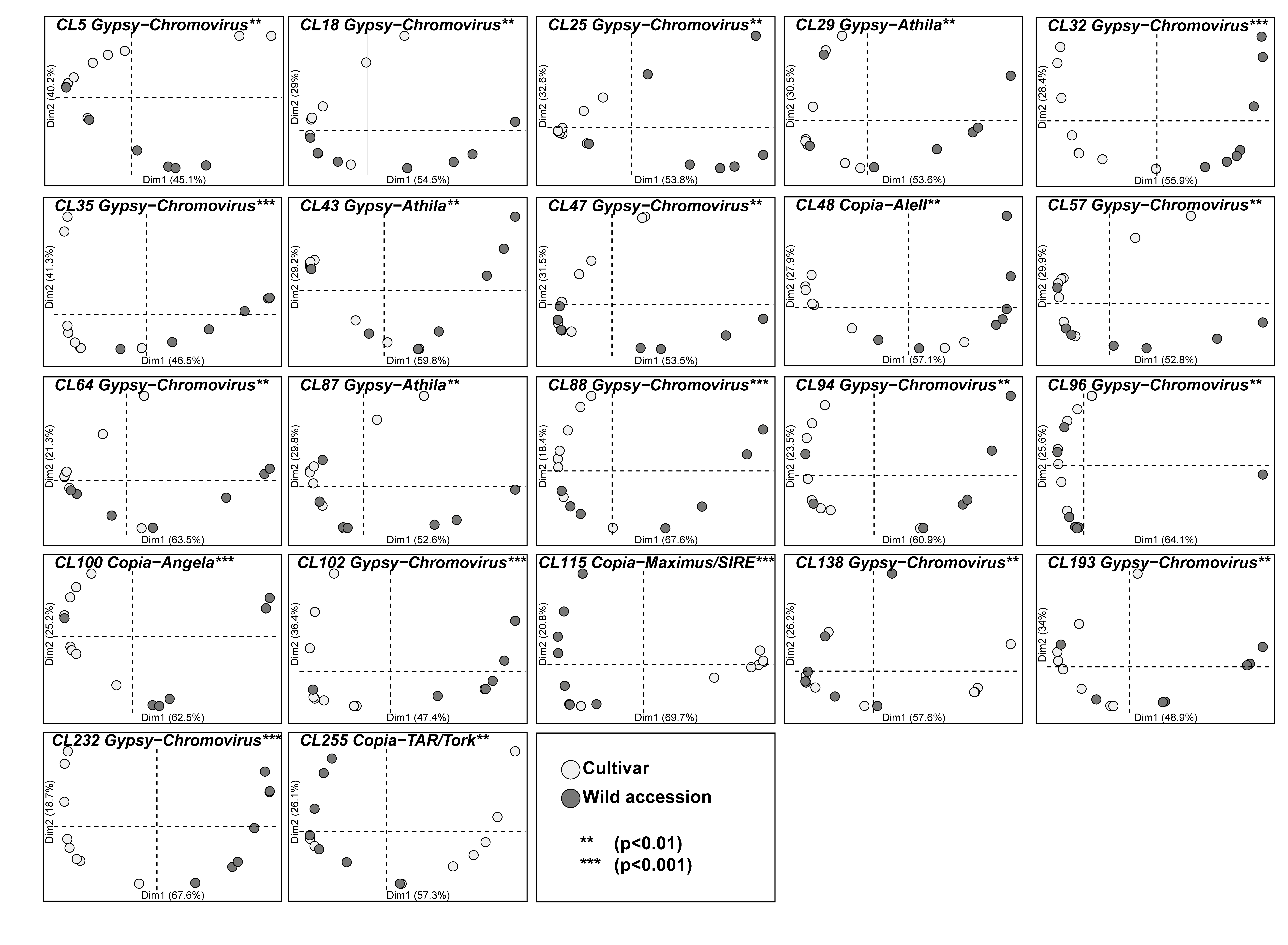
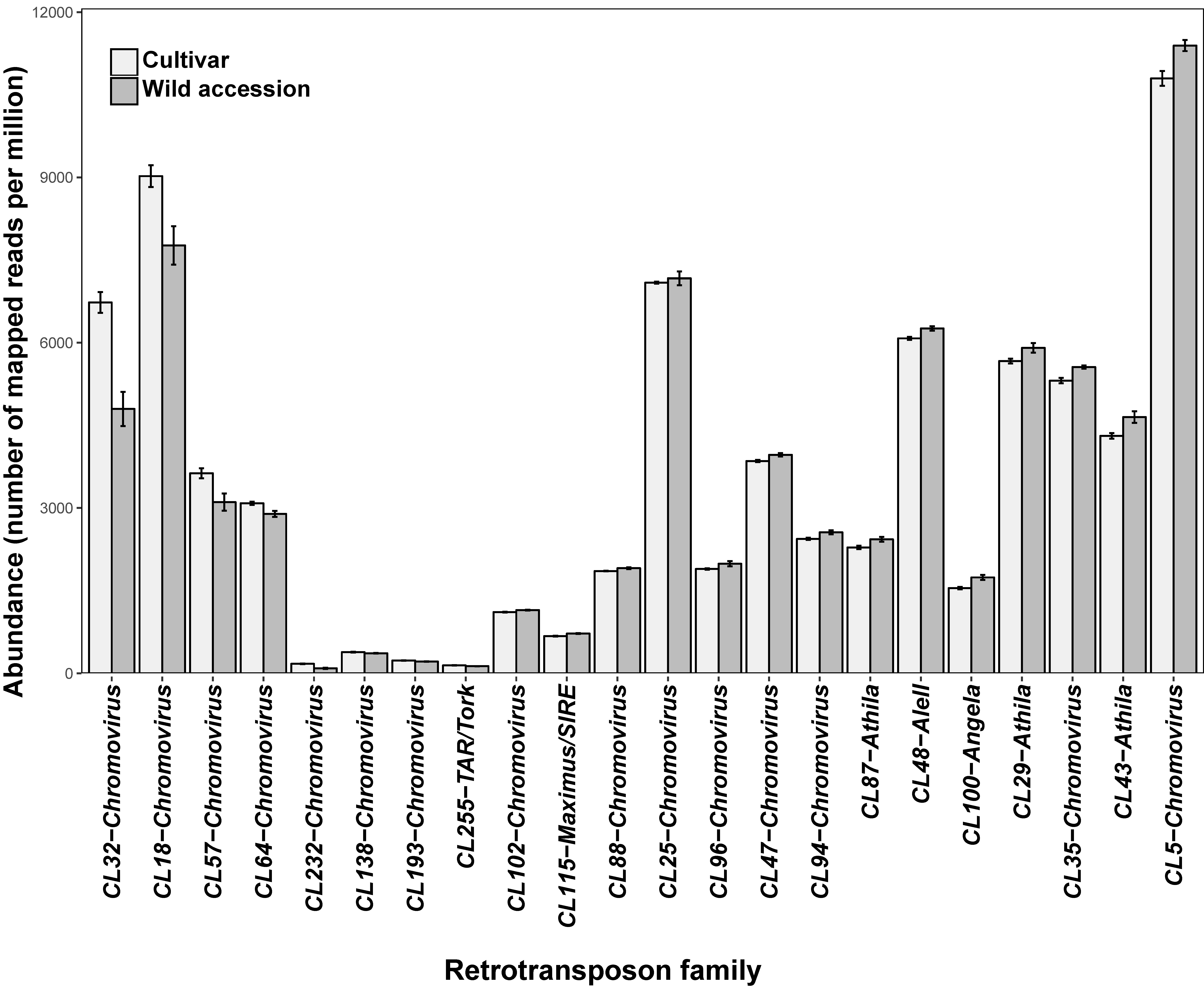
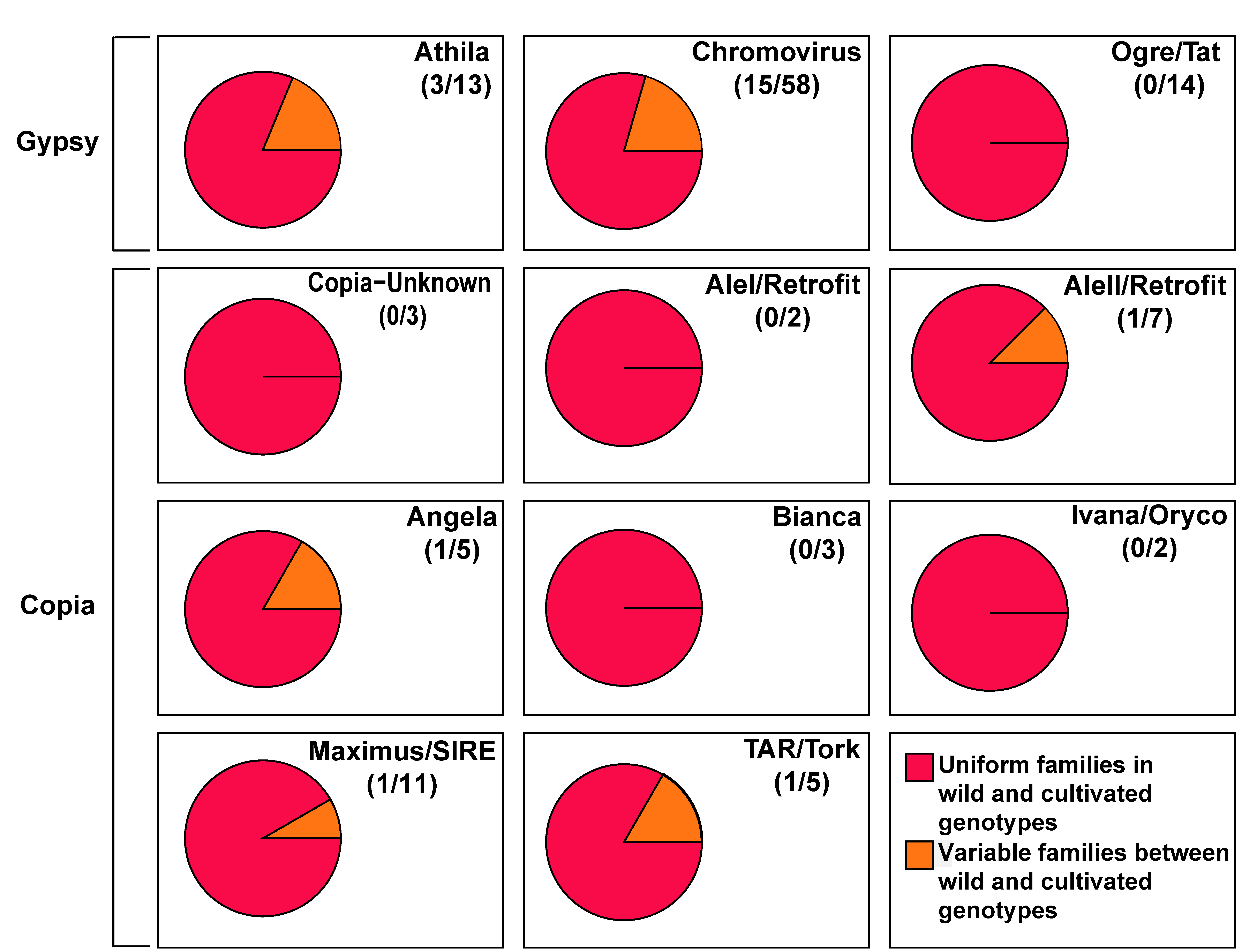
3.1. Some Long Terminal Repeats-Retrotransposon Families Show Significant Differences in Abundance between Wild and Cultivated Genotypes
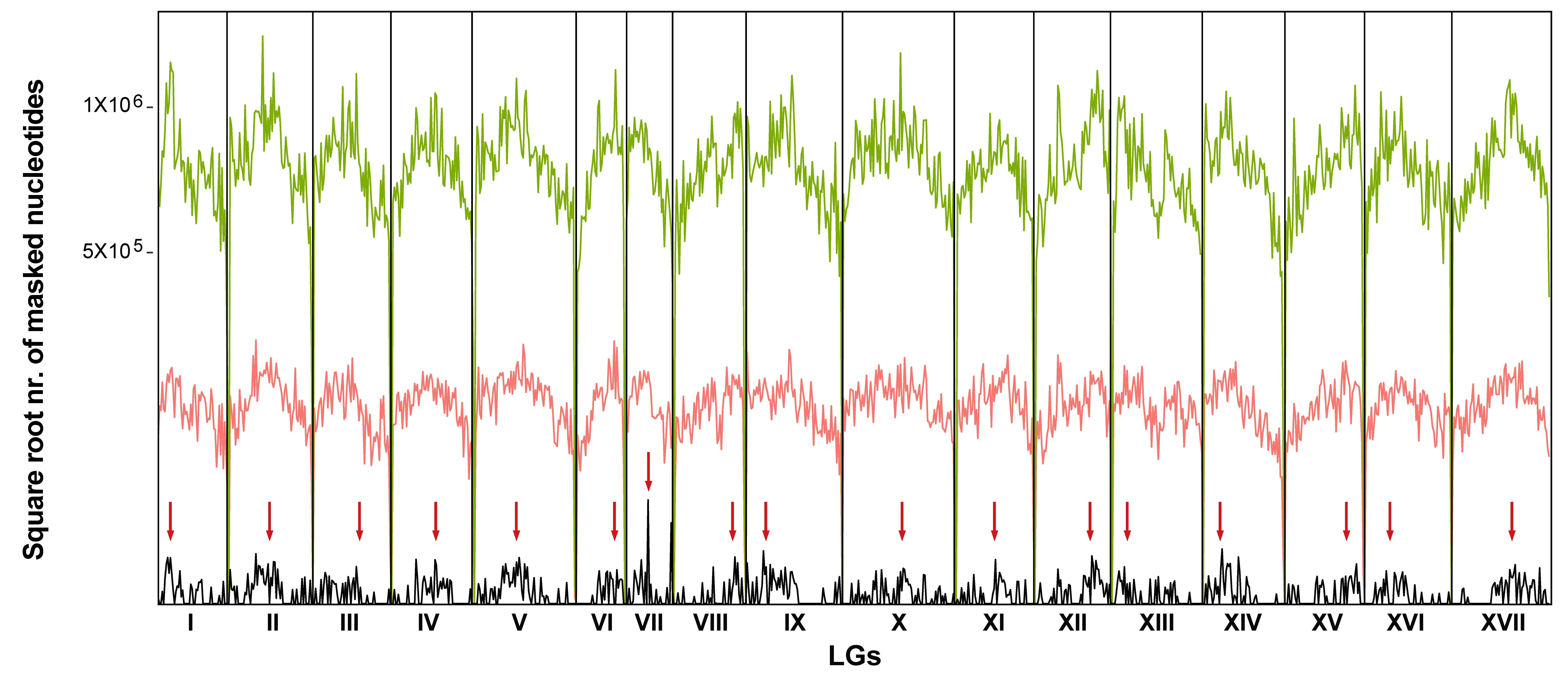
3.2. Chromosomal Localization of Long Terminal Repeats-Retrotransposons Families
3.3. Proximity of Retrotransposons to Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Llorens, C.; Futami, R.; Covelli, L.; Domínguez-Escribá, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodríguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucl. Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Cossu, R.M.; Mascagni, F.; Giordani, T.; Cavallini, A. A survey of Gypsy and Copia LTR-retrotransposon superfamilies and lineages and their distinct dynamics in the Populustrichocarpa (L.) genome. Tree Genet. Genomes 2015, 11, 107. [Google Scholar] [CrossRef]
- Vitte, C.; Bennetzen, J.L. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc. Natl. Acad. Sci. USA 2006, 103, 17638–17643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Dooner, H.K. Remarkable variation in maize genome structure inferred from haplotype diversity at the bz locus. Proc. Natl. Acad. Sci. USA 2006, 103, 17644–17649. [Google Scholar] [CrossRef] [PubMed]
- Devos, K.M.; Brown, J.K.; Bennetzen, J.L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice (Oryza sativa L.). Mol. Biol. Evol. 2003, 20, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Brunner, S.; Fengler, K.; Morgante, M.; Tingey, S.; Rafalski, A. Evolution of DNA sequence nonhomologies among maize inbreds. Plant Cell 2005, 17, 343–360. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Luo, X.; Tian, F.; Li, K.; Zhu, Z.; Su, W.; Qian, X.; Fu, Y.; Wang, X.; Sun, C.; et al. Haplotype variation in structure and expression of a gene cluster associated with a quantitative trait locus for improved yield in rice. Genome Res. 2006, 16, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Dubin, M.J.; Mittelsten Scheid, O.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Biol. 2018, 42, 23–29. [Google Scholar] [CrossRef] [PubMed]
- VanDriel, R.; Fransz, P.F.; Verschure, P.J. The eukaryotic genome: A system regulated at different hierarchical levels. J. Cell Sci. 2003, 116, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sui, A.; Garen, A. Binding of mouse VL30 retrotransposon RNA to PSF protein induces genes repressed by PSF: Effects on steroidogenesis and oncogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Fustier, M.A.; Alix, K.; Tenaillon, M.I. The bright side of transposons in crop evolution. Brief. Funct. Genom. 2014, 13, 276–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer, N.M.; Ying, K.; Fu, Y.; Ji, T.; Yeh, C.T.; Jia, Y.; Wu, W.; Richmond, T.; Kitzman, J.; Rosenbaum, H.; et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009, 5, e1000734. [Google Scholar] [CrossRef] [PubMed]
- Albert, P.S.; Gao, Z.; Danilova, T.V.; Birchler, J.A. Diversity of chromosomal karyotypes in maize and its relatives. Cytogenet. Genome Res. 2010, 129, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Cho, E.; Yang, G.; Campbell, M.A.; Yano, K.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Dramatic amplification of a rice transposable element during recent domestication. Proc. Natl. Acad. Sci. USA 2006, 103, 17620–17625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascagni, F.; Barghini, E.; Giordani, T.; Rieseberg, L.H.; Cavallini, A.; Natali, L. Repetitive DNA and plant domestication: Variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biol. Evol. 2015, 7, 3368–3382. [Google Scholar] [CrossRef] [PubMed]
- Schilling, E.E. Phylogenetic analysis of Helianthus (Asteraceae) based on chloroplast DNA restriction site data. Theor. Appl. Genet. 1997, 94, 925–933. [Google Scholar] [CrossRef]
- Schilling, E.E.; Linder, C.R.; Noyes, R.D.; Rieseberg, L.H. Phylogenetic relationships in Helianthus (Asteraceae) based on nuclear ribosomal DNA internal transcribed spacer region sequence data. Syst Bot. 1998, 23, 177–187. [Google Scholar] [CrossRef]
- Lentz, D.L.; Pohl, M.D.; Alvarado, J.L.; Tarighat, S.; Bye, R. Sunflower (Helianthus annuus L.) as a pre-Columbian domesticate in Mexico. Proc. Natl. Acad. Sci. USA 2008, 105, 6232–6237. [Google Scholar] [CrossRef] [PubMed]
- Harter, A.V.; Gardner, K.A.; Falush, D.; Lentz, D.L.; Bye, R.A.; Rieseberg, L.H. Origin of extant domesticated sunflowers in eastern North America. Nature 2004, 430, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Blackman, B.K.; Scascitelli, M.; Kane, N.C.; Luton, H.H.; Rasmussen, D.A.; Bye, R.A.; Lentz, D.L.; Rieseberg, L.H. Sunflower domestication alleles support single domestication center in eastern North America. Proc. Natl. Acad. Sci. USA 2011, 108, 14360–14365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zukovsky, P.M. Cultivated Plants and Their Wild Relatives; Commonwealth Agriculture Bureau: Farnham Royal, UK, 1950. [Google Scholar]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Olsen, K.M.; Wendel, J.F. A bountiful harvest: Genomic insights into crop domestication phenotypes. Ann. Rev. Plant Biol. 2013, 64, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Semelczi-Kovacs, A. Acclimatization and dissemination of the sunflower in Europe. Acta Ethnogr. Acad. Sci. Hung. 1975, 24, 47–88. [Google Scholar]
- Korell, M.; Mosges, G.; Friedt, W. Construction of a sunflower pedigree map. Helia 1992, 15, 7–16. [Google Scholar]
- Burke, J.M.; Tang, S.; Knapp, S.J.; Rieseberg, L.H. Genetic analysis of sunflower domestication. Genetics 2002, 161, 1257–1267. [Google Scholar] [PubMed]
- Rogers, C.; Thompson, T.; Seiler, G.J. Sunflower Species of the United States; National Sunflower Association: Bismarck, ND, USA, 1982. [Google Scholar]
- Leclercq, P. Une stérilité male cytoplasmique chez le tournesol. Annales de l’Amelioration des Plantes 1969, 19, 99–106. (In French) [Google Scholar]
- Blackman, B.K.; Rasmussen, D.A.; Strasburg, J.L.; Raduski, A.R.; Burke, J.M.; Knapp, S.J.; Michaels, S.D.; Rieseberg, L.H. Contributions of flowering time genes to sunflower domestication and improvement. Genetics 2011, 187, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Burke, J.M. Evidence of selection on fatty acid biosynthetic genes during the evolution of cultivated sunflower. Theor. Appl. Genet. 2012, 125, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.R.; Nambeesan, S.; Bowers, J.E.; Marek, L.F.; Ebert, D.; Rieseberg, L.H.; Knapp, S.J.; Burke, J.M. Association mapping and the genomic consequences of selection in sunflower. PLoS Genet. 2013, 9, e1003378. [Google Scholar] [CrossRef] [PubMed]
- Mandel, J.R.; McAssey, E.V.; Nambeesan, S.; Garcia-Navarro, E.; Burke, J.M. Molecular evolution of candidate genes for crop-related traits in sunflower (Helianthus annuus L.). PLoS ONE 2014, 9, e99620. [Google Scholar] [CrossRef] [PubMed]
- Baute, G.J.; Kane, N.C.; Grassa, C.; Lai, Z.; Rieseberg, L.H. Genome scans reveal candidate domestication and improvement genes in cultivated sunflower, as well as post-domestication introgression with wild relatives. New Phytol. 2015, 206, 830–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.C.R.; Tittes, S.; Mendieta, J.P.; Collier-zans, E.; Rowe, H.C.; Rieseberg, L.H.; Kane, N.C. Genetics of alternative splicing evolution during sunflower domestication. Proc. Natl. Acad. Sci. USA 2018, 115, 6768–6773. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Cavallini, A.; Natali, L.; Minelli, S.; Maggini, F.; Cionini, P.G. Ty1/copia-and Ty3/gypsy-like DNA sequences in Helianthus species. Chromosoma 2002, 111, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Santini, S.; Giordani, T.; Minelli, S.; Maestrini, P.; Cionini, P.G.; Cavallini, A. Distribution of Ty3-gypsy- and Ty1-copia-like DNA sequences in the genus Helianthus and other Asteraceae. Genome 2006, 49, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Natali, L.; Cossu, R.M.; Barghini, E.; Giordani, T.; Buti, M.; Mascagni, F.; Morgante, M.; Gill, N.; Kane, N.C.; Rieseberg, L.H.; et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genom. 2013, 14, 686. [Google Scholar] [CrossRef] [PubMed]
- Staton, S.E.; Bakken, B.H.; Blackman, B.K.; Chapman, M.A.; Kane, N.C.; Tang, S.; Ungerer, M.C.; Knapp, S.J.; Rieseberg, L.H.; Burke, J.M. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012, 72, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Giordani, T.; Cavallini, A.; Natali, L. The repetitive component of the sunflower genome. Curr. Plant Biol. 2014, 1, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Vukich, M.; Schulman, A.H.; Giordani, T.; Natali, L.; Kalendar, R.; Cavallini, A. Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor. Appl. Genet. 2009, 119, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.C.; Strakosh, S.C.; Stimpson, K.M. Proliferation of Ty3/Gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 2009, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Giordani, T.; Cattonaro, F.; Cossu, R.M.; Pistelli, L.; Vukich, M.; Morgante, M.; Cavallini, A.; Natali, L. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor. Appl. Genet. 2011, 123, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Vukich, M.; Giordani, T.; Natali, L.; Cavallini, A. Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 2009, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Macas, J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinform. 2010, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genom. 2017, 18, 634. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package ‘Vegan’, Community Ecology Package. R Package, version 2.0-10. 2013.
- Cavallini, A.; Natali, L.; Zuccolo, A.; Giordani, T.; Jurman, I.; Ferrillo, V.; Vitacolonna, N.; Sarri, V.; Cattonaro, F.; Ceccarelli, M.; et al. Analysis of transposons and repeat composition of the sunflower (Helianthus annuus L.) genome. Theor. Appl. Genet. 2010, 120, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.C.; Rieseberg, L.H. Genome-scale transcriptional analyses of first-generation interspecific sunflower hybrids reveals broad regulatory compatibility. BMC Genom. 2013, 14, 342. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, K.; Varala, K.; Hudson, M.E. Global repeat discovery and estimation of genomic copy number in a large, complex genome using a high-throughput 454 sequence survey. BMC Genom. 2007, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, M.I.; Hufford, M.B.; Gaut, B.S.; Ross-Ibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zealuxurians. Genome Biol. Evol. 2011, 3, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Barghini, E.; Natali, L.; Cossu, R.M.; Giordani, T.; Pindo, M.; Cattonaro, F.; Scalabrin, S.; Velasco, R.; Morgante, M.; Cavallini, A. The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol. Evol. 2014, 6, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Barghini, E.; Natali, L.; Giordani, T.; Cossu, R.M.; Scalabrin, S.; Cattonaro, F.; Šimková, H.; Vrána, J.; Doležel, J.; Morgante, M.; et al. LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Res. 2015, 22, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, F.; Cavallini, A.; Giordani, T.; Natali, L. Different histories of two highly variable LTR retrotransposons in sunflower species. Gene 2017, 634, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Navrátilová, A.; Koblížková, A.; Kejnovský, E.; Hřibová, E.; Hobza, R.; Widmer, A.; Doležel, J.; Macas, J. Plant centromeric retrotransposons: A structural and cytogenetic perspective. Mob. DNA 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.L.; Stec, A.; Hey, J.; Lukens, L.; Doebley, J. The limits of selection during maize domestication. Nature 1999, 398, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Gepts, P.; Papa, R. Evolution during domestication. In Encyclopedia of Life Sciences; Nature Publishing Group: London, UK, 2002. [Google Scholar]
- Olsen, K.M.; Purugganan, M.D. Molecular evidence on the origin and evolution of glutinous rice. Genetics 2002, 162, 941–950. [Google Scholar] [PubMed]
- Doebley, J. The genetics of maize evolution. Annu. Rev. Genet. 2004, 38, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Innan, H.; Kim, Y. Pattern of polymorphism after strong artificial selection in a domestication event. Proc. Natl. Acad. Sci. USA 2004, 101, 10667–10672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyre-Walker, A.; Gaut, R.L.; Hilton, H.; Feldman, D.L.; Gaut, B.S. Investigation of the bottleneck leading to the domestication of maize. Proc. Natl. Acad. Sci. USA 1998, 95, 4441–4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellucci, E.; Bitocchi, E.; Ferrarini, A.; Benazzo, A.; Biagetti, E.; Klie, S.; Minio, A.; Rau, D.; Rodriguez, M.; Panziera, A.; et al. Decreased nucleotide and expression diversity and modified coexpression patterns characterize domestication in the common bean. Plant Cell 2014, 26, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [PubMed]
- Falchi, R.; Vendramin, E.; Zanon, L.; Scalabrin, S.; Cipriani, G.; Verde, I.; Vizzotto, G.; Morgante, M. Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach. Plant J. 2013, 76, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.H.; Eubel, H.; Jansch, L.; Kruft, V.; Heazlewood, J.L.; Braun, H.P. Mitochondrial cytochrome c oxidase and succinate dehydrogenase complexes contain plant specific subunits. Plant Mol. Biol. 2004, 56, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. Biochemistry of sulfur-containing amino acids. Ann. Rev. Biochem. 1983, 52, 187–222. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Wachter, A. Sulfur metabolism: A versatile platform for launching defence operations. Trends Plant Sci. 2005, 10, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.X.; Wirtz, M.; Phua, S.Y.; Estavillo, G.M.; Pogson, B.J. Balancing metabolites in drought: The sulfur assimilation conundrum. Trends Plant Sci. 2013, 18, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Name | Id Code | Area of Cultivation | Raw Reads | Trimmed Reads (as Single Ends, 90 nt) | Trimmed Reads (as Paired Ends) |
|---|---|---|---|---|---|---|
| Domesticated | Hata | Ames 22503 | Argentina | 32,100,390 | 31,085,284 | 31,624,960 |
| Dussol | Ames 22499 | France | 25,678,406 | 24,988,640 | 25,375,912 | |
| Argentario | Ames 1842 | Italy | 10,759,866 | 10,134,402 | 10,566,652 | |
| Karlik | Ames 3454 | Spain | 23,499,752 | 22,938,364 | 23,087,458 | |
| Zelenka | Ames 22530 | Russia | 9,048,276 | 8,824,270 | 8,858,154 | |
| Roman “A” | PI531386 | Romania | 19,408,888 | 18,621,244 | 19,095,974 | |
| HOPI | PI369359 | USA | 15,768,198 | 15,254,502 | 15,437,790 | |
| Seneca | PI369360 | USA | 13,911,506 | 13,334,732 | 13,667,436 | |
| Wild | Arizona (AZ) | Ames14400 | - | 14,641,510 | 14,013,588 | 14,357,666 |
| Colorado (CO) | PI586840 | - | 23,335,576 | 21,965,694 | 22,916,284 | |
| Illinois (IL) | PI 435540 | - | 18,577,580 | 17,366,768 | 18,145,470 | |
| Kentucky (KY) | PI 435613 | - | 14,853,802 | 13,845,748 | 14,580,828 | |
| Mississippi (MS) | PI 435608 | - | 22,921,544 | 21,376,594 | 22,226,864 | |
| North Dakota (ND) | PI586811 | - | 51,681,332 | 47,906,352 | 49,574,892 | |
| Washington (WA) | PI 531018 | - | 6,996,658 | 6,479,624 | 6,724,410 |
| Superfamily | Lineage | Family | Mean nr. of Gene-RE Mapping Paired Reads per Million Reads | ||
|---|---|---|---|---|---|
| Cultivars | Wild accessions | PERMANOVA | |||
| Gypsy | Chromovirus | CL5 | 2.95 | 4.05 | |
| Gypsy | Chromovirus | CL18 | 1.61 | 1.64 | |
| Gypsy | Chromovirus | CL25 | 4.37 | 6.53 | * |
| Gypsy | Chromovirus | CL32 | 1.07 | 0.80 | |
| Gypsy | Chromovirus | CL35 | 0.80 | 0.90 | |
| Gypsy | Chromovirus | CL47 | 0.84 | 1.57 | *** |
| Gypsy | Chromovirus | CL57 | 0.73 | 1.04 | * |
| Gypsy | Chromovirus | CL64 | 0.47 | 0.65 | |
| Gypsy | Chromovirus | CL88 | 0.47 | 0.81 | * |
| Gypsy | Chromovirus | CL94 | 0.25 | 0.32 | |
| Gypsy | Chromovirus | CL96 | 0.76 | 0.82 | |
| Gypsy | Chromovirus | CL102 | 0.14 | 0.18 | |
| Gypsy | Chromovirus | CL138 | 0.17 | 0.14 | |
| Gypsy | Chromovirus | CL193 | 0.03 | 0.05 | |
| Gypsy | Chromovirus | CL232 | 0.03 | 0.01 | |
| Gypsy | Athila | CL29 | 1.16 | 1.34 | |
| Gypsy | Athila | CL43 | 1.31 | 1.90 | * |
| Gypsy | Athila | CL87 | 0.42 | 0.64 | * |
| Mean Gypsy | 0.98 | 1.38 | |||
| Copia | AleII | CL48 | 1.08 | 1.08 | |
| Copia | Maximus/SIRE | CL115 | 0.12 | 0.21 | |
| Copia | Angela | CL100 | 0.27 | 0.29 | |
| Copia | TAR/Tork | CL255 | 0.18 | 0.17 | |
| Mean Copia | 0.41 | 0.44 | |||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mascagni, F.; Vangelisti, A.; Giordani, T.; Cavallini, A.; Natali, L. Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes 2018, 9, 433. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090433
Mascagni F, Vangelisti A, Giordani T, Cavallini A, Natali L. Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes. 2018; 9(9):433. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090433
Chicago/Turabian StyleMascagni, Flavia, Alberto Vangelisti, Tommaso Giordani, Andrea Cavallini, and Lucia Natali. 2018. "Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers" Genes 9, no. 9: 433. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090433