Complemented Palindromic Small RNAs First Discovered from SARS Coronavirus
by
 ,
,
Chang Liu
1,† ,
,
Ze Chen
2,3,†,
Yue Hu
1,
Haishuo Ji
4,5,
Deshui Yu
4,
Wenyuan Shen
1,
Siyu Li
4,
Jishou Ruan
6,
Wenjun Bu
4,* and
Shan Gao
4,5,* 1
Laboratory of Medical Molecular Virology, School of Medicine, Nankai University, Tianjin 300071, China
2
State Key Laboratory of Veterinary Etiological Biology and Key Laboratory of Veterinary Parasitology of Gansu Province, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Science, Lanzhou 730046, China
3
Co-Innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou 225009, China
4
College of Life Sciences, Nankai University, Tianjin 300071, China
5
Institute of Statistics, Nankai University, Tianjin 300071, China
6
School of Mathematical Sciences, Nankai University, Tianjin 300071, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Genes 2018, 9(9), 442; https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090442
Submission received: 4 July 2018
/
Revised: 21 August 2018
/
Accepted: 22 August 2018
/
Published: 5 September 2018
(This article belongs to the Special Issue microRNA Regulation in Health and Disease)
Abstract
:In this study, we report for the first time the existence of complemented palindromic small RNAs (cpsRNAs) and propose that cpsRNAs and palindromic small RNAs (psRNAs) constitute a novel class of small RNAs. The first discovered 19-nt cpsRNA UUAACAAGCUUGUUAAAGA, named SARS-CoV-cpsR-19, was detected from a 22-bp DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA in the severe acute respiratory syndrome coronavirus (SARS-CoV) genome. The phylogenetic analysis supported that this DNA complemented palindrome originated from bat betacoronavirus. The results of RNA interference (RNAi) experiments showed that one 19-nt segment corresponding to SARS-CoV-cpsR-19 significantly induced cell apoptosis. Using this joint analysis of the molecular function and phylogeny, our results suggested that SARS-CoV-cpsR-19 could play a role in SARS-CoV infection or pathogenesis. The discovery of cpsRNAs has paved a way to find novel markers for pathogen detection and to reveal the mechanisms underlying infection or pathogenesis from a different point of view. Researchers can use cpsRNAs to study the infection or pathogenesis of pathogenic viruses when these viruses are not available. The discovery of psRNAs and cpsRNAs, as a novel class of small RNAs, also inspire researchers to investigate DNA palindromes and DNA complemented palindromes with lengths of psRNAs and cpsRNAs in viral genomes.
1. Introduction
Small RNA sequencing (small RNA-seq or sRNA-seq) is used to obtain thousands of short RNA sequences with lengths that are usually less than 50 bp. With sRNA-seq, many novel non-coding RNAs (ncRNAs) have been discovered. For example, two featured series of ribosomal RNA (rRNA)-derived RNA fragments (rRFs) constitute a novel class of small RNAs [1]. Small RNA-seq has also been used for virus detection in plants [2,3,4] and invertebrates [5]. In 2016, Wang et al. first used sRNA-seq data from the National Center for Biology Information Sequence Read Archive database (NCBI SRA) to show that sRNA-seq can be used to detect and identify human viruses [6], however the detection results were not as robust as those of plant or invertebrate viruses. To improve virus detection in mammals, our strategy was to detect and compare featured RNA fragments in plants, invertebrates, and mammals using sRNA-seq data. In a previous study [7], we detected siRNA duplexes that were induced by plant viruses and analyzed these small interfering RNA (siRNA) duplexes as an important class of featured RNA fragments. In this study, we aimed to investigate siRNA duplexes that were induced by invertebrate and mammalian viruses and unexpectedly discovered another important class of featured RNA fragments: complemented palindromic small RNAs (cpsRNAs). Among all of the detected cpsRNAs, the first discovered cpsRNA, named SARS-CoV-cpsR-19, from the severe acute respiratory syndrome coronavirus (SARS-CoV) strain MA15 merited further study, because mice infected with SARS-CoV MA15 died from an overwhelming viral infection with viral-mediated destruction of pneumocytes and ciliated epithelial cells [8]. SARS-CoV-cpsR-19 was detected from a DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA in the SARS-CoV genome. In our previous study of mitochondrial genomes, we reported palindromic small RNAs (psRNAs) for the first time [9]. Both of psRNAs and cpsRNAs could comprise a novel class of small RNAs due to their special sequence structures.
In this study, we compared the features of the siRNA duplexes that were induced by mammalian viruses with those that were induced by plant and invertebrate viruses. We found that the detected siRNA duplexes that were induced by mammalian viruses had significantly lower percentages of total sequenced reads than those that were induced by plant and invertebrate viruses, and it seemed that they were produced only from a few sites in the viral genomes. One possible reason could be that a large proportion of the sRNA-seq data is from other small RNA fragments, owing to the presence of a number of double-stranded RNA (dsRNA)-triggered nonspecific responses, such as type I interferon (IFN) synthesis [10]. Another possible reason could be that the missing siRNA duplexes or siRNA fragments function in cells by interacting with host RNAs or proteins. Based on this idea, we hypothesized that SARS-CoV-cpsR-19 or other cpsRNAs from SARS-CoV MA15 could play a role in SARS-CoV infection or pathogenesis.
To test our hypothesis, we conducted the joint analysis of the molecular function and phylogeny. First, we investigated the origins of SARS-CoV-cpsR-19 by the phylogenetic analysis of coronavirus genome sequences that were associated with bats, palm civets, rats, mice, monkeys, dogs, bovines, hedgehogs, giraffes, waterbucks, and equines. Subsequently, we performed RNA interference (RNAi) experiments to test the possible cellular effects that were induced by SARS-CoV-cpsR-19. The phylogenetic analysis supported that the DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA could originate from bat betacoronaviruses. The results of the RNAi experiments showed that one 19-nt segment corresponding to SARS-CoV-cpsR-19 significantly induced cell apoptosis. This study aimed to provide a different point of view for pathogen detection and pathogenesis studies.
2. Materials and Methods
2.1. Datasets and Data Analysis
All sRNA-seq data were downloaded from the NCBI SRA database. In our previous study, 6 mammalian viruses (HPV-18, HBV, HCV, HIV-1, SMRV, and EBV) were detected from 36 runs of sRNA-seq data [6]. In this study, 11 invertebrate viruses were detected from 51 runs of sRNA-seq data (Supplementary file 1) and 2 mammalian viruses (H1N1 and SARS-CoV) were detected from 20 runs of sRNA-seq data (NCBI SRA: SRP012018) [11]. In total, 11 invertebrate viruses and 8 mammalian viruses were detected from 107 runs of sRNA-seq data using VirusDetect [4], and their genome sequences were downloaded from the NCBI GenBank database. Among 107 runs of sRNA-seq data, four runs (NCBI SRA: SRR452404, SRR452406, SRR452408, and SRR452410) had been sequenced from lung tissue in mice that were infected with SARS-CoV MA15 [11] and they were used to detect SARS-CoV. The cleaning and quality control of the sRNA-seq data were performed using pipeline Fastq_clean [12], which was optimized to clean the raw reads from the Illumina platforms. Using the software Bowtie v0.12.7 [13] with one mismatch, we aligned all of the cleaned sRNA-seq reads to viral genome sequences and obtained alignment results in sequence alignment map (SAM) format for the detection of siRNA duplexes using the program duplexfinder [7]. Statistical computation and plotting were performed using the software R v2.15.3 (R Core Team, Vienna, Austria) with the Bioconductor packages [14]. The ORF3b gene from human betacoronavirus (GenBank: DQ497008.1), 20 homologous sequences from the bat betacoronaviruses, and nine homologous sequences from the civet betacoronaviruses (Supplementary file 1) were aligned using ClustalW2 [15] with curation. After the removal of the identical sequences, the ORF3b gene from human betacoronavirus, eight homologous sequences from bat betacoronaviruses, and two homologous sequences from civet betacoronaviruses were used for phylogenetic analysis. Since these homologous sequences had high identities (from 85.16% to 99.78%) to the ORF3b gene from DQ497008, the Neighbor Joining (NJ) method was used for phylogenetic analysis.
2.2. RNAi and Cellular Experiments
Based on the short hairpin RNA (shRNA) design protocol [1], the 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments from the DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA and their control “CGTACGCGGAATACTTCGA” were selected as the target sequences for pSIREN-RetroQ vector construction (Clontech, Mountain View, CA, USA). PC-9 cells were provided by Dr. Qingsong Wang from Tianjin Medical University and they were divided into six groups—namely, 16, 18, 19, 20, 22, and control—for transfection using plasmids containing the 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments and the control sequences. Each group had three replicate samples for plasmid transfection and cell apoptosis measurement. Each sample was processed following the procedure described below. The PC-9 cells were washed with phosphate buffer saline (PBS) and were trypsinized 12 h prior to transfection. Gbico RPMI-1640 medium (Thermo Fisher Scientific, Wattham, MA, USA) was added to the cells, which were then centrifuged at 1000 rpm for 10 min at 4 °C to remove the supernatant. Gbico RPMI-1640 medium containing 10% fetal bovine serum was added to adjust the solvent to reach a volume of 2 μL and contain 2 × 105 cells. These cells were seeded into one well of a 6-well plate for plasmid transfection. Transfection of 2 μg of plasmid was performed using 5 μL Lipofectamine 2000 (Life technology, Carlsbad, CA, USA), following the manufacturer’s instructions. Cell apoptosis was measured with the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, Franklin Lakes, NJ, USA), following the procedure described below. The cells were washed with PBS, were trypsinized, and were collected using a 5-mL culture tube 48 or 72 h after transfection. The culture tube was then centrifuged at 1000 rpm for 10 min at 4 °C to remove the supernatant. The cells were washed twice with cold PBS and were resuspended in 1X Binding Buffer at a concentration of 1 × 106 cells/mL. Then, 100 μL of the solution (1 × 105 cells) was transferred to a new culture tube with 5 μL of FITC-Annexin V and 5 μL PI. The cells were gently vortexed and were incubated for 15 min at room temperature in the dark. 1X Binding Buffer (400 μL) was added to the tube. Finally, the sample was analyzed using a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) within 1 h. Apoptotic cells were quantified by summing the count of the early apoptotic cells (FITC-Annexin V+/PI−) and the late apoptotic cells (FITC-Annexin V+/PI+). To confirm the results using Annexin V/PI staining and detection, the expression levels of three cell-apoptosis marker genes (BAX, BCL2, and CASP3) were also measured by quantitatice PCR (qPCR) assays using 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments to produce siRNA duplexes by pSIREN-RetroQ plasmid transfection. The cells were collected 24 h after transfection. For each gene, three RNAi samples and three control samples were tested for relative quantification. After transfection, RNA extraction, complementary DNA (cDNA) synthesis, and cDNA amplification were performed following the same procedure described below. For each sample, total RNA was isolated using RNAiso Plus Reagent (TaKaRa Bio Inc., Kusatsu, Shiga, Japan) and the cDNA was synthesized by M-MuLV (New England Biolabs, Hitchin, UK). The cDNA product was amplified by qPCR (Eppendorf, Hamburg, Germany) using GAPDH as internal control and the qPCR reaction mixture using Real Time PCR Easy (Foregene, Chengdu, China) was incubated at 95 °C for 10 min, followed by 35 PCR cycles (10 s at 95 °C, 20 s at 55 °C, and 30 s at 72 °C for each cycle). The primers of five genes are listed in Table 1.
3. Results
3.1. Comparison of siRNA Duplexes Induced by Plant, Invertebrate, and Mammalian Viruses
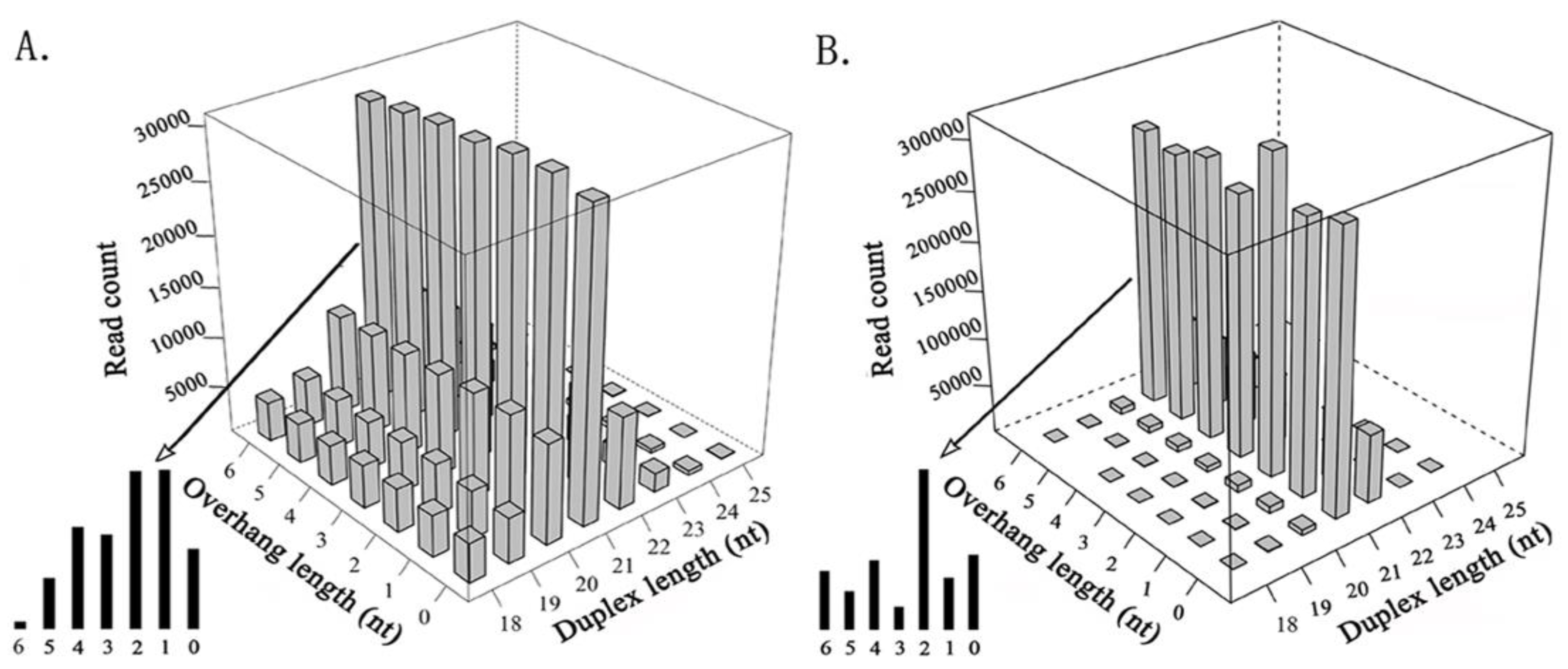
In total, 11 invertebrate and 8 mammalian viruses (HPV-18, HBV, HCV, HIV-1, SMRV, and EBV that were detected in the previous study, and H1N1 and SARS-CoV that were detected in this study) were used to detect siRNA duplexes (see Materials and Methods). Next, we compared the features of the siRNA duplexes that were induced by invertebrate viruses (Figure 1A) with those that were induced by plant viruses (Figure 1B). The results showed that duplex length was the principal factor that determined the read count in both plants and invertebrates. The 21-nt siRNA duplexes were the most abundant duplexes in both plants and invertebrates, followed by the 22-nt siRNA duplexes in plants, whereas it was the 20-nt siRNA duplexes in invertebrates. The 21-nt siRNA duplexes with 2-nt overhangs were the most abundant 21-nt duplexes in plants, whereas the 21-nt siRNA duplexes with 1-nt overhangs were the most abundant 21-nt duplexes in invertebrates, however they had a very similar read count to that of the 21-nt siRNA duplexes with 2-nt overhangs. The 18-nt, 19-nt, 20-nt, and 22-nt siRNA duplexes in invertebrates had much higher percentages of total sequenced reads than those in plants. In addition, 18-nt and 19-nt siRNA duplexes had very similar read counts, and the 20-nt and 22-nt siRNA duplexes had very similar read counts in invertebrates. As the siRNA duplexes that were induced by mammalian viruses had significantly lower percentages of total sequenced reads, comparison of the siRNA-duplex features between mammals and invertebrates or plants could not provide meaningful results using our data.
However, as an unexpected result of the comparison, we discovered cpsRNAs from both invertebrate and mammalian viruses. As this study was to focus on the study of cpsRNAs from SARS-CoV, we did not include cpsRNAs from other animal viruses. One 19-nt cpsRNA UUAACAAGCUUGUUAAAGA from a DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA (DQ497008: 25962-25983), located in the ORF3b gene of the SARS-CoV MA15 genome (GenBank: DQ497008.1), was detected in four runs of sRNA-seq data (see Materials and Methods). This 19-nt cpsRNA was named SARS-CoV-cpsR-19 and the DNA complemented palindrome contained 22 nucleotides which perfectly matched its reverse complement sequence. From this DNA complemented palindrome, we also detected one 18-nt and one 21-nt cpsRNA named SARS-CoV-cpsR-18 and SARS-CoV-cpsR-21 (Figure 2A), respectively. however we did not detect cpsRNAs of other lengths (e.g., 16-nt, 20-nt, or 22-nt) using our data. We speculated that 18-nt, 19-nt, and one 21-nt cpsRNA could be derived from siRNA duplexes (Figure 2B), as the presence of these overhanging nucleotides is a hallmark of small RNAs that are produced by silencing-related ribonucleases (e.g., Drosha or Dicer). However, we did not have any evidence to prove the existence of siRNA duplexes.
3.2. Discovery of psRNAs and cpsRNAs
Palindromes have been discovered in the published genomes of most species and play important roles in biological processes. The well-known samples of DNA palindromes include restriction enzyme sites, methylation sites, and palindromic motifs in T cell receptors [16]. In this study, we classified DNA palindromes into DNA palindromes and DNA complemented palindromes, from which psRNAs and cpsRNAs could be produced, respectively. A DNA palindrome is classically defined as a nucleic acid sequence that is reverse complementary to itself, while small RNAs that are reverse complementary to themselves are defined as cpsRNAs in this study. Accordingly, a typical psRNA should have a sequence that is 100% identical to its reverse sequence, however most psRNAs are semipalindromic or heteropalindromic, such as hsa-tiR-MDL1-16 AAAGACACCCCCCACA from a DNA palindrome AAAGACACCCCCCACAGTTT (NC_012920: 561-580) [9]. As a heteropalindromic psRNA, hsa-tiR-MDL1-16 is derived by cleavage after it starts transcription of the long heavy strand (H-strand) primary transcript at the position 561 of the human mitochondrial genome (Figure 2A). All cpsRNAs that are discovered from SARS-CoV MA15 are semipalindromic or heteropalindromic, such as SARS-CoV-cpsR-19 and SARS-CoV-cpsR-21. Although SARS-CoV-cpsR-21 contains almost 100% of the total nucleotides which contribute to the matches (Figure 2A), most cpsRNAs have mismatches or insertions/deletions (InDels). One example is a new Epstein-Barr virus (EBV) microRNA precursor (pre-miRNA) with a length of 89 nt, as was reported in our previous study [6]. This pre-miRNA sequence contains only 87.64% (78/89) of the total nucleotides which contribute to the matches.
In this study, we also found that DNA palindromes with sizes ranging from 14 to 31 nt and DNA complemented palindromes with sizes ranging from 14 to 53 nt existed ubiquitously in the animal virus genomes, however only a few of them were detected as transcribed or processed into psRNAs or cpsRNAs. For example, we only detected psRNAs from two (CTACTGACCAGTCATC and AAGGTCTCCCTCTGGAA) of 14 DNA palindromes and cpsRNA from two (GCAAATTGCACAATTTGC and TCTTTAACAAGCTTGTTAAAGA) of 29 DNA complemented palindromes (Table 2) in the SARS-CoV genome (GenBank: DQ497008.1) using four runs of sRNA-seq data (see Materials and Methods). One possible reason could be that only a few of cpsRNAs had been ligated to adapters during the sRNA-seq library preparation process as they could form hairpins (Figure 2A) at room temperature (~20 °C). From Table 2, it can be seen that the Tm (melting temperature) of cpsRNA hairpins in the SARS-CoV genome is distributed ranging from 14 °C to 26 °C. This finding provided a clue to explain why the detected siRNA duplexes that were induced by mammalian viruses had significantly lower percentages of total sequenced reads than those that were induced by plant and invertebrate viruses, and could help to improve the virus detection of mammalian viruses.
3.3. Clues to Origins of SARS-CoV-cpsR-19
The previously unknown SARS virus generated widespread panic in 2002 and 2003 when it caused 774 deaths and more than 8000 cases of illness. Scientists immediately suspected that civet cats, which are only distant relatives of house cats, may have been the springboard for the transmission of SARS-CoV to humans [17]. Later, scientists concluded that civets were not the original source of SARS. Further investigation showed that the genetic diversity of coronaviruses in bats increased the possibility of variants crossing the species barrier to cause disease outbreaks in the human population [18]. To investigate the origins of SARS-CoV-cpsR-19, we obtained coronavirus genome sequences that were associated with bats, palm civets, rats, mice, monkeys, dogs, bovines, hedgehogs, giraffes, waterbucks, and equines from the NCBI GenBank database. The results of the sequence analysis showed that the DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA was only located in the ORF3b genes of betacoronaviruses. Next, we blasted the ORF3b gene of human betacoronavirus (GenBank: DQ497008.1) with all of the obtained betacoronavirus genomes, except for those that were obtained from experiments with mice and monkeys. The results showed that the ORF3b gene from human betacoronavirus had homologous genes from the betacoronaviruses of bats and palm civets (Supplementary file 1) rather than from those of other species. The DNA complemented palindrome also had 22-nt homologous sequences in the bat and civet betacoronavirus genomes. All of the 22-nt homologous sequences in the civet betacoronavirus genomes were identical to the DNA complemented palindrome, whereas four genotypes of 22-nt homologous sequences were detected in the bat betacoronavirus genomes, however only one of them was identical to it (Figure 2C). Four genotypes had no, one, two, and three mismatches with the DNA complemented palindrome and their corresponding ORF3b homologous sequences had identities of 96.77%, 96.13%, 87.96%, and 85.16%, respectively. This suggested that one betacoronavirus variant containing the DNA complemented palindrome could have originated from bats and was then passed onto palm civets and finally to humans. This was consistent with the results of the phylogenetic analysis using the ORF3b homologous sequences from the bat and civet betacoronavirus genomes (Figure 2D). In the phylogenetic tree, all of the human and civet betacoronaviruses containing the DNA complemented palindrome were grouped into one clade. The nearest relative of the human and civet clade was the bat betacoronavirus (GenBank: JX993988.1) containing the DNA complemented palindrome, and the next nearest relative was the bat betacoronavirus (GenBank: DQ412042.1) containing a homologous 22-nt sequence with one mismatch with the DNA complemented palindrome.
3.4. Preliminary Studies on Biological Functions of SARS-CoV-cpsR-19
Our previous study showed that the psRNA hsa-tiR-MDL1-16 contained the Transcription Initiation Site (TIS) of the human mitochondrial H-strand and could be involved in mtDNA transcription regulation [9]. This inspired us to speculate that cpsRNAs could also have specific biological functions and we investigated SARS-CoV-cpsR-19 using RNAi and Annexin V/PI staining and detection. 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments from the DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA were used to produce siRNA duplexes by pSIREN-RetroQ plasmid transfection (see Materials and Methods). As a result, the 19-nt and 20-nt segments significantly induced cell apoptosis 2.76- and 1.48-fold 48 h after their transfection, respectively. Particularly, the 19-nt segment significantly induced cell apoptosis 7.94-fold (36.04%/4.54%) 72 h after its transfection into PC-9 cells (Figure 3). Using the 19-nt segment, we also tested cell apoptosis in five other human cell lines and one mouse cell line. The results showed that the 19-nt segment significantly induced cell apoptosis in the A549, MCF-7, and H1299 cell lines, however it did not in the MB231, H520, and L929 (mouse) cell lines. The siRNA duplexes that were induced by the 19-nt segment could silence cell-specific transcripts to induce cell apoptosis through RNAi. These results suggested that SARS-CoV-cpsR-19 had significant biological functions and could play a role in SARS-CoV infection or pathogenesis.
To confirm the results using Annexin V/PI staining and detection, the expression levels of three cell-apoptosis marker genes (BAX, BCL2, and CASP3) were measured by qPCR assays using the same RNAi protocol in the Hela cell line (see Materials and Methods). The results showed 1.08-, 1.65-, 1.60-, 1.24-, and 1.30-fold increases in BAX/BCL2 ratios which were caused by 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments, respectively (Figure 3E). As a low BAX/BCL2 ratio (<1) and a high BAX/BCL2 ratio (>1) is against and in favor of cell apoptosis, it suggested that 18-nt and 19-nt segments significantly induced cell apoptosis. The RNAi using 18-nt, 19-nt, and 20-nt segments also caused significantly increased expression of CASP3 by 1.72, 2.58, and 1.89 fold changes in Hela cells. Therefore, the experimental results by qPCR assays were consistent with those using Annexin V/PI staining and detection. In addition, the expression levels of two novel long non-coding RNAs (lncRNAs) (MDL1 and MDL1AS) from mitochondrial genome were measured to investigate their expression changes that were caused by RNAi using 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments. MDL1 and MDL1AS were recently discovered [9] and were predicted to be markers which can indicate the activities of mitochondria and even the whole cells. The RNAi using 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt segments also caused increased relative expression levels of MDL1 by 1.28, 5.43, 1.58, 9.25, and 1.41 fold changes in Hela cells (Figure 3E). It suggested that qPCR of MDL1 produce higher sensitivities than that of BAX/BCL2 and CASP3 in the detection of cell apoptosis.
4. Conclusions and Discussion
SARS-CoV-cpsR-19 from the DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA (DQ497008: 25962-25983) is located in the ORF3b gene of the SARS-CoV MA15 genome. In one previous study, this 22-nt DNA palindrome was studied using two mathematics models (M0 and M1) to conclude that this DNA palindrome was quite unlikely to occur by chance [19]. These authors also found that the underrepresentation of 4-nt DNA palindromes existed in all of the coronaviruses and the significant underrepresentation of 6-nt DNA palindromes existed only in SARS but not in the other six coronaviruses. Then, they hypothesized that this avoidance of 6-nt DNA palindromes in the SARS genome would offer a protective effect on the virus, making it comparatively more difficult to be destroyed by some host-defence mechanisms (e.g., restriction enzymes). In this study, we unexpectedly discovered cpsRNAs from the DNA palindrome TCTTTAACAAGCTTGTTAAAGA and reported that SARS-CoV-cpsR-19 could play a role in SARS-CoV infection or pathogenesis. We hypothesized that cpsRNAs, as a novel class of small RNAs, were produced by some host-defence mechanisms (e.g., RNAi) and that they could interact with the immune system of the host cells to help virus survival or infection.
Another previous study reported that another sRNA AGGAACUGGCCCAGAAGCUUC in the SARS-CoV genome, named small viral RNA-N (svRNA-N), contributed to SARS-CoV pathogenesis [20]. SARS-CoV-cpsR-19 was detected in very low abundance using four runs of sRNA-seq data (see Materials and Methods) in this study but was not detected using the sRNA-seq data (NCBI SRA: SRP094035) in that study [20]. One possible reason could be that SARS-CoV-cpsR-19 can form one hairpin and the Tm (melting temperature) of this hairpin is about 20 °C. Therefore, most of SARS-CoV-cpsR-19 cannot be ligated to adapters during the sRNA-seq library preparation process at room temperature (~20 °C). As the optimal secondary structure of svRNA-N (Minimum Free Energy (MFE) = −1.7) is less stable than those of SARS-CoV-cpsR-19 (MFE = −2.6) and all the other possible cpsRNAs (Table 2), svRNA-N is more easily captured during the sRNA-seq library preparation process. SARS-CoV-cpsR-19 induced cell apoptosis, whereas svRNA-N caused pulmonary inflammation. In Vivo experiments revealed that the biogenesis pathway that was responsible for svRNA-N’s synthesis was not dependent on Dicer or Drosha as the canonical biogenesis of miRNAs and svRNA-N were able to silence mRNA expression by targeting its 3’ UTR. However, the mechanisms underlying infection or pathogenesis of both SARS-CoV-cpsR-19 and svRNA-N are still unknown.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4425/9/9/442/s1, Table S1: 51 runs of sRNA-seq data for invertebrate virus detection, Table S2: Homologous sequences of ORF3b.
Author Contributions
S.G. conceived this project. S.G. and W.B. supervised this project. S.G., Z.C., and D.Y. analyzed the data. H.J. and S.L. curated the sequences and prepared all of the figures, tables, and additional files. C.L., Y.H., and W.S. performed the experiments. S.G. drafted the main manuscript. S.G. and J.R. revised the manuscript. All of the authors have read and approved the manuscript.
Funding
This work was supported by grants from the National Key Research and Development Program of China (2016YFC0502304-03) to Defu Chen, Central Public-Interest Scientific Institution Basal Research Fund of Lanzhou Veterinary Research Institute of CAAS to Ze Chen, Natural Science Foundation of China (81472052) to Qingsong Wang, and Internationalization of Outstanding Postdoctoral Training Program from Tianjin government to Shan Gao.
Acknowledgments
We thank Dr. Qingsong Wang from Tianjin Medical University for her cell lines, Yangguang Han and Jinke Huang from TianJin KeYiJiaXin Technology Co., Ltd. for his professional guidance on the RNAi and cellular experiments. This manuscript has been released as a pre-print at https://www.biorxiv.org/content/early/2017/12/13/185876.
Conflicts of Interest
Non-financial competing interests are claimed in this study.
References
- Chen, Z.; Sun, Y.; Yang, X.; Wu, Z.; Guo, K.; Niu, X.; Wang, Q.; Ruan, J.; Bu, W.; Gao, S. Two featured series of rRNA-derived RNA fragments (rRFs) constitute a novel class of small RNAs. PLoS ONE 2017, 12, e0176458. [Google Scholar] [CrossRef] [PubMed]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Gao, S.; Hernandez, A.G.; Wechter, W.P.; Fei, Z.; Ling, K. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS ONE 2012, 7, e37127. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gao, S.; Chellappan, P.; Li, R.; Marco, G.; Dina, G.; Segundo, F.; Ling, K.; Jan, K.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Tassetto, M.; Kunitomi, M.; Andino, R. RNA Interference-Mediated Intrinsic Antiviral Immunity in Invertebrates; Springer: Berlin/Heidelberg, Germany, 2013; Volume 371, pp. 183–200. [Google Scholar]
- Wang, F.; Sun, Y.; Ruan, J.; Chen, R.; Chen, X.; Chen, C.; Kreuze, J.F.; Fei, Z.; Zhu, X.; Gao, S. Using small RNA deep sequencing to detect human viruses. BioMed Res. Int. 2016, 2016, 2596782. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Sun, Y.; Chen, Z.; Li, R.; Padmanabhan, C.; Ruan, J.; Kreuze, J.F.; Ling, K.; Fei, Z.; Gao, S. Using small RNA-seq data to detect siRNA duplexes induced by plant viruses. Genes 2017, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Deming, D.; Paddock, C.D.; Cheng, A.; Yount, B.; Vogel, L.; Herman, B.D.; Sheahan, T.; Heise, M.; Genrich, G.L. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Tian, X.; Chang, H.; Sun, Y.; Wu, Z.; Cheng, Z.; Dong, P.; Zhao, Q.; Ruan, J.; Bu, W. Two novel lncRNAs discovered in human mitochondrial DNA using PacBio full-length transcriptome data. Mitochondrion 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Weber, K.; Tuschl, T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002, 26, 199–213. [Google Scholar] [CrossRef]
- Peng, X.; Gralinski, L.; Ferris, M.T.; Frieman, M.B.; Thomas, M.J.; Proll, S.; Korth, M.J.; Tisoncik, J.R.; Heise, M.; Luo, S. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. Mbio 2011, 2, e00198-11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhan, F.; Sun, H.; Gong, X.; Fei, Z.; Gao, S. Fastq_clean: An optimized pipeline to clean the Illumina sequencing data with quality control. In Proceedings of the 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Belfast, UK, 2–5 November 2014. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Ou, J.; Xiao, K. R Language and Bioconductor in Bioinformatics Applications, Chinese ed.; Tianjin Science and Technology Translation Publishing Co., Ltd.: Tianjin, China, 2014. [Google Scholar]
- Clustal Omega. Available online: http://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 18th May 2014).
- Srivastava, S.K.; Robins, H.S. Palindromic nucleotide analysis in human T cell receptor rearrangements. PLoS ONE 2012, 7, e52250. [Google Scholar] [CrossRef] [PubMed]
- Enserink, M. Clues to the animal origins of SARS. Science 2003, 300, 1351. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Chew, D.S.H.; Choi, K.P.; Heidner, H.; Leung, M.Y. Palindromes in SARS and other coronaviruses. Informs J. Comput. 2004, 16, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Morales, L.; Oliveros, J.C.; Fernandez-Delgado, R.; Tenoever, B.R.; Enjuanes, L.; Sola, I. SARS-CoV-encoded small RNAs contribute to infection-associated lung pathology. Cell Host Microbe 2017, 21, 344–355. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison of small interfering RNA (siRNA) duplexes that were induced by plant and invertebrate viruses. All of the cleaned small RNA sequencing (sRNA-seq) reads were aligned to viral genome sequences using the software Bowtie v0.12.7 [13] with one mismatch. The detection of siRNA duplexes was performed using the program duplexfinder [7]. (A). The read count of siRNA duplexes varies with the duplex length and the overhang length, using data from 11 invertebrate viral genomes. (B). The read count of siRNA duplexes varies with the duplex length and the overhang length, using data from seven plant viral genomes [7].
Figure 1.
Comparison of small interfering RNA (siRNA) duplexes that were induced by plant and invertebrate viruses. All of the cleaned small RNA sequencing (sRNA-seq) reads were aligned to viral genome sequences using the software Bowtie v0.12.7 [13] with one mismatch. The detection of siRNA duplexes was performed using the program duplexfinder [7]. (A). The read count of siRNA duplexes varies with the duplex length and the overhang length, using data from 11 invertebrate viral genomes. (B). The read count of siRNA duplexes varies with the duplex length and the overhang length, using data from seven plant viral genomes [7].

Figure 2.
Clues to the origins of SARS-CoV-cpsR-19. All of the genome sequences are represented by their GenBank accession numbers (e.g., DQ497008). (A). hsa-tiR-MDL1-16 is derived by cleavage after it starts transcription of the long H-strand primary transcript at the position 561 of human mitochondrial genome (left). SARS-CoV-cpsR-21 can form a hairpin (right). (B). 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt siRNA duplexes were used for RNA interference (RNAi) experiments. * 16-nt, 20-nt, and 22-nt palindromic small RNAs (cpsRNAs) were not detected in this study. (C). SARS-CoV-cpsR-19 was detected from a DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA in the SARS-CoV genome. All of the 22-nt homologous sequences in the civet betacoronavirus genomes were identical to the DNA complemented palindrome, whereas four genotypes of 22-nt homologous sequences were detected in the bat betacoronavirus genomes, however only one of them was identical to it. (D). The phylogenetic tree was built by the Neighbor Joining (NJ) method using the ORF3b gene from human betacoronavirus, eight homologous sequences from bat betacoronaviruses, and two homologous sequences from civet betacoronaviruses. The branch’s length corresponds to an average number of nucleotide changes per 100 nucleotides.
Figure 2.
Clues to the origins of SARS-CoV-cpsR-19. All of the genome sequences are represented by their GenBank accession numbers (e.g., DQ497008). (A). hsa-tiR-MDL1-16 is derived by cleavage after it starts transcription of the long H-strand primary transcript at the position 561 of human mitochondrial genome (left). SARS-CoV-cpsR-21 can form a hairpin (right). (B). 16-nt, 18-nt, 19-nt, 20-nt, and 22-nt siRNA duplexes were used for RNA interference (RNAi) experiments. * 16-nt, 20-nt, and 22-nt palindromic small RNAs (cpsRNAs) were not detected in this study. (C). SARS-CoV-cpsR-19 was detected from a DNA complemented palindrome TCTTTAACAAGCTTGTTAAAGA in the SARS-CoV genome. All of the 22-nt homologous sequences in the civet betacoronavirus genomes were identical to the DNA complemented palindrome, whereas four genotypes of 22-nt homologous sequences were detected in the bat betacoronavirus genomes, however only one of them was identical to it. (D). The phylogenetic tree was built by the Neighbor Joining (NJ) method using the ORF3b gene from human betacoronavirus, eight homologous sequences from bat betacoronaviruses, and two homologous sequences from civet betacoronaviruses. The branch’s length corresponds to an average number of nucleotide changes per 100 nucleotides.

Figure 3.
RNAi and cellular experiments for validation. PC-9 cells were divided into six groups named 16, 18, 19, 20, 22, and control for transfection using plasmids containing 16-nt, 18-nt, 19-nt, 20-nt, 22-nt segments, and their controls (Figure 2B). Each group had three replicate samples for plasmid transfection and cell apoptosis measurement. The 19-nt and 20-nt segments significantly induced cell apoptosis, whereas the 16-nt, 18-nt, and 22-nt did not show significantly positive results. The samples in this figure were selected randomly from the control (A), 18 (B), 19 (C), and 20 (D) group. (E). The experimental results by qPCR assays were consistent with those using Annexin V/PI staining and detection. The primers of MDL1 were used to amplify both MDL1 and MDL1AS, as they are sense–antisense transcripts from the same loci [9] and are predicted to be markers which can indicate the activities of mitochondria and even the whole cells.
Figure 3.
RNAi and cellular experiments for validation. PC-9 cells were divided into six groups named 16, 18, 19, 20, 22, and control for transfection using plasmids containing 16-nt, 18-nt, 19-nt, 20-nt, 22-nt segments, and their controls (Figure 2B). Each group had three replicate samples for plasmid transfection and cell apoptosis measurement. The 19-nt and 20-nt segments significantly induced cell apoptosis, whereas the 16-nt, 18-nt, and 22-nt did not show significantly positive results. The samples in this figure were selected randomly from the control (A), 18 (B), 19 (C), and 20 (D) group. (E). The experimental results by qPCR assays were consistent with those using Annexin V/PI staining and detection. The primers of MDL1 were used to amplify both MDL1 and MDL1AS, as they are sense–antisense transcripts from the same loci [9] and are predicted to be markers which can indicate the activities of mitochondria and even the whole cells.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Primers for quantitative PCR (qPCR) assays.
| Gene Symbol | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | ACATCGCTCAGACACCATG | TGTAGTTGAGGTCAATGAAGGG |
| BAX | AGTAACATGGAGCTGCAGAG | AGTAGAAAAGGGCGACAACC |
| BCL2 | GTGGATGACTGAGTACCTGAAC | GCCAGGAGAAATCAAACAGAGG |
| CASP3 | ACTGGACTGTGGCATTGAG | GAGCCATCCTTTGAATTTCGC |
| MDL1 * | CCCAATCCACATCAAAACCC | GGACGAGAAGGGATTTGACTG |
* The primers of MDL1 were used to amplify both MDL1 and MDL1AS as they are sense–antisense transcripts from the same loci [9] and were predicted to be markers which can indicate the activities of mitochondria and even the whole cells.
Table 2.
DNA complemented palindromes in the severe acute respiratory syndrome coronavirus (SARS-CoV) genome.
Table 2.
DNA complemented palindromes in the severe acute respiratory syndrome coronavirus (SARS-CoV) genome.
| DNA Complemented Palindrome | Start | End | Length | GC% | Tm | MFE |
|---|---|---|---|---|---|---|
| GGTAACTATAAAGTTACC | 1783 | 1800 | 18 | 33 | 20 | −6.9 |
| AATGTGAGAATCACATT | 2779 | 2795 | 17 | 29 | 18 | −4.4 |
| AAGAAACTAAGTTTCTT | 3923 | 3939 | 17 | 24 | 18 | −3.5 |
| ATGGTAAGCTTTACCAT | 3971 | 3987 | 17 | 35 | 18 | −4.5 |
| AAATGCAAATCTGCATTT | 4234 | 4251 | 18 | 28 | 18 | −4.9 |
| ATATGTCTATGACATAT | 4949 | 4965 | 17 | 24 | 18 | −4.3 |
| CCTCATGTAAATCATGAGG | 5020 | 5038 | 19 | 42 | 22 | −9.0 |
| ATAACAATTGTTAT | 5207 | 5220 | 14 | 14 | 12 | −0.9 |
| ACTTCAACAGCTTGAAGT | 5241 | 5258 | 18 | 39 | 18 | −4.7 |
| ACTTCAAATTCATTTGAAGT | 6256 | 6275 | 20 | 25 | 20 | −5.5 |
| GTACTTTTACTAAAAGTAC | 6734 | 6752 | 19 | 26 | 20 | −4.6 |
| ATCTACCAGTGGTAGAT | 9189 | 9205 | 17 | 41 | 20 | −6.5 |
| TTACCTTCCAAGGTAA | 10,892 | 10,907 | 16 | 38 | 16 | −4.0 |
| CCACTTATTAAGTGG | 14,160 | 14,174 | 15 | 40 | 18 | −4.7 |
| CCCATTTAATAAATGGG | 14,882 | 14,898 | 17 | 35 | 20 | −5.7 |
| CAGTGACAATGTCACTG | 16,463 | 16,479 | 17 | 47 | 22 | −7.7 |
| CACCTTTGAAAAAGGTG | 16,760 | 16,776 | 17 | 41 | 20 | −5.6 |
| TGTAAGAGAATTTCTTACA | 17,651 | 17,669 | 19 | 26 | 18 | −5.3 |
| TGAATATGACTATGTCATATTCA | 17,783 | 17,805 | 23 | 26 | 26 | −9.7 |
| CTACTTTAAGAAAGTAG | 20,081 | 20,097 | 17 | 29 | 18 | −3.6 |
| AGCATTCTTGGAATGCT | 21,106 | 21,122 | 17 | 41 | 20 | −6.1 |
| TTCCTCTTAAATTAAGAGGAA | 21,337 | 21,357 | 21 | 29 | 22 | −9.1 |
| GCATTACTACAGAAGTAATGC | 23,599 | 23,619 | 21 | 38 | 22 | −7.9 |
| AGCCCTTTATAAGGGCT | 25,480 | 25,496 | 17 | 47 | 22 | −8.7 |
| TCTTTAACAAGCTTGTTAAAGA * | 25,962 | 25,983 | 22 | 27 | 20 | −7.2 |
| CAACGGTACTATTACCGTTG | 26,406 | 26,425 | 20 | 45 | 24 | −9.0 |
| ACCTTCATGAAGGT | 28,048 | 28,061 | 14 | 43 | 14 | −2.6 |
| GCAAATTGCACAATTTGC * | 29,028 | 29,045 | 18 | 39 | 18 | −4.0 |
| TAAAATTAATTTTA | 29,668 | 29,681 | 14 | 0 | NA | NA |
In total, 29 DNA complemented palindromes were identified in the SARS-CoV genome (GenBank: DQ497008.1). * In this study, we only detected psRNAs from two of 29 DNA complemented palindromes. Minimum Free Energy (MFE) was calculated using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Tm (melting temperature) of cpsRNA hairpins was calculated by the formula: Tm = 4*(C+G) + 2*(A+T) and only using the nucleic acids in the stems of DNA complemented palindromes.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, C.; Chen, Z.; Hu, Y.; Ji, H.; Yu, D.; Shen, W.; Li, S.; Ruan, J.; Bu, W.; Gao, S. Complemented Palindromic Small RNAs First Discovered from SARS Coronavirus. Genes 2018, 9, 442. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090442
AMA Style
Liu C, Chen Z, Hu Y, Ji H, Yu D, Shen W, Li S, Ruan J, Bu W, Gao S. Complemented Palindromic Small RNAs First Discovered from SARS Coronavirus. Genes. 2018; 9(9):442. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090442
Chicago/Turabian StyleLiu, Chang, Ze Chen, Yue Hu, Haishuo Ji, Deshui Yu, Wenyuan Shen, Siyu Li, Jishou Ruan, Wenjun Bu, and Shan Gao. 2018. "Complemented Palindromic Small RNAs First Discovered from SARS Coronavirus" Genes 9, no. 9: 442. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9090442
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.