Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1)
by
 , ,
, ,
Mike Dyall-Smith
1,2 ,
,
Felicitas Pfeifer
3,
Angela Witte
4,
Dieter Oesterhelt
1 and
Friedhelm Pfeiffer
1,* 1
Computational Biology Group, Max-Planck-Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany
2
Veterinary Biosciences, Faculty of Veterinary and Agricultural Sciences, University of Melbourne, Parkville, VIC 3052, Australia
3
Department of Biology; Microbiology and Archaea, TU Darmstadt, Schnittspahnstrasse 10, 64287 Darmstadt, Germany
4
Department of Microbiology, Immunobiology and Genetics, MFPL Laboratories, University of Vienna, Dr. Bohr-Gasse 9, Vienna 1030, Austria
*
Author to whom correspondence should be addressed.
Genes 2018, 9(10), 493; https://0-doi-org.brum.beds.ac.uk/10.3390/genes9100493
Submission received: 11 September 2018
/
Revised: 5 October 2018
/
Accepted: 8 October 2018
/
Published: 12 October 2018
(This article belongs to the Special Issue Genetics of Halophilic Microorganisms)
Abstract
:The halophilic myohalovirus Halobacterium virus phiH (ΦH) was first described in 1982 and was isolated from a spontaneously lysed culture of Halobacterium salinarum strain R1. Until 1994, it was used extensively as a model to study the molecular genetics of haloarchaea, but only parts of the viral genome were sequenced during this period. Using Sanger sequencing combined with high-coverage Illumina sequencing, the full genome sequence of the major variant (phiH1) of this halovirus has been determined. The dsDNA genome is 58,072 bp in length and carries 97 protein-coding genes. We have integrated this information with the previously described transcription mapping data. PhiH could be classified into Myoviridae Type1, Cluster 4 based on capsid assembly and structural proteins (VIRFAM). The closest relative was Natrialba virus phiCh1 (φCh1), which shared 63% nucleotide identity and displayed a high level of gene synteny. This close relationship was supported by phylogenetic tree reconstructions. The complete sequence of this historically important virus will allow its inclusion in studies of comparative genomics and virus diversity.
Keywords:
halovirus; virus; halophage; Halobacterium salinarum; Archaea; haloarchaea; halobacteria; genome inversion1. Introduction
The temperate myovirus Halobacterium virus phiH (ΦH) infects the extremely halophilic archaeon Halobacterium salinarum strain R1 (DSM 671) and was isolated after the spontaneous lysis of a culture of its host [1]. Purified virions require 3.5 M NaCl for stability, have an isometric head of 64 nm diameter and a long, contractile tail (170 × 18 nm) with short tail fibres [1,2]. Virus preparations contain 3 major and 10 minor proteins [3]. The virus genome is linear dsDNA with a G+C content of 64%, contains a pac site, is about 3% terminally redundant and partially circularly permuted, and estimated to be 59 kb in length [4,5]. In the provirus state, the genome is extrachromosomal, covalently closed and circular, and 57 kb in length [4]. While always classified within the Myoviridae, the genus name has changed over the years from phiH-like viruses to Phihlikevirus, and most recently to Myohalovirus [6,7]. The species name itself has changed from Halobacterium phage phiH to Halobacterium virus phiH [6,7] but for convenience we will refer to it from here onwards simply as phiH, and the analysed variant as phiH1 or halovirus phiH1.
The original lysate of phiH was found to consist of a mixture of several distinct variants that appeared to have arisen from the activity of insertion sequences. The predominant variant, phiH1, was plaque-purified and a restriction map determined [5]. This was used for further study [3]. PhiH1 became a key model in the study of gene expression and regulation in haloarchaea and was instrumental in the development of genetic tools and methods in these extremophiles. Examples include the polyethylene glycol (PEG)-mediated transfection method [8], the pUBP1 cloning/expression vector [9], the identification of archaeal promoters, mapping transcription start and stop sites [10] and the analysis of gene regulation via repression [11,12]. The presence and function of antisense RNA in haloarchaea was first described in this virus [13]. An 11 kb invertible segment of the virus genome, called the L-region, was found to be flanked on one or both sides by the insertion sequence ISH1.8, and could also circularize to form a 12 kb plasmid (including one copy of ISH1.8), with subsequent loss of the remaining phage DNA [14]. A strain carrying this plasmid was immune to infection [14].
Unfortunately, work on phiH stopped in 1994 [15,16] but a related virus, Natrialba virus phiCh1 (φCh1), was described a few years later [17] and continues to be studied in the Witte laboratory [18,19]. PhiCh1 infects a haloalkaliphilic archaeon, Natrialba magadii, and the genomes of both host and virus are fully sequenced [18,20]. The provirus state of phiCh1 corresponds to plasmid pNMAG03 carried by Nab. magadii. A full comparison between phiCh1 and phiH1 was prevented as only parts of the phiH genome were ever determined. This deficit also prevented the inclusion of phiH in broad-ranging studies of virus diversity, taxonomy, and evolution. The aim of this study was to complete the phiH1 genome sequence and provide a thorough annotation. This will not only provide a better understanding of the results from previous studies on this virus but also allow complete genomic comparisons with a wealth of other datasets, including other sequenced viruses, haloarchaeal proviruses, metaviromic/metagenomic and environmental RNA sequences.
2. Materials and Methods
2.1. Virus DNA and Sequencing Methods
Purified phiH1 DNA [1] was originally provided to F. Pfeifer by Hans-Peter Klenk while both were working in the department of W. Zillig [21]. The DNA was stored frozen at −80 °C until use. Sequencing was performed in two stages. For the first stage, all available sequences of phiH1 were downloaded from National Center for Biotechnology Information (NCBI) [22] and imported into the Phred–Phrap–Consed package [23]. Overlapping sequences were assembled and primers designed to gather additional sequences using Sanger technology. This consisted either of primer-walking directly on virus DNA, or on polymerase chain reaction (PCR) amplimers, or PCR-sequencing across gaps. The resulting sequence reads were progressively assembled into contigs, base calls inspected manually and corrected where needed, and new primers designed for further rounds of sequencing until all gaps were closed. Except for overlaps, this approach left most of the previously published sequences unchecked.
In the second stage, short-read Illumina HiSeq sequencing of phiH1 DNA was performed (Max-Planck Genome Centre, Cologne, Germany). This returned 243 Mb of high quality sequence data (coverage = 4200-fold). De-novo assembly did not produce a single contig, due to short read-lengths and the presence of repeat sequences within the viral genome, but reads could be confidently mapped to the genome sequence obtained in the first stage (Map to Reference option; Geneious mapper method) in order to improve the sequence reliability.
2.2. CRISPR Spacer Searches
The crass v0.3.12 software [24] was used to extract CRISPR spacer sequences from genomic/metagenomic data available at the NCBI SRA database (accessed 27 July 2018) [25], as described previously [26]. These included all available genomes of members of the class Halobacteria, and metagenomes of hypersaline environments. CRISPR direct repeats (DR) identified by crass were used to search the CRISPRfinder database (accessed 25 July 2018) [27] for haloarchaea with matching or closely matching DR.
2.3. Bioinformatic Methods
Gene annotation used a combination of gene prediction with GeneMarkS-2 [28] and manual refinement using database searches (BLASTp/BLASTn; nr databases) at the NCBI webserver [29]. Repeats were identified by BLASTn, dot-plot comparison in Yass [30], and with tools within the Geneious software suite [31]. Circos plots were performed via the circoletto webserver [32]. Plots are coloured by the ‘score/max’ ratio of tBLASTx bitscores (real score/maximal score). Colours are: blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75, red > 0.75. Sequence mapping, alignments, editing and phylogenetic tree reconstructions were performed with Geneious software version 10.2 [31]. For phylogenetic tree reconstructions, protein sequences were first aligned using CLUSTALW, and trees inferred using the Neighbor-Joining algorithm (within Geneious). Consensus trees were determined after 100 bootstrap repetitions. Protein structural modelling used the I-Tasser webserver [33]. Identification of the pac site utilised the program PhageTerm [34] as implemented on the CPT Phage Galaxy [35]. The VIRFAM webserver [36] uses proteins of the phage head-neck-tail module to cluster phages into related groups, and was used to classify phiH1.
2.4. Data Availability
The phiH1 genome sequence has been deposited at Genbank under the accession MK002701. Raw reads were submitted to the SRA archive under accession SRP159490.
3. Results and Discussion
3.1. Sequence and Annotation of PhiH1
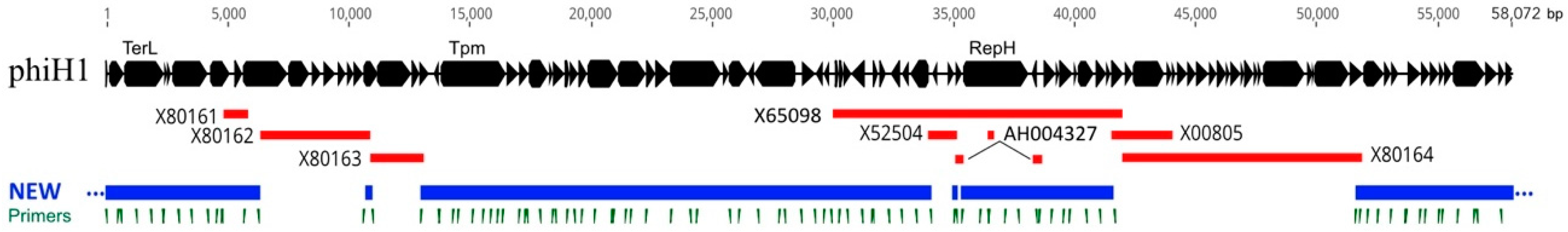
The previously sequenced regions of the phiH1 genome represented about 50% of the complete sequence (Figure 1, red lines). Using virus DNA as template, the gaps between these sequences were PCR amplified and Sanger sequenced. However, the quality of the previously sequenced regions was of uncertain reliability. High-coverage Illumina sequencing (ca. 4200-fold) was then used to enhance sequence confidence. Sequence revisions were only found to be required in previously deposited sequences but not to Sanger sequencing results of the first stage of the project. While the virus DNA found in capsid particles is linear, the head-full packaging process produces a population of molecules that are terminally redundant and partially circularly permuted [1]. The complete genome sequence determined in the current study is represented as the provirus form; a circular sequence of 58,072 bp. This value is close to the published size of 57 kb, estimated from restriction fragment sizes [4,37]. The G+C content of the genome was 63.7%, almost identical to the published value of 64% [3] but slightly lower than that of the host chromosome (68.0%) [38].
The original restriction map of phiH1 DNA, as determined by [5], corresponded closely with the in silico map inferred from the phiH1 genome sequence (Figure S1). The pac site located at the left end of the restriction map matched closely to the corresponding pac sequence of phiCh1. While the pac site of phiCh1 had been localized by restriction mapping [18], it had not been precisely mapped. For consistency, the start point of phiH1 was set to the corresponding start of phiCh1 even though this splits the terS gene. Using this numbering, the program PhageTerm [34] was used to analyse the mapping of Illumina reads to the phiH1 genome, and this located the pac site terminal base at nt 46, with high probability (p = 2.5 × 10−238). This is within the terS coding sequence (CDS) close to the stop codon and within a GC-rich region that is strongly conserved between phiH1 and phiCh1.
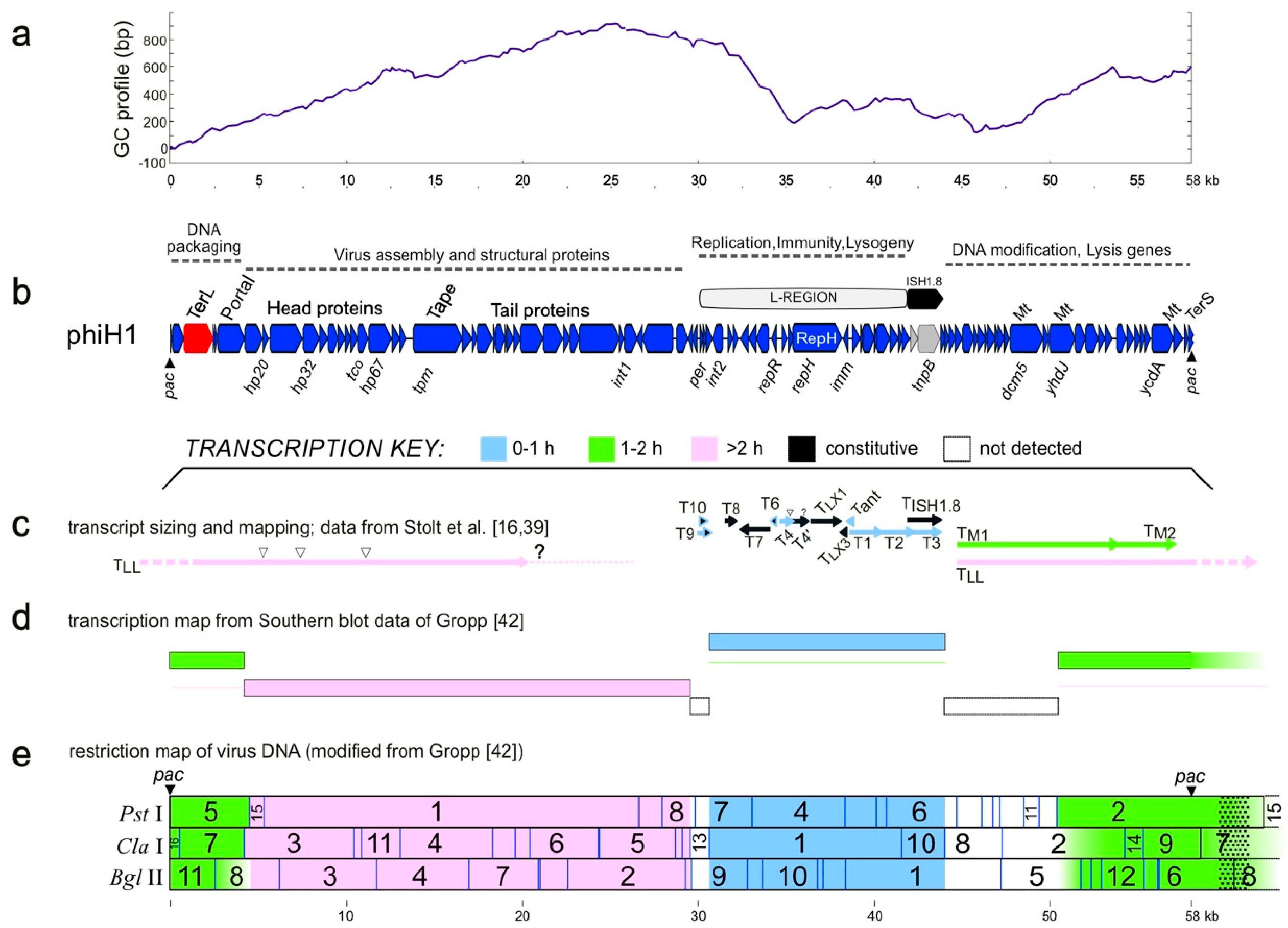
Annotation of the phiH1 genome resulted in 97 CDS (Table 1), most of which were encoded on the plus strand (86/97, Figure 2 panel b), and were frequently closely spaced, with 45 overlapping at start/stop codons and 23 separated by ≤8 nt. Many genes were in functional groupings typical of bacteriophages (Figure 2 panel b). The left end of the genome encodes DNA packaging proteins (e.g., terminase, portal protein), then virus assembly and structural proteins (e.g., major capsid protein, tape-measure protein, tail proteins). The three main proteins of purified virus were originally labelled by their estimated sizes on sodium dodecylsufate (SDS)-polyacrylamide gels (22, 53 and 80 kDa) [1], which were later revised to 27, 46 and 80 kDa [3] but in 1994, Stolt et al. [39] determined their N-terminal amino acid sequences and used this information to map the proteins (HP20, HP32 and HP67) to their genes (hp20, hp32, hp67) and sequence them. The inferred molecular weights (MWs) of proteins HP20 and HP67 were noted by these authors to be much smaller than previous estimates. In the present study, the locations of these genes on the full genome sequence have been resolved, an error in the hp20 (accession X80161) coding sequence was corrected, and the MWs of the inferred proteins calculated (11.6, 35.4 and 45.5 kDa). For consistency we have retained the original gene names (Table 1).
The next genomic region is a replication/regulatory module (the L-region) that encodes RepR (repressor), a ParA-family protein (partition) and RepH (replication). There is also a VapC-like protein that together with the small overlapping upstream CDS may form a toxin‒antitoxin pair that could be involved in plasmid maintenance [40]. The right end of the genome carries many genes with unknown function but includes genes specifying DNA methylases and cell lysis proteins. The taxonomic position of phiH1 was assessed using the VIRFAM webserver [36], which classifies bacteriophages and archaeal viruses based on the order and similarity of capsid assembly/structural proteins. Consistent with previous studies [6], phiH1 was classified by this system as a member of the Myoviridae (Type1, Cluster 4).
A GC-profile plot [41] of the phiH1 genome shows a major low point inflection within the L-region (Figure 2, panel a), indicating a potential replication origin. The L-region is ~12 kb in length, can replicate as a plasmid in Halobacterium [14], and carries genes encoding a replication protein (RepH), and a DNA-binding repressor (RepR). It can also provide cells with immunity to infection by phiH1 virus. The transcription program of phiH1 during lytic growth (panels c, d and e) has been summarized from previous studies, and shows temporal changes (early, middle and late transcripts). The broad directions of transcription reflect the closely spaced and similarly directed gene clusters as well as the correspondence with functional gene groupings (panel b). The lowest two panels (d, e) summarize the results of hybridizing labelled transcripts from infected cells to Southern blots of restricted phiH1 DNA [42], so mapping transcripts to fragments of the virus genome. Panel c shows a summary of the virus-specific transcripts that were sized by agarose gel electrophoresis and had 5′ start sites mapped. While transcription across the L-region has been examined in more detail compared to the rest of the genome, there remains much that is incomplete or uncertain. For example, the 3′ end of the late transcript labelled TLL, which is depicted ending in a dotted line and question mark (at ~21 kb), has not been determined. This transcript could potentially extend for another 5.5 kb. Counter-transcripts are commonly produced by prokaryotes and their viruses and play important roles in gene regulation. Their presence and activity in phiH1 gene expression has been studied and was one of the first reports of antisense RNA in Archaea [13]. However, this interesting topic remains to be fully explored.
Corrections to the previously sequenced regions resulted in significant changes to several coding sequences. For example, the tnpB gene of transposon ISH1.8 (nt 41,906–43,789) was thought to be inactive as it was split into three CDS by multiple mutations [43]. The high-quality Illumina sequence data show, however, that the gene is intact and that the previously reported transposon ISH1.8 (X00805) is actually an exact copy of transposon ISH12 from the host Hbt. salinarum strain R1 [44]. The element plays a key role in the mobilisation of the L-region of the genome to form the 12 kb plasmid, pΦHL [14,43]. Another case is the Dcm5 cytosine methylase, which was also reported as being split [39]. The revised sequence shows that the gene codes for a single, probably functional protein (PhiH1_405, nt 47,732–49,618) and not for the two parts (dcm5a, dcm5b) as previously reported. Although phiH1 carries three potentially active DNA methylase genes (dcm5, yhdJ and ycdA), the presence of modified bases in phiH1 DNA was not detected in the chromatographic (high-pressure liquid chromatography) profiles of deoxyribonucleosides released by enzymatic hydrolysis [45]. In that study, the genomes of phiH and another, unrelated virus (phiN) were analysed, and while unmodified dC was detected in phiH, the phiN genome contained only methylated dC (Figure 3 in [45]). The related halovirus phiCh1 carries homologs of two of the phiH1 methylases, and one of them, N6-adenine methylase (ORF94/M·φCh1-I, corresponding to YcdA of phiH1) has been shown to methylate DNA at GATC motifs [46] but the proportion of sites found to be modified in virus DNA by M·φCh1-I varies from 5% to 50%, depending upon the infection conditions. Modifying only some of the available sites is presumably advantageous to avoid host restriction, as distinct enzymes may target either unmethylated or methylated sites.
3.2. Matches to CRISPR Spacers
The phiH1 genome was used to search for matching CRISPR spacers among metagenomic datasets of hypersaline environments downloaded from the NCBI sequence read archive (SRA; see methods). Only four spacers showing close to moderate similarity to phiH1 were detected (Table 2). These spacers match to virus genes encoding structural and non-structural proteins, and the DRs of these spacers show that they are carried by haloarchaea. The datasets include metagenomes from the USA and Iran, as well as an isolate from the Andaman Islands, India. The results suggest that phiH1-like viruses are geographically widespread.
3.3. Relatives and Phylogeny of PhiH1
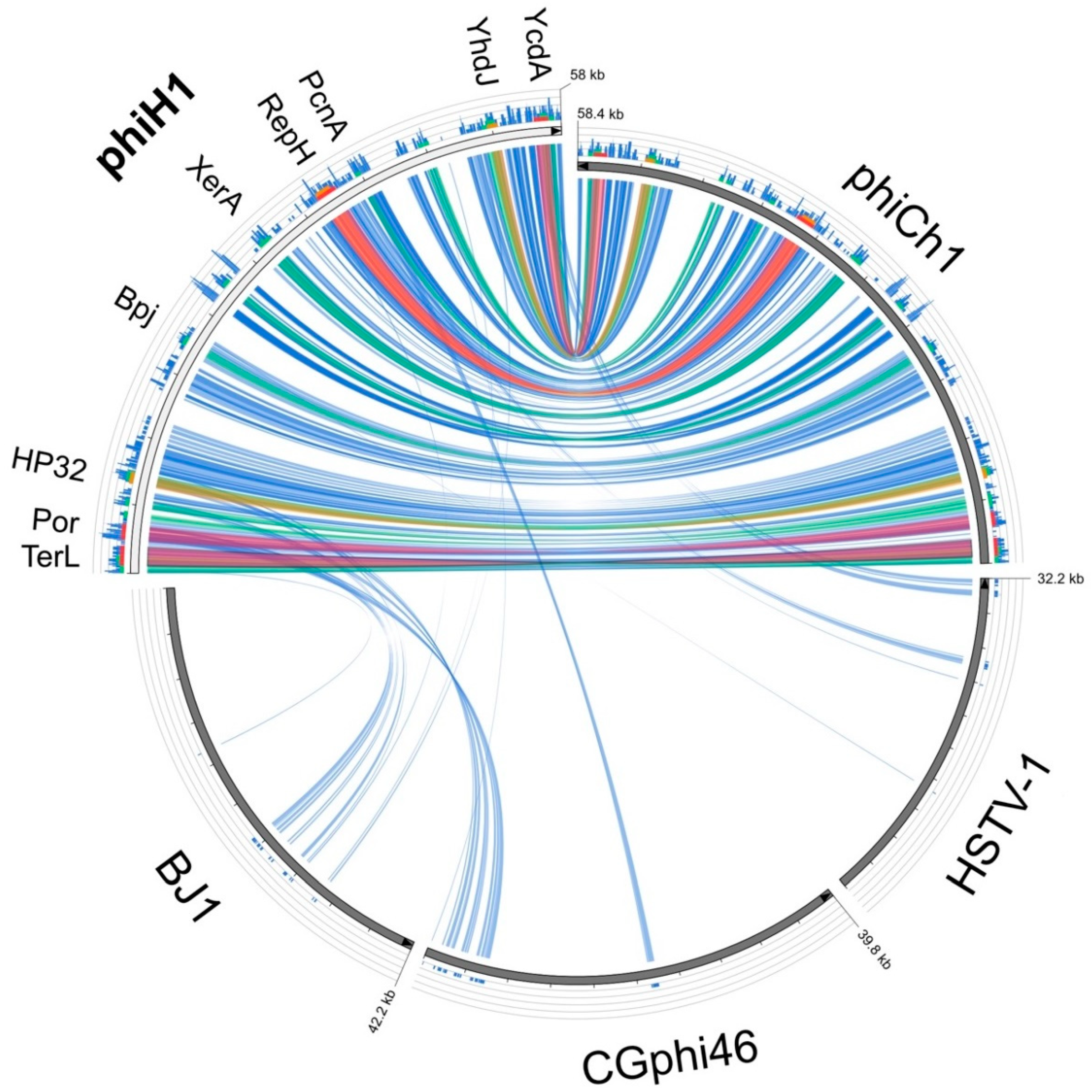
The only close matches to the phiH1 genome in the GenBank database were phiCh1 and the corresponding Nab. magadii plasmid pNMAG03 (BLASTn, accessed 20 July 2018). A dot-plot comparison of phiH with phiCh1 (Figure S2) revealed a largely colinear relationship (green line) and an overall nucleotide similarity of 63%. The plot also highlights several indels (line gaps) and two regions showing inversions (red lines). Inversion 1 (nt 24,227–27,767) corresponds closely in sequence and arrangement to the invertible region described in phiCh1 (ORF34-36) that has a central XerD type integrase/recombinase gene flanked by inverted repeats, and facilitates switching between two related tail fibre genes, each containing numerous short repeats [18]. The phiH1 orthologous integrase is PhiH1_175. In the current sequence version, PhiH1_165 is active while PhiH1_185 is uncoupled from a start codon and thus is inactivated. Upon inversion of the genome segment, PhiH1_185 is activated while PhiH1_165 is inactivated. Overall, this results in tail fibre protein switching, which may affect receptor binding specificity and host range of phiH1. The similarity in the tail fibre protein repeats is high enough to be detectable at the DNA level, which results in the X-shaped pattern for this region in the dot-plot. Inversion 2 (nt 31,932–34,126) occurs within the phiH1 L-segment, encompasses four CDS including a ParA-domain protein, and is nearby a different integrase/recombinase gene (Int2, PhiH1_240). Protein searches (BLASTp) of the phiH1 genome returned matches to phiCh1, a limited number of haloarchaeal genomes (often 5–10, which may flag proviral regions) and the haloarchaeal caudoviruses BJ1 [47] and CGphi46 (NC_021537), both of which infect Halorubrum spp. Pairwise alignments (BLASTp) between all phiH1 and matching phiCh1 proteins gave an average protein sequence identity of 70% (range 39–95%; with a few exceptions, see footnote 5, Table 1). Figure 3 is a graphical comparison of phiH1 proteins (tBLASTx) with those of phiCh1, BJ1, CGphi46 and, as an outlier, HSTV-1. The Haloarcula caudovirus HSTV-1 [48] shows very low similarity to phiH1. The figure summarizes the close similarity of phiH1 and phiCh1 proteins. BJ1 and CGphi46 show far fewer matching regions, mainly to proteins encoded near the left end of the phiH1 genome, a region specifying portal and capsid proteins. The three significant matches to HSTV-1 were to a methyltransferase (HSTV1_52), a hypothetical protein (HSTV1_53), and a DNA polymerase sliding clamp protein (HSTV1_40).
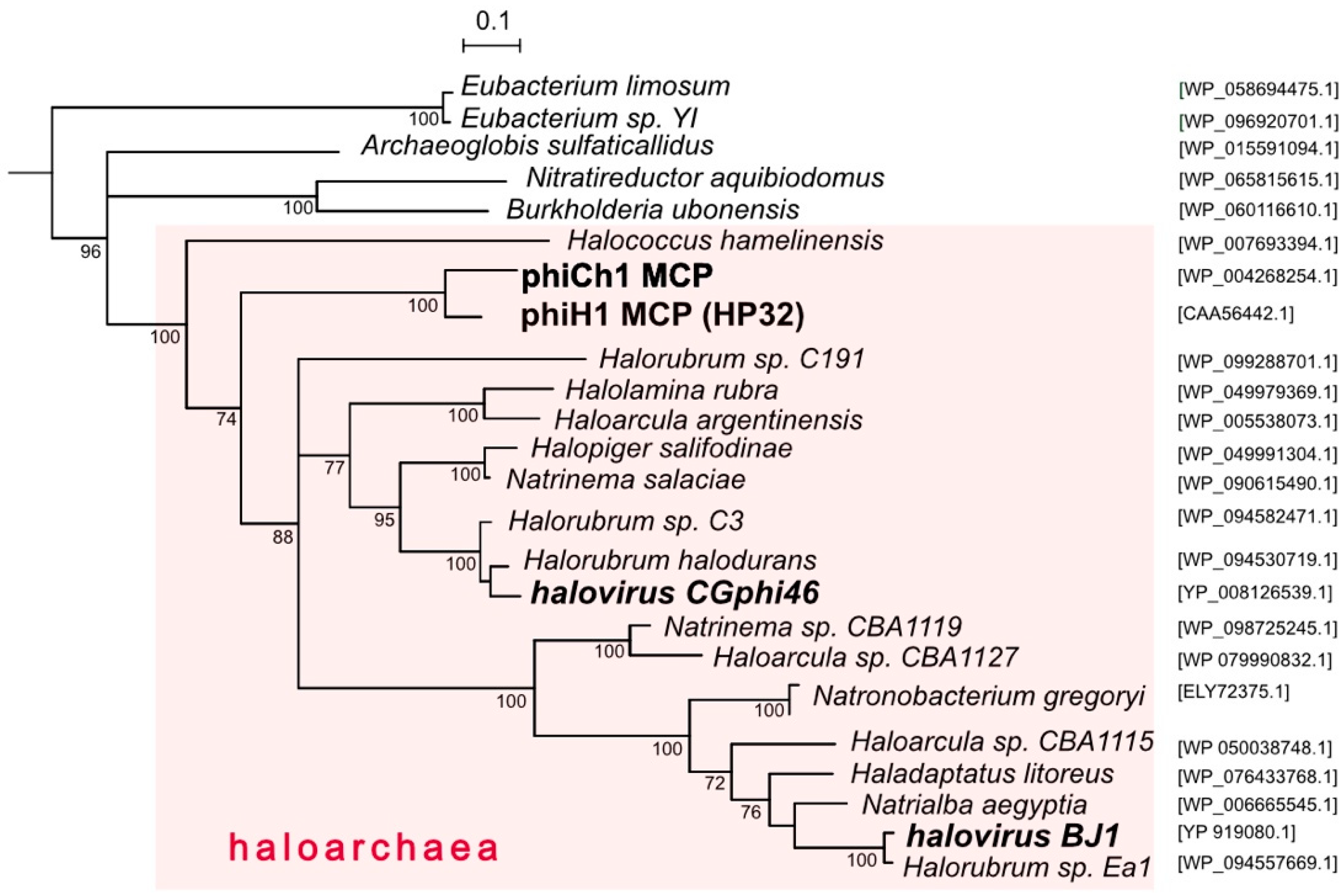
While several caudovirus proteins have been used to infer virus phylogenies, the major capsid protein (MCP) is often used because of its functional constraints maintaining a conserved structure [49]. Figure 4 shows a tree reconstruction using an alignment of phiH1 MCP (HP32, 35.4 kDa) and related sequences. Haloarchaeal proteins are seen to branch together (pink shading) and within this cluster the phiH1 and phiCh1 MCPs form a distinct and closely branching clade. These two proteins share 82% amino acid identity. The MCPs of CGphi46 and BJ1 branch at distant locations from each other and from phiH1 MCP. Structures of close homologs of phiH1 HP32 have not yet been determined. However, the major capsid proteins of bacterial caudoviruses and eukaryotic herpesviruses share a common folding structure, the archetype of which is the phage HK97 MCP (gp5) [50]. Consistent with this, modelling of the phiH1 MCP (I-Tasser) returned bacteriophage HK97 gp5 (PDB 2fs3A) as the closest matching structure (Template Modeling (TM)-score = 0.848, Root-Mean-Square Deviation (RMSD) = 1.17). Based on structure prediction and homology modelling, the HK97-fold may also be present in the MCP of phiCh1 [49]. The structure of the MCP of the haloarchaeal podovirus, HSTV-1, has recently been shown to be of the HK97 type [48].
PhiH1 and phiCh1 display a close sequence similarity across most of their genomes yet infect physiologically and biochemically different haloarchaeal hosts. Hbt. salinarum is a widely distributed neutrophilic heterotroph with glycolipid-containing membranes, and has often been isolated from spoilage of salted products while Nab. magadii is a haloalkaliphile (optimum pH 9.5) that lacks glycolipids [51] and is restricted in its distribution to highly alkaline salt lakes [52]. Looking more widely, the presence of phiH1 MCP homologs in diverse genera of haloarchaea and two haloviruses (Figure 4) indicates that the Myohalovirus genus and related viruses are a highly successful group, the reasons for which are worthy of more detailed study, particularly when large-scale cultivation of Halobacterium becomes more common [53]. PhiH1 has been well studied in the past, and the completion of its genome sequence now allows it to be included in much of the sequence-based studies used today, including comparative virology, detection of proviruses in archaeal genomes, virus evolution and the microbial ecology of hypersaline environments.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4425/9/10/493/s1, Figure S1: Original phiH1 restriction map compared to in silico map from genome sequence. Figure S2: Dot plot sequence comparison of phiH1 and phiCh1 genomes.
Author Contributions
Conceptualization, visualization, investigation, M.D.-S.; data curation, Fr.P., M.D.-S.; funding acquisition, D.O., Fr.P.; project administration, Fr.P.; resources, A.W., Fe.P.; writing—original draft, Fr.P., M.D.-S.; writing—review & editing, A.W., D.O., Fe.P., Fr.P., M.D.-S.; validation, A.W.
Funding
This research was funded by the Max Planck Society, Germany, to Dieter Oesterhelt, emeritus Director of the Department of Membrane Biochemistry, Max Planck Institute, Martinsried, Germany.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schnabel, H.; Zillig, W.; Pfaffle, M.; Schnabel, R.; Michel, H.; Delius, H. Halobacterium halobium phage ΦH. EMBO J. 1982, 1, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Zillig, W.; Gropp, F.; Henschen, A.; Neumann, H.; Palm, P.; Reiter, W.D.; Rettenberger, M.; Schnabel, H.; Yeats, S. Archaebacteria virus host systems. Syst. Appl. Microbiol. 1986, 7, 58–66. [Google Scholar] [CrossRef]
- Zillig, W.; Reiter, W.-D.; Palm, P.; Gropp, F.; Neumann, H.; Rettenberger, M. Viruses of Archaebacteria. In The Bacteriophages; Calendar, R., Ed.; Plenum Publishing Corpn: New York, NY, USA, 1988. [Google Scholar]
- Schnabel, H.; Zillig, W. Circular structure of the genome of phage ΦH in a lysogenic Halobacterium halobium. Mol. Gen. Genet. 1984, 193, 422–426. [Google Scholar] [CrossRef]
- Schnabel, H.; Schramm, E.; Schnabel, R.; Zillig, W. Structural variability in the genome of phage ΦH of Halobacterium halobium. Mol. Gen. Genet. 1982, 188, 370–377. [Google Scholar] [CrossRef]
- ICTV Report, C. ICTV Online (10th) Report on Virus Taxonomy. Available online: https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=20170459 (accessed on 19 March 2018).
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: Update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.W.; Doolittle, W.F. Efficient transfection of the archaebacterium Halobacterium halobium. J. Bacteriol. 1987, 169, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Blaseio, U.; Pfeifer, F. Transformation of Halobacterium halobium: Development of vectors and investigation of gas vesicle synthesis. Proc. Natl. Acad. Sci. USA 1990, 87, 6772–6776. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo studies on the effects of immunity genes on early lytic transcription in the Halobacterium salinarium phage ϕH. Mol. Gen. Genet. 1992, 235, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Ken, R.; Hackett, N.R. Halobacterium halobium strains lysogenic for phage phiH contain a protein resembling coliphage repressors. J. Bacteriol. 1991, 173, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. In vivo and in vitro analysis of transcription of the L region from the Halobacterium salinarium phage ϕH: Definition of a repressor-enhancing gene. Virology 1993, 195, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Antisense RNA mediates transcriptional processing in an archaebacterium, indicating a novel kind of RNase activity. Mol. Microbiol. 1993, 7, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, H. An immune strain of Halobacterium halobium carries the invertible L segment of phage ΦH as a plasmid. Proc. Natl. Acad. Sci. USA 1984, 81, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Zillig, W. Transcription of the halophage ΦH repressor gene is abolished by transcription from an inversely oriented lytic promoter. FEBS Lett. 1994, 344, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Stolt, P.; Zillig, W. Gene regulation in halophage ΦH; more than promoters. Syst. Appl. Microbiol. 1993, 16, 591–596. [Google Scholar] [CrossRef]
- Witte, A.; Baranyi, U.; Klein, R.; Sulzner, M.; Luo, C.; Wanner, G.; Krüger, D.H.; Lubitz, W. Characterization of Natronobacterium magadii phage ϕCh1, a unique archaeal phage containing DNA and RNA. Mol. Microbiol. 1997, 23, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Baranyi, U.; Rössler, N.; Greineder, B.; Scholz, H.; Witte, A. Natrialba magadii virus ϕCh1: First complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol. Microbiol. 2002, 45, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Selb, R.; Derntl, C.; Klein, R.; Alte, B.; Hofbauer, C.; Kaufmann, M.; Beraha, J.; Schoner, L.; Witte, A. The viral gene ORF79 encodes a repressor regulating induction of the lytic life cycle in the haloalkaliphilic virus phiCh1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Siddaramappa, S.; Challacombe, J.F.; Decastro, R.E.; Pfeiffer, F.; Sastre, D.E.; Gimenez, M.I.; Paggi, R.A.; Detter, J.C.; Davenport, K.W.; Goodwin, L.A.; et al. A comparative genomics perspective on the genetic content of the alkaliphilic haloarchaeon Natrialba magadii ATCC 43099T. BMC Genom. 2012, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Zillig, W.; Palm, P.; Reiter, W.D.; Gropp, F.; Puhler, G.; Klenk, H.P. Comparative evaluation of gene expression in Archaebacteria. Eur. J. Biochem. 1988, 173, 473–482. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/ (accessed on 11 October 2018).
- Gordon, D. Viewing and editing assembled sequences using Consed. Curr. Protoc. Bioinform. 2003, 2. [Google Scholar] [CrossRef]
- Skennerton, C.T.; Imelfort, M.; Tyson, G.W. Crass: Identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic Acids Res. 2013, 41, e105. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall-Smith, M.; Pfeiffer, F. The PL6-family plasmids of Haloquadratum are virus-related. Front. Microbiol. 2018, 9, 1070. [Google Scholar] [CrossRef] [PubMed]
- CRISPRs Web Server. Available online: http://crispr.i2bc.paris-saclay.fr/ (accessed on 11 October 2018).
- Lomsadze, A.; Gemayel, K.; Tang, S.; Borodovsky, M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018, 28, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 11 October 2018).
- YASS Genomic Similarity Search Tool. Available online: http://bioinfo.lifl.fr/yass/index.php (accessed on 11 October 2018).
- Geneious. Available online: https://www.geneious.com/geneious/ (accessed on 11 October 2018).
- Circoletto. Available online: http://tools.bat.infspire.org/circoletto/ (accessed on 11 October 2018).
- I-Tasser, Protein Structure & Function Predictions. Available online: https://zhanglab.ccmb.med.umich.edu/I-TASSER (accessed on 11 October 2018).
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- CPT Phage Galaxy. Available online: https://cpt.tamu.edu/galaxy-pub/ (accessed on 11 October 2018).
- VIRFAM, Remote Homology Detection of Viral Protein Families. Available online: http://biodev.cea.fr/virfam/ (accessed on 11 October 2018).
- Schnabel, H.; Schnabel, R.; Yeats, S.; Tu, J.; Gierl, A.; Neumann, H.; Zillig, W. Genome organization and transcription in Archaebacteria. Folia Biol. (Praha) 1984, 30, 2–6. [Google Scholar] [PubMed]
- Pfeiffer, F.; Schuster, S.C.; Broicher, A.; Falb, M.; Palm, P.; Rodewald, K.; Ruepp, A.; Soppa, J.; Tittor, J.; Oesterhelt, D. Evolution in the laboratory: The genome of Halobacterium salinarum strain R1 compared to that of strain NRC-1. Genomics 2008, 91, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Grampp, B.; Zillig, W. Genes for DNA cytosine methyltransferases and structural proteins, expressed during lytic growth by the phage ΦH of the archaebacterium Halobacterium salinarium. Biol. Chem. Hoppe Seyler 1994, 375, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Pavelka, M.S., Jr.; Butler, J.S. Structure-function analysis of VapB4 antitoxin identifies critical features of a minimal VapC4 toxin-binding module. J. Bacteriol. 2015, 197, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhang, C.T. GC-Profile: A web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acids Res. 2006, 34, W686–W691. [Google Scholar] [CrossRef] [PubMed]
- Gropp, F. Genexpression im Archaebakterium Halobacterium halobium: Der Phage ΦH und die DNA-abhängige RNA-Polymerase. Ph.D. Thesis, Ludwig-Maximilians-Universitaet Muenchen, Munich, Germany, 26 July 1989. [Google Scholar]
- Gropp, F.; Grampp, B.; Stolt, P.; Palm, P.; Zillig, W. The immunity-conferring plasmid pϕHL from the Halobacterium salinarium phage ϕH: Nucleotide sequence and transcription. Virology 1992, 190, 45–54. [Google Scholar] [CrossRef]
- ISfinder. Available online: https://isfinder.biotoul.fr/ (accessed on 11 October 2018).
- Vogelsang-Wenke, H.; Oesterhelt, D. Isolation of a halobacterial phage with a fully cytosine-methylated genome. MGG Mol. Gen. Genet. 1988, 211, 407–414. [Google Scholar] [CrossRef]
- Baranyi, U.; Klein, R.; Lubitz, W.; Kruger, D.H.; Witte, A. The archaeal halophilic virus-encoded Dam-like methyltransferase M. ϕCh1-I methylates adenine residues and complements dam mutants in the low salt environment of Escherichia coli. Mol. Microbiol. 2000, 35, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Pagaling, E.; Haigh, R.D.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Sequence analysis of an archaeal virus isolated from a hypersaline lake in Inner Mongolia, China. BMC Genom. 2007, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. Proc. Natl. Acad. Sci. USA 2013, 110, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P.; Bamford, D.H. Comparative analysis of the mosaic genomes of tailed archaeal viruses and proviruses suggests common themes for virion architecture and assembly with tailed viruses of bacteria. J. Mol. Biol. 2010, 397, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef] [PubMed]
- Kamekura, M.; Dyall-Smith, M. Taxonomy of the family Halobacteriaceae and the description of two new genera Halorubrobacterium and Natrialba. J. Gen. Appl. Microbiol. 1995, 41, 333–350. [Google Scholar] [CrossRef]
- Tindall, B.J.; Ross, H.N.M.; Grant, W.D. Natronobacterium gen. nov. and Natronococcus gen. nov. Two new genera of haloalkaliphilic archaebacteria. Syst. Appl. Microbiol. 1984, 5, 41–57. [Google Scholar] [CrossRef]
- Kalenov, S.V.; Baurina, M.M.; Skladnev, D.A.; Kuznetsov, A.Y. High-effective cultivation of Halobacterium salinarum providing with bacteriorhodopsin production under controlled stress. J. Biotechnol. 2016, 233, 211–218. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of the phiH1 genome with lines below showing regions previously sequenced (red) along with their database accessions. The blue lines (NEW) indicate regions sequenced in the present study by Sanger sequencing. Tick marks (dark green) below the blue lines show the positions of oligonucleotide primers used for PCR and primer-walking. Dots at the right and left contig ends indicate sequence continuity between them. Scale bar at top shows position in bp.
Figure 1.
Diagram of the phiH1 genome with lines below showing regions previously sequenced (red) along with their database accessions. The blue lines (NEW) indicate regions sequenced in the present study by Sanger sequencing. Tick marks (dark green) below the blue lines show the positions of oligonucleotide primers used for PCR and primer-walking. Dots at the right and left contig ends indicate sequence continuity between them. Scale bar at top shows position in bp.

Figure 2.
PhiH1 GC-profile, genetic map, and corresponding transcription program (adapted from [16,42]). (a) GC-profile of the phiH1 genome. (b) Genetic map of the phiH1 genome, showing coding sequences as red, blue or grey arrows. Dotted lines above indicate gene clusters involved in particular functions. Some CDS are labelled above the map, e.g., TerL, terminase large subunit; Portal, portal protein; Tape, tape-measure protein; RepH, replicase (label within CDS arrow); Mt, DNA methylases; TerS, terminase small subunit. Some genes are shown below the map, such as hp32, encoding the major capsid protein HP32. Panels c, d and e summarise transcription data from previously published studies, and above them is a colour key that indicates the time of appearance of early (0–1 h, blue), middle (1–2 h, green) and late (>2 h, pink) transcripts. (c) Precise mapping of viral transcripts, including start and termination sites [16,39]. (d) Summary transcription program of lytic infection based on hybridisation of labelled infected-cell transcripts to restriction fragments of virus DNA [42]. Thin coloured lines indicate whether continuing transcription persists over time. (e) The transcription map data of [42] are shown, projected onto the in silico restriction map of phiH1, as determined from the complete genome sequence (this study). Enzymes are indicated at the left. Numbers on the restriction map refer to those of the original publication of [42] (see also Figure S1). Coloured shading follows that of panels c and d. Dotted pattern shown beyond the right-hand pac site indicates terminal redundancy of virus DNA. Scale bars (in kb) are shown below panels a and e.
Figure 2.
PhiH1 GC-profile, genetic map, and corresponding transcription program (adapted from [16,42]). (a) GC-profile of the phiH1 genome. (b) Genetic map of the phiH1 genome, showing coding sequences as red, blue or grey arrows. Dotted lines above indicate gene clusters involved in particular functions. Some CDS are labelled above the map, e.g., TerL, terminase large subunit; Portal, portal protein; Tape, tape-measure protein; RepH, replicase (label within CDS arrow); Mt, DNA methylases; TerS, terminase small subunit. Some genes are shown below the map, such as hp32, encoding the major capsid protein HP32. Panels c, d and e summarise transcription data from previously published studies, and above them is a colour key that indicates the time of appearance of early (0–1 h, blue), middle (1–2 h, green) and late (>2 h, pink) transcripts. (c) Precise mapping of viral transcripts, including start and termination sites [16,39]. (d) Summary transcription program of lytic infection based on hybridisation of labelled infected-cell transcripts to restriction fragments of virus DNA [42]. Thin coloured lines indicate whether continuing transcription persists over time. (e) The transcription map data of [42] are shown, projected onto the in silico restriction map of phiH1, as determined from the complete genome sequence (this study). Enzymes are indicated at the left. Numbers on the restriction map refer to those of the original publication of [42] (see also Figure S1). Coloured shading follows that of panels c and d. Dotted pattern shown beyond the right-hand pac site indicates terminal redundancy of virus DNA. Scale bars (in kb) are shown below panels a and e.

Figure 3.
Circos plot of amino acid similarity (tBLASTx) between phiH1 and the haloviruses phiCh1, BJ1, CGphi46 and HSTV-1. The threshold for connecting lines was E-value ≤ 10−40, with line colours reflecting the ratio of actual tBLASTx bitscore to the maximal score (using ‘score/max’ ratio colouring with blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75, red > 0.75). The outer histogram counts how many times each colour has hit the specific part of the sequence and uses an equivalent colouring scheme. The distance between successive tick marks shown along each virus genome represents 0.1 of the full genome length. Protein names shown along the phiH1 genome indicate the positions of the corresponding genes.
Figure 3.
Circos plot of amino acid similarity (tBLASTx) between phiH1 and the haloviruses phiCh1, BJ1, CGphi46 and HSTV-1. The threshold for connecting lines was E-value ≤ 10−40, with line colours reflecting the ratio of actual tBLASTx bitscore to the maximal score (using ‘score/max’ ratio colouring with blue ≤ 0.25, green ≤ 0.50, orange ≤ 0.75, red > 0.75). The outer histogram counts how many times each colour has hit the specific part of the sequence and uses an equivalent colouring scheme. The distance between successive tick marks shown along each virus genome represents 0.1 of the full genome length. Protein names shown along the phiH1 genome indicate the positions of the corresponding genes.

Figure 4.
Phylogenetic tree reconstruction (NJ method) of major capsid proteins (MCP) of phiH1, other haloviruses and related proteins of haloarchaea. Species names of haloarchaeal species are shown, with accession numbers given at the right side. Bootstrap confidence values (100 repetitions) are shown at branch points. The pink shading highlights taxa belonging to the class Halobacteria. Scale bar (expected changes per site) is shown at top. The outgroup (not shown) consisted of distantly related MCP sequences of Bacillus spp. (WP_001060157.1, WP_098773561.1, WP_001064748.1 and WP_000178926.1).
Figure 4.
Phylogenetic tree reconstruction (NJ method) of major capsid proteins (MCP) of phiH1, other haloviruses and related proteins of haloarchaea. Species names of haloarchaeal species are shown, with accession numbers given at the right side. Bootstrap confidence values (100 repetitions) are shown at branch points. The pink shading highlights taxa belonging to the class Halobacteria. Scale bar (expected changes per site) is shown at top. The outgroup (not shown) consisted of distantly related MCP sequences of Bacillus spp. (WP_001060157.1, WP_098773561.1, WP_001064748.1 and WP_000178926.1).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Annotated coding sequences (CDS) of halovirus phiH1.
| Start (nt) | Stop (nt) | Locus_tag | Length (bp) | Direction | Gene | Product | Homologs1: phiCh1, ORF pNMAG03 [Other] |
|---|---|---|---|---|---|---|---|
| 115 | 717 | PhiH1_005 | 603 | + | - | uncharacterized protein | PhiCh1p02, ORF1 Nmag_4251 |
| 710 | 2371 | PhiH1_010 | 1662 | + | terL | terminase large subunit TerL | PhiCh1p03, ORF2 Nmag_4252 |
| 2377 | 2505 | PhiH1_015 | 129 | + | - | uncharacterized protein | Nmag_4253 |
| 2498 | 2689 | PhiH1_020 | 192 | + | - | uncharacterized protein | PhiCh1p05, ORF4 Nmag_4255 |
| 2686 | 4242 | PhiH1_025 | 1557 | + | por | portal protein Por | PhiCh1p07, ORF6 Nmag_4257 |
| 4246 | 5187 | PhiH1_030 | 942 | + | - | head morphogenesis protein | PhiCh1p08, ORF7 Nmag_4258 |
| 5261 | 5587 | PhiH1_035 | 327 | + | hp20 | capsid protein HP20 | [AJF28118.1] |
| 5667 | 7466 | PhiH1_040 | 1800 | + | - | prohead protease | 4 PhiCh1p09, ORF8 4 PhiCh1p10, ORF9 Nmag_4259 |
| 7506 | 8468 | PhiH1_045 | 963 | + | hp32 | major capsid protein HP32 | PhiCh1p12, ORF11 Nmag_4260 |
| 8481 | 8933 | PhiH1_050 | 453 | + | - | uncharacterized protein | PhiCh1p13, ORF12 Nmag_4261 |
| 8940 | 9542 | PhiH1_055 | 603 | + | ada | head-tail adaptor protein Ada | PhiCh1p14, ORF13 Nmag_4262 |
| 9539 | 9919 | PhiH1_060 | 381 | + | hco | head closure protein type 1 Hco | PhiCh1p15, ORF14 Nmag_4263 |
| 9921 | 10,202 | PhiH1_065 | 282 | + | - | uncharacterized protein | PhiCh1p16, ORF15 Nmag_4264 |
| 10,202 | 10,636 | PhiH1_070 | 435 | + | nep | probable neck protein type 1 Nep | PhiCh1p17, ORF16 Nmag_4265 |
| 10,643 | 11,239 | PhiH1_075 | 597 | + | tco | tail completion protein type 1 Tco | PhiCh1p18, ORF17 Nmag_4266 |
| 11,259 | 12,557 | PhiH1_080 | 1299 | + | hp67 | tail sheath protein HP67 | PhiCh1p19, ORF18 Nmag_4267 |
| 12,607 | 13,002 | PhiH1_085 | 396 | + | - | probable structural protein | PhiCh1p20, ORF19 Nmag_4268 |
| 13,006 | 13,407 | PhiH1_090 | 402 | + | - | uncharacterized protein | PhiCh1p21, ORF20 Nmag_4269 |
| 13,572 | 13,745 | PhiH1_095 | 174 | − | - | DUF4177 domain protein | [SEH60446.1] |
| 13,792 | 16,581 | PhiH1_100 | 2790 | + | tpm | tape-measure tail protein Tpm | 4 PhiCh1p23, ORF22 4 PhiCh1p24, ORF23 Nmag_4272 |
| 16,583 | 17,104 | PhiH1_105 | 522 | + | - | uncharacterized protein | PhiCh1p25, ORF24 Nmag_4273 |
| 17,108 | 17,446 | PhiH1_110 | 339 | + | - | uncharacterized protein | PhiCh1p26, ORF25 Nmag_4274 |
| 17,450 | 18,298 | PhiH1_115 | 849 | + | - | uncharacterized protein | PhiCh1p27, ORF26 Nmag_4275 |
| 18,306 | 18,446 | PhiH1_120 | 141 | + | - | CxxC motif protein | [SEH61109.1] |
| 18,443 | 18,988 | PhiH1_125 | 546 | + | - | uncharacterized protein | PhiCh1p29, ORF28 Nmag_4276 |
| 18,988 | 19,146 | PhiH1_130 | 159 | + | - | uncharacterized protein | - |
| 19,143 | 19,508 | PhiH1_135 | 366 | + | - | virus-related protein | [AGM10900.1] |
| 19,505 | 19,867 | PhiH1_140 | 363 | + | - | uncharacterized protein | PhiCh1p30, ORF29 Nmag_4277 |
| 19,874 | 21,148 | PhiH1_145 | 1275 | + | bpj | baseplate J family protein Bpj | PhiCh1p31, ORF30 Nmag_4278 |
| 21,135 | 22,277 | PhiH1_150 | 1143 | + | - | uncharacterized protein | PhiCh1p32, ORF31 Nmag_4279 |
| 22,295 | 22,678 | PhiH1_155 | 384 | + | - | virus-related protein | [AFH21897.1] |
| 22,683 | 23,249 | PhiH1_160 | 567 | + | - | virus-related protein | [AFH21653.1] |
| 23,252 | 25,504 | PhiH1_165 | 2253 | + | - | repeat-containing tail fibre protein | PhiCh1p37, ORF36 Nmag_4282 PhiCh1p35, ORF34 Nmag_4286 |
| 25,506 | 25,787 | PhiH1_170 | 282 | + | - | uncharacterized protein | Nmag_4285 |
| 25,825 | 26,499 | PhiH1_175 | 675 | + | int1 | tyrosine integrase/recombinase Int1 | PhiCh1p36, ORF35 Nmag_4284 |
| 26,490 | 26,792 | PhiH1_180 | 303 | − | - | uncharacterized protein | Nmag_4283 |
| 26,798 | 27,766 | PhiH1_185 | 969 | − | - | repeat-containing tail fibre protein 2 | PhiCh1p37, ORF36 Nmag_4282 PhiCh1p35, ORF34 Nmag_4286 |
| 27,803 | 28,150 | PhiH1_190 | 348 | + | - | YncB-like endonuclease | [AGM11801.1] |
| 28,153 | 28,386 | PhiH1_195 | 234 | + | - | virus-related protein | [AGC34510.1] |
| 28,379 | 28,675 | PhiH1_200 | 297 | + | - | uncharacterized protein | [EMA49173.1] |
| 28,682 | 28,783 | PhiH1_205 | 102 | + | - | uncharacterized protein | - |
| 28,788 | 29,357 | PhiH1_210 | 570 | + | - | transmembrane domain protein | - |
| 29,394 | 29,642 | PhiH1_215 | 249 | − | - | uncharacterized protein | - |
| 29,651 | 29,941 | PhiH1_220 | 291 | − | - | uncharacterized protein | PhiCh1p40, ORF39 Nmag_4289 |
| 30,104 | 30,244 | PhiH1_225 | 144 | + | - | uncharacterized protein | - |
| 30,250 | 30,414 | PhiH1_230 | 165 | + | - | uncharacterized protein | PhiCh1p44, ORF43 Nmag_4292 |
| 30,411 | 30,806 | PhiH1_235 | 396 | + | - | VapC family toxin | PhiCh1p45, ORF44 Nmag_4293 |
| 30,803 | 31,465 | PhiH1_240 | 663 | − | int2 | tyrosine integrase/recombinase Int2 | PhiCh1p46, ORF45 Nmag_4294 |
| 31,680 | 31,934 | PhiH1_245 | 255 | + | - | uncharacterized protein | - |
| 31,939 | 32,271 | PhiH1_250 | 333 | + | - | uncharacterized protein | Nmag_4297 |
| 32,420 | 32,857 | PhiH1_255 | 438 | − | - | HNH-type endonuclease | PhiCh1p48, ORF47 Nmag_4296 |
| 32,854 | 33,255 | PhiH1_260 | 402 | − | - | uncharacterized protein | [ELY96531.1] |
| 33,248 | 34,024 | PhiH1_265 | 777 | − | - | parA domain protein | PhiCh1p47, ORF46 Nmag_4295 |
| 34,161 | 34,430 | PhiH1_270 | 270 | − | repR | repressor protein RepR | 5 PhiCh1p49, ORF48 5 Nmag_4298 [ELZ06324.1] |
| 34,730 | 35,071 | PhiH1_275 | 342 | + | - | uncharacterized protein | - |
| 35,068 | 35,424 | PhiH1_280 | 357 | + | - | uncharacterized protein | PhiCh1p50, ORF49 |
| 35,381 | 38,167 | PhiH1_285 | 2787 | + | repH | plasmid replication protein RepH | 4 PhiCh1p54, ORF53 4 PhiCh1p55, ORF54 Nmag_4299 |
| 38,262 | 38,489 | PhiH1_290 | 228 | − | imm | probable immunity protein Imm | PhiCh1p56, ORF55 Nmag_4300 |
| 38,733 | 39,263 | PhiH1_295 | 531 | + | - | transcriptional regulator, PadR-like family | PhiCh1p57, ORF56 Nmag_4301 |
| 39,260 | 39,385 | PhiH1_300 | 126 | + | - | CxxC motif protein | - |
| 39,382 | 39,978 | PhiH1_305 | 597 | + | - | uncharacterized protein | PhiCh1p59, ORF58 Nmag_4303 |
| 39,975 | 40,133 | PhiH1_310 | 159 | + | - | uncharacterized protein | - |
| 40,153 | 40,902 | PhiH1_315 | 750 | + | pcnA | DNA polymerase sliding clamp PcnA | PhiCh1p60, ORF59 Nmag_4211 |
| 40,908 | 41,339 | PhiH1_320 | 432 | + | - | uncharacterized protein | PhiCh1p61, ORF60 Nmag_4212 |
| 41,339 | 41,554 | PhiH1_325 | 216 | + | - | uncharacterized protein | PhiCh1p62, ORF61 Nmag_4213 |
| 41,547 | 42,041 | PhiH1_330 | 495 | + | - | uncharacterized protein | - |
| 42,098 | 42,490 | PhiH1_335 | 393 | + | tnpA | IS200-type transposase TnpA | [CAP12925.1] |
| 42,492 | 43,748 | PhiH1_340 | 1257 | + | tnpB | IS1341-type transposase TnpB | [CAP12926.1] |
| 43,808 | 44,014 | PhiH1_345 | 207 | + | - | uncharacterized protein | - |
| 44,007 | 44,234 | PhiH1_350 | 228 | + | - | uncharacterized protein | PhiCh1p66, ORF65 Nmag_4217 |
| 44,231 | 44,656 | PhiH1_355 | 426 | + | - | CxxC motif protein | PhiCh1p68, ORF67 Nmag_4219 |
| 44,646 | 45,026 | PhiH1_360 | 381 | + | - | uncharacterized protein | PhiCh1p69, ORF68 Nmag_4220 |
| 45,023 | 45,646 | PhiH1_365 | 624 | + | - | HNH-type endonuclease | [KYG11427.1] |
| 45,639 | 45,926 | PhiH1_370 | 288 | + | - | uncharacterized protein | PhiCh1p71, ORF70 Nmag_4222 |
| 45,919 | 46,350 | PhiH1_375 | 432 | + | - | DUF4326 domain protein | PhiCh1p72, ORF71 Nmag_4223 |
| 46,343 | 46,441 | PhiH1_380 | 99 | + | - | uncharacterized protein | - |
| 46,438 | 46,884 | PhiH1_385 | 447 | + | - | CxxC motif protein | PhiCh1p74, ORF73 Nmag_4225 |
| 46,865 | 47,038 | PhiH1_390 | 174 | + | - | uncharacterized protein | 5 PhiCh1p73, ORF72 5 Nmag_4224 |
| 47,031 | 47,447 | PhiH1_395 | 417 | + | - | uncharacterized protein | - |
| 47,440 | 47,739 | PhiH1_400 | 300 | + | - | NTPase protein | [PLX87675.1] |
| 47,732 | 49,618 | PhiH1_405 | 1887 | + | dcm5 | C-5 cytosine-specific DNA methylase Dcm5 | 5 PhiCh1p81, ORF80 [PCR88664.1] |
| 49,611 | 49,931 | PhiH1_410 | 321 | + | - | uncharacterized protein | PhiCh1p82, ORF81 Nmag_4234 |
| 49,918 | 50,037 | PhiH1_415 | 120 | + | - | CxxC motif protein | - |
| 50,091 | 51,452 | PhiH1_420 | 1362 | + | yhdJ | DNA methylase N-4/N-6 domain protein YhdJ | PhiCh1p83, ORF82 Nmag_4235 |
| 51,449 | 52,024 | PhiH1_425 | 576 | + | - | uncharacterized protein | PhiCh1p84, ORF83 Nmag_4236 |
| 52,021 | 52,791 | PhiH1_430 | 771 | + | - | uncharacterized protein | PhiCh1p85, ORF84 Nmag_4237 |
| 52,784 | 53,152 | PhiH1_435 | 369 | + | - | uncharacterized protein | PhiCh1p88, ORF87 Nmag_4240 |
| 53,145 | 53,504 | PhiH1_440 | 360 | + | - | uncharacterized protein | PhiCh1p89, ORF88 Nmag_4241 |
| 53,788 | 54,369 | PhiH1_445 | 582 | + | - | CxxC motif protein | PhiCh1p90, ORF89 Nmag_4242 |
| 54,403 | 54,771 | PhiH1_450 | 369 | + | - | uncharacterized protein | PhiCh1p91, ORF90 Nmag_4243 |
| 54,794 | 55,147 | PhiH1_455 | 354 | + | - | uncharacterized protein | - |
| 55,144 | 55,401 | PhiH1_460 | 258 | + | - | transmembrane domain protein | PhiCh1p93, ORF92 Nmag_4244 |
| 55,394 | 55,729 | PhiH1_465 | 336 | + | - | transmembrane domain protein 3 | PhiCh1p94, ORF93 Nmag_4245 |
| 55,794 | 57,053 | PhiH1_470 | 1260 | + | ycdA | DNA methylase N-4/N-6 domain protein YcdA | PhiCh1p95, ORF94 Nmag_4246 |
| 57,046 | 57,564 | PhiH1_475 | 519 | + | - | uncharacterized protein | PhiCh1p96, ORF95 Nmag_4247 |
| 57,621 | 57,830 | PhiH1_480 | 210 | + | - | CxxC motif protein | PhiCh1p98, ORF97 Nmag_4249 |
| 57,827> | <63 | PhiH1_485 | 309 | + | terS | terminase small subunit TerS | PhiCh1p01, ORF98 Nmag_4250 |
1 PhiCh1/pNMAG03 homologs of phiH1 proteins show BLASTp E-values < 10−20. For phiCh1 proteins, both the PhiCh1p and originally assigned ORF codes (ORF for open reading frame) are shown (e.g., PhiCh1p02, ORF1). Codes starting with ORF represent the original annotation of the phiCh1 genome [17] (GB accession AF440695.1); and codes starting with PhiCh1p represent the RefSeq version of the annotation of the same genome sequence (GB accession NC_004084). The number shift is due to the terS gene, the N-terminal part being encoded at the end of the genome, and the C-terminal part at its beginning. This ORF is complete in the provirus state due to circularization and in the linear virus state due to terminal redundancy. This gene is ORF98 in the original annotation and PhiCh1p01 in the RefSeq annotation. Codes starting with Nmag_ represent the annotation of the Natrialba magadii plasmid pNMAG03 [20] (accession CP001935.1). The point of ring opening in pNMAG03 was set between Nmag_4303 and Nmag_4211. Codes in square brackets represent NCBI accessions referring to homologous proteins (BLASTp E-values ≤ 10−11), which are from other sources. 2 Gene PhiH1_185 is encoded on an invertible segment. In the current sequence version, it is inactivated because it is uncoupled from a start codon. By genome inversion, it becomes activated while its partner gene PhiH1_165 becomes inactivated. Overall, this results in tail fibre protein switching. 3 This protein (PhiH1_465) has three predicted transmembrane domains and has been suspected to function as a holin [18]. 4 In these cases, the phiCh1 gene is split into two CDS but is continuous in phiH1. 5 These proteins are more distantly related (show less than 39% sequence identity or fall above BLASTp E-values of 10−20). In these cases, a similar genetic context supports their stated relationship.
Table 2.
CRISPR spacers matching phiH1.
| No. | CRISPR Spacer Matches to phiH1 1 | Translation 2 |
|---|---|---|
 | ||
1 The matching spacer sequences were found in the following NCBI bioprojects using the crass program: PRJNA337743, (SRA SRR4030040; Alviso Ponds, San Francisco, CA, USA; metagenome); PRJNA245787 (Halostagnicola sp. A56 26 genome; Andaman Islands, India); PRJEB18068 (Lake Meyghan, Iran; metagenome). Aligned sequences show nt positions for phiH1, and asterisks indicated identical bases. DR: direct repeat (with haloarchaea containing most closely matching DR shown in brackets). 2 Symbols under alignment (*:.) indicate identical, similar and weakly similar residues, respectively (based on Gonnet PAM 250 matrix).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dyall-Smith, M.; Pfeifer, F.; Witte, A.; Oesterhelt, D.; Pfeiffer, F. Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1). Genes 2018, 9, 493. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9100493
AMA Style
Dyall-Smith M, Pfeifer F, Witte A, Oesterhelt D, Pfeiffer F. Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1). Genes. 2018; 9(10):493. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9100493
Chicago/Turabian StyleDyall-Smith, Mike, Felicitas Pfeifer, Angela Witte, Dieter Oesterhelt, and Friedhelm Pfeiffer. 2018. "Complete Genome Sequence of the Model Halovirus PhiH1 (ΦH1)" Genes 9, no. 10: 493. https://0-doi-org.brum.beds.ac.uk/10.3390/genes9100493
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.