Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms


Department of Internal Medicine, Section of Digestive Diseases, Yale University, New Haven, CT 06519, USA
Gastrointest. Disord. 2020, 2(3), 246-266; https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2030024
Submission received: 30 May 2020
/
Revised: 27 July 2020
/
Accepted: 20 August 2020
/
Published: 22 August 2020
(This article belongs to the Special Issue Aspects of the Pathogenesis and Management of the Inflammatory Bowel Diseases)
{kind=link}
{kind=link}
Abstract
:The pathogenesis of inflammatory bowel disease (IBD) involves perturbation of intestinal immune homeostasis in genetically susceptible individuals. A mutual interplay between intestinal epithelial cells (IECs) and gut resident microbes maintains a homeostatic environment across the gut. An idiopathic gastrointestinal (GI) complication triggers aberrant physiological stress in the epithelium and peripheral myeloid cells, leading to a chronic inflammatory condition. Indeed, events in the endoplasmic reticulum (ER) and mitochondria contribute to orchestrating intracellular mechanisms such as the unfolded protein response (UPR) and oxidative stress, respectively, to resolve aberrant cellular stress. This review highlights the signaling cascades encrypted within ER and mitochondria in IECs and/or myeloid cells to dissipate chronic stress in maintaining intestinal homeostasis.
1. Introduction
Human gastrointestinal (GI) tract mucosa is a multicellular interface that separates the host from intruding commensal/pathogenic microbes and potentially inimical contents derived from food materials. The intestinal epithelium cells (IECs) construct the gut mucosa and form a “barrier” against intruding microbes across the lumen. Indeed, the specific lineage of IECs, including Goblet cells, Paneth cells, enteroendocrine cells, absorptive enterocytes, and tuft cells, assists in the maintenance of the integrity of the mucosal barrier [1,2,3].
Goblet cells (GCs) are specialized secretory cells that synthesize and release mucins to protect the mucosa from intruding proinflammatory microbes [4,5,6]. In line with this, altered expression of mucin leads to the spontaneous development of chronic colitis in mice [7,8]. An earlier study showed that goblet cells coordinate with the NLRP6 inflammasome and activate Ca2+ ion-dependent Muc2 mucin secretion to protect the colonic crypt from microbial intrusion [9]. A unique aspect of this study is the identification of a distinct GC (termed “sentinel” GC) population that activates the inflammasome-dependent secretion of Muc2 and, in turn, triggers an alarm to adjacent GCs for more mucin secretion to expel bacteria. Besides mucin secretion, goblet cells also contribute, in antigen presentation, to CX3CR1+ dendritic cells in the small intestine [10] and secretion of cytokines such as interleukin (IL)-7 for immune regulation in intestinal mucosa [11].
Paneth cells are enriched with intracellular vesicles filled with microbicidal proteins and peptides, including lysozyme, α-defensins, C-type lectins, phospholipase A2, cryptdin, and angiogenin 4 [12,13,14,15]. Upon sensing microbial invasion, Paneth cells release the microbicidal contents into the lumen, which provides a defensive coating against infection [16,17,18]. Paneth cells also acquire stem cell-like characteristics and secrete growth factors, which help in the differentiation of IECs [19,20]. Furthermore, enteroendocrine cells representing a lower proportion (nearly 1%) of IECs secrete hormones and peptides contributing to gut barrier function and other metabolic activities [8,21].
Together, the functional outcomes of IECs are dedicated to protecting the mucosal epithelial layer. Recruitment of healthy IECs to the gut epithelium is one of the critical processes in maintaining intestinal homeostasis. The intestinal crypt is a niche to a unique set of Lgr5+ intestinal stem cells (ISCs), which constantly differentiate into IEC subsets and monitor the distribution and localization of newly generated cells. Interestingly, enterocytes, goblet cells, and enteroendocrine cells migrate upward, turning over every 3–4 days, and cover the majority of the villus. However, Paneth cells migrate to the bottom of the crypt and live for 6–8 weeks [22,23,24]. Accumulating evidence emphasizes that the integrity of the intestinal epithelium is critical to human health, and an unbidden breach in the epithelium protective barrier leads to the pathogenesis of several GI disorders, including inflammatory bowel disease (IBD) [2,25,26].
An unexpected alteration in the functioning of IECs may cause perturbation in the mucosal integrity that poses a risk to the GI tract. Indeed, dysregulation in IEC subsets has been shown in the pathogenesis of IBD. Consistently, histological examination of the rectal biopsies of patients with IBD has revealed reduced levels of mucin in goblet cells, demonstrating impaired goblet cell functioning, independent of disease state [27]. In addition, the mucus layer was found to be thin in intestinal tissues from patients with ulcerative colitis (UC), highlighting goblet cell perturbation in the pathogenesis of IBD [28,29].
There are multiple causes of IBD, including polymorphism in genetic coding, leading to impaired cellular outcomes [30,31,32,33]. Earlier studies have examined that a missense mutation in Atg16L1 (Autophagy Related 16 Like 1) leads to impaired Paneth cell function and microbial clearance and is associated with the prevalence of Crohn’s disease (CD) [34,35,36]. In line with the following observations, another CD-susceptible gene, IRGM (immunity-related GTPase family M), has impaired Paneth cell outcomes and intestinal inflammation [37,38].
In addition to genetic susceptibility, environmental factors can also influence intestinal permeability and the mucosal immune system, which, in turn, contributes to the etiology and/or pathogenesis of chronic diseases, including IBD [39,40]. Along those lines, several environmental factors reportedly modulate immune outcomes, thereby creating a susceptibility to IBD [41,42,43]. Environmental stresses associated with inflammatory flares are smoking, oral contraceptives, nonsteroidal anti-inflammatory drugs (NSAIDs), hygiene, and microbial metabolites [44,45]. Particularly, cigarette smoking may have a different effect on UC (ulcerative colitis) than on CD, as cigarette smoking provides protection against UC [46,47]. However, it may cause a two-fold higher risk of developing CD [48,49]. Notably, IL-10−/− mice treated with NSAIDs spontaneously developed colitis as compared to wild-type mice, with characteristic infiltration of macrophages and CD4+ T-cells in the lamina propria [50]. It is well established that metabolites from intestinal microbiota also contribute as environmental factors, which confer susceptibility to IBD [51,52]. Though environmental factors are significantly associated with disease development, more comprehensive studies are warranted to define the mechanisms through which these factors modulate intestinal inflammation.
Considering the importance of IEC subsets in maintaining the integrity of the epithelial barrier and protecting the GI tract against pathogenic intrusion, a rapid physiological transition in the GI tract (due to nutrient deprivation, pH imbalance, hypoxia, cellular differentiation, and cell death) imposes aberrant stress to IECs, which, in turn, leads to the alteration of intestinal permeability [26,53]. These observations are consistent with the fact that unresolved IEC stress and a breach in the mucosal barrier have been implicated in various GI disorders, including IBD [25]. In this context, an elevated serum zonulin (a tight junction regulatory protein) has been correlated with increased intestinal permeability in patients with IBD [54,55]. Indeed, the intracellular stress response mechanisms have consequently evolved as protective events to restore homeostasis in the intestinal epithelium [20]. Mechanistically, in multicellular organisms, a coordinated stress-response mechanism emanating from subcellular organelles, such as the endoplasmic reticulum (ER) and mitochondrion, contributes to restoring cellular homeostasis [56,57,58]. ER and mitochondria are vital intracellular organelles involved in various signaling pathways, including protein synthesis and folding. Moreover, ER and mitochondria have conserved intuitive response mechanisms to surmount cellular stress, termed as the unfolded protein response (UPR) [53,59,60,61,62,63]. It is well established that the secretory goblet and Paneth cells instigate UPR signaling for proper folding and secretion of various proteins [56]. Indeed, mitochondrial matrix encrypts a well-organized stress response system, including protein folding chaperons, ATP machinery, and reactive oxygen species (ROS) production [58,64], highlighting that mitochondrial UPR (mitoUPR) serves as an additional arm to respond to cellular stress and has been implicated in the regulation of intestinal inflammation [65]. This review highlights the mechanisms encrypted within ER and mitochondria to resolve cellular stress in maintaining gastrointestinal homeostasis.
2. ER-Associated UPR Activation and IBD
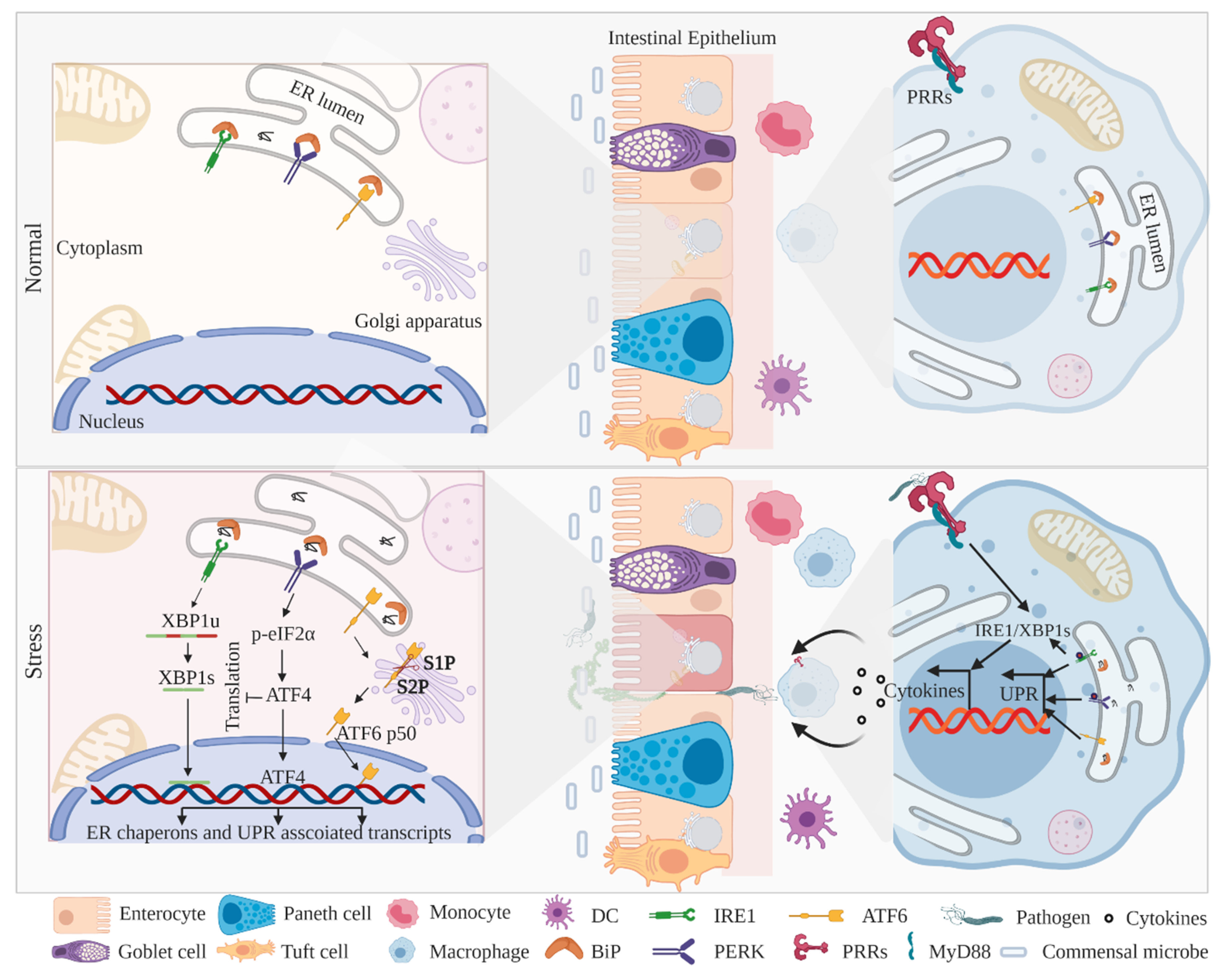
ER-associated UPR (ER-UPR) signaling has long been proposed as an organized response mechanism to resolve cellular stress, augmented by misfolded protein aggregation and/or pathogenic stress [59,61]. Earlier in-vivo studies with genetic or pharmacological manipulation of the ER-UPR genes have implicated these in intestinal inflammation and the pathogenesis of IBD [27,66,67]. The mammalian ER-UPR machinery is constituted by three transmembrane stress sensors: IRE1/Ern1 (inositol-requiring enzyme 1; α and β isoforms), PERK/Eif2ak3 (protein kinase related-like ER kinase), and ATF6 (activating transcription factor 6; α and β isoforms). All three UPR sensors remain inactive through interaction with ER luminal protein chaperon BiP (immunoglobulin heavy chain-binding protein)/GRP78 (glucose-regulated protein 78) [60,68,69]. Accumulated unfolded proteins in the ER lumen then sequester BiP, with concomitant release of UPR sensors followed by activation of downstream signaling [70,71,72].
Apart from professional secretory cells (Goblet and Paneth cells), the GI tract encompasses myeloid cells (macrophages and dendritic cells) that are also involved in the UPR-mediated maintenance of intestinal homeostasis [73,74,75]. Of note is the activation of myeloid cell-specific pathogen recognition receptors (PRRs), which are the vanguard of inflammatory priming against microbial incursion, although some interesting studies have shown that PRR signaling intermediates coordinate with UPR components and induce inflammatory responses against microbes (Figure 1) to restore intestinal homeostasis [73,76]. However, limited studies have investigated ER-UPR signaling in myeloid cells, and the in-depth underlying mechanisms still remain obscure. Furthermore, some interesting studies have revealed that PRR stimulation translocates innate immune mediators to ER and, in turn, activates UPR signaling for optimal cytokine secretion and microbial clearance [77,78]. Importantly, GWAS (genome-wide association studies) have identified several single nucleotide polymorphisms (SNPs) leading to perturbations in ER-UPR signaling that confers susceptibility to IBD [38,79]. Although signaling outcomes emanating from all three UPR branches coordinate to maintain intestinal homeostasis, genetic alteration minimizes the effectiveness of the outcomes, leading to the pathogenesis of IBD.
2.1. IRE1–XBP1 Signaling in IECs and Myeloid Cells
The IRE1α–XBP1 (X-box binding protein 1) signaling axis is the most evolutionarily conserved branch of ER-UPR signaling to restore ER homeostasis. Mechanistically, the aggregated unfolded proteins in the ER lumen titrate off BiP from IRE1α, which leads to IRE1α activation [71,72,80]. The activated IRE1α triggers autophosphorylation and undergoes a conformational change at the C-terminal to expose its endoribonuclease domain [81]. The endoribonuclease activity of IRE1α excises the 26-nucleotide mRNA sequence of XBP1, followed by the release of a spliced XBP1 (XBP1s), which further transactivates UPR target genes such as chaperones, ER translocases, glycosylases, and components of ER-associated degradation (ERAD) pathways [82,83,84] (Figure 1). In contrast, the truncated unspliced XBP1 (XBP1u) is highly unstable and rapidly degraded through the ubiquitin proteasome system [85]. Interestingly, prolonged ER stress constitutively activates IRE1α endoribonuclease activity to degrade several ER-localized mRNAs and alleviate the post-translational protein folding burden in ER. This process is termed regulated IRE1α-dependent decay (RIDD) [81,86]. Indeed, in metazoans, two isoforms of IRE1 exist, e.g., IRE1α/Ern1 and IRE1β/Ern2; the kinase and RNase domains of both the isoforms share 80% and 61% identity, respectively [63,87]. The human IRE1α is ubiquitously expressed in all types of tissues, whereas the expression of IRE1β is limited to IECs and [63,68] airway mucous cells [88]. In mice, the deletion of IRE1β [68] and the IEC-specific deletion of IRE1α [57] demonstrated perturbed UPR signaling, leading to IBD-like colitis. In line with the observation that IRE1α is more effective in XBP1 splicing and UPR activation compared to IRE1β [89], some interesting studies have investigated that IRE1β negatively regulates IRE1α activity and the related UPR signaling [67]. A recent in-vitro study showed that IRE1β directly interacts with IRE1α and inhibits its endonuclease activity [90]. Moreover, compared to IRE1α conditional knockout mice, IRE1β−/− mice showed exacerbated mucin secretion and enhanced ER stress signaling in goblet cells, leading to impaired intestinal barrier function [91]. Furthermore, IEC-specific deletion of IRE1α results in the reduction of goblet cell count and loss of epithelial integrity, leading to spontaneous colitis in mice [57]. The Streptomyces sp.-derived metabolite trierixin has an inhibitory effect on XBP1 splicing, highlighting the effect of environmental factors in the regulation of XBP1 downstream signaling [43]. Besides UPR-specific outcomes, the IRE1α/XBP1 branch of UPR signaling further provides an integrative platform for several intracellular molecules such as BAK, BAX, BI-1, and AIP-1 to orchestrate a coordinated physiological response [70].
Myeloid lineage displays a new dimension of restoring intestinal homeostasis in coordination with UPR signaling. Studies in murine macrophages showed that toll-like receptor (TLR) stimulation activates the IRE1α–XBP1 axis and induces the production of proinflammatory cytokine IL-6, independent of UPR activation [77]. In addition, murine macrophages infected with pathogenic strains provoke UPR signaling, leading to activation of apoptosis [92,93] and innate immune responses [73,76] to restore homeostasis. An interesting study showed that the activation of IRE1α coordinates with glycogen synthase kinase 3β (GSK3β) to modulate IL1β levels, independent of XBP1 processing [94], highlighting a differential outcome from the IRE1α/XBP1 branch in the regulation of inflammatory pathways during ER stress. A constitutively activated XBP1 was examined in antigen-presenting dendritic cells (DCs) required for cellular development and differentiation of DC subsets [75]. Furthermore, silencing XBP1 in murine DCs failed to induce TLR-dependent activation of IFN-β, suggesting an important role of XBP1 in mediating TLR-induced activation of the immune response [74]. An interesting study showed that constitutively active XBP1 in tumor-associated DCs impedes host antitumor immunity, leading to the progression of metastases [95]. The assorted functions of XBP1, in a cell-specific manner, further highlight its role in tuning various signaling pathways to maintain cellular homeostasis. Furthermore, GWAS have identified several genetic polymorphisms that confer susceptibility to IBD; one such polymorphism is located on human chromosome 22q12, encompassing XBP1 [96,97]. In addition, deep sequencing analysis has identified hypomorphic XBP1 variants in IBD patients [31,96]. Moreover, several IBD-associated SNPs have been identified in the XBP1 region, leading to the impaired activation of XBP1 [98]. Furthermore, a hypomorphic, ATG16L1, has been implicated in the defective clearance of IRE1α aggregates in IECs, leading to CD-like inflammation [67]. Collectively, all these studies demonstrate that the activation of the IRE1a–XBP1 axis in epithelial and myeloid cells orchestrate UPR downstream signaling to protect intestinal homeostasis.
2.2. PERK–eIF2α–CHOP Signaling in IECs and Myeloid Cells
PERK is a type I transmembrane protein kinase that shares relevant structural identity with IRE1 to encrypt a common mode of activation and downstream signaling [99,100]. Similar to IRE1, BiP disassociation from PERK causes a structural reorientation at C-terminal kinases, leading to autophosphorylation and autoactivation [100] (Figure 1). The activated PERK directly phosphorylates the Ser51 residue of eukaryotic translation initiation factor 2 (eIF2α-P) on the alpha subunit, leading to global attenuation of de novo translation, thereby imposing a transient pause on ER protein folding machinery [101,102]. An interesting study in mice showed that colonic epithelial deletion of IRE1α induces phosphorylation of eIF2α, governing the autonomy of the PERK–eIF2α axis among the UPR branches [57]. Furthermore, in mice, an impediment of eIF2α phosphorylation in Paneth cells leads to dysregulated protein machinery, which confers susceptibility to colitis [103]. This illustrates, apart from PERK autophosphorylation, the importance of downstream eIF2α phosphorylation in intestinal homeostasis and host immune responses. Indeed, PERK-activated eIF2α phosphorylation induces the translation of selective subsets of mRNAs, including bZIP transcription factor ATF4 (activating transcription factor 4) [104,105]. ATF4 transcription factor, downstream to the PERK–eIF2α axis, differently activates genes involved in glutathione biosynthesis, oxidative stress, and transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP) [59,61]. The transcription factor CHOP interacts with ATF4 and transactivates downstream genes implicated in energy metabolism, ER stress, and apoptosis [104]. Mice carrying a conditional ablation of IRE1α in colonic epithelial cells exhibited an increased CHOP expression, leading to aggravated ER stress and apoptosis [57,98,106]. Activated PERK–eIF2α–CHOP signaling simultaneously induces apoptosis and an inflammatory cascade [107]. Apart from transcription regulatory roles, CHOP expression also regulates mechanisms such as infiltration of macrophages, induction of ROS, and IL1β and apoptotic instigation in intestinal mucosa. The genetic ablation of CHOP has been shown to reduce apoptosis in colonic mucosa and protect mice from DSS (dextran sodium sulfate)-induced colitis [108]. Furthermore, microbial infection to myeloid cells induces CHOP expression and promotes its binding to the interleukin 23 (IL-23) promoter, leading to enhanced IL-23p19 expression, a cytokine associated with the pathogenesis of IBD [30,109]. Suppressing the expression of CHOP enhances the viability of the macrophages during immune responses [73,110].
Earlier studies have examined robust phosphorylation of eIF2α and activation of CHOP in the intestinal epithelia of patients with IBD [111,112]. Furthermore, inflamed colonic samples from patients with IBD demonstrated an increased level of phosphorylated eIF2α [113]. In contrast, the colonic tissue of patients with UC exhibited reduced phosphorylation of eIF2α, leading to attenuated translation of target genes in the epithelial cells [114]. A recent report demonstrated that ATF4 levels were reduced in the inflamed intestinal mucosa of patients with IBD, and mice lacking ATF4 exhibited exasperated intestinal inflammation by reducing the uptake of glutamine and expression of antimicrobial peptides in IECs [115]. Notably, cigarette smoke induces the activation of the PERK–eIF2α UPR branch [116,117], phosphorylation of eIF2α, and induction of CHOP and BiP [118,119]. Altogether, activation of the PERK–eIF2α axis attenuates global translation machinery to pause the protein folding burden on ER and activates CHOP to regulate the expression of genes required to restore cellular homeostasis.
2.3. ATF6 Signaling in IECs and Myeloid Cells
The UPR branch of ATF6 is a type II ER transmembrane glycoprotein composed of cytosolic protruding N-terminal basic leucine zipper (bZIP) transcription activator and ER lumen facing C-terminal [120]. Unlike PERK and IRE1α, upon the aggregation of unfolded proteins in the ER lumen, ATF6 is relieved from BiP and exposes its two Golgi-localization motifs (GLS1 and GLS2) that facilitate in translocation to the Golgi apparatus [121,122,123]. Upon reaching to the Golgi apparatus, the luminal domain of ATF6 undergoes processing by resident Site-1 (S1P) and Site-2 (S2P) proteases, and fragmented N-terminal ATF6 (ATF6 p50) is liberated in the cytoplasm [120,121]. The active ATF6 p50 subsequently translocates to the nucleus and transactivates target genes, including BiP, glucose-regulated protein 94 (Grp94), CHOP, and XBP1, to restore ER homeostasis [60,63,122] (Figure 1).
An interesting study examined evidence that genetically perturbed Mbtps1 (encoding S1P protease) mice exhibit severe DSS-induced colitis and that the S1P→ATF6 axis is essential to avoid DSS-induced colitis [124]. Furthermore, the S1P→ATF6 axis regulates bZIP transcription factor OASIS (old astrocyte specifically induced substance), which is critical for the differentiation and maturation of goblet cells in the large intestine [125], and OASIS−/− mice have been shown to exhibit severe DSS-induced colitis, leading to robust ER stress and apoptosis in intestinal epithelial cells [126].
ATF6 has two isoforms, ATF6α and ATF6β, both of which have partial sequence similarity. However, ATF6β has reduced ability to transactivate genes and may repress ATF6α-dependent activation during ER stress [127,128]. In line with these observations, an interesting study showed that the individual genetic ablation of ATF6α or ATF6β has no effect on the viability of mice, but ATF6 double knockout in mice causes embryonic lethality, suggesting that both ATF6α and ATF6β possess at least an overlapping function for transcriptional induction of UPR chaperones [129]. In mice, the conditional deletion of ATF6α exhibited impaired activation of chaperones such as BiP, Grp94, and P58IPK, leading to a defective inflammatory response in intestinal epithelium. Unresolved ER stress in ATF6α−/− mice intensified the DSS-induced colitis in colonic IECs [106]. The cell-specific response emanating from the ATF6–S1P axis is vital for the required downstream signaling to restore ER homeostasis. Notably, liver macrophages primed with prolonged ischemia triggered the ATF6 branch and increased secretion of proinflammatory cytokines TNF-α and IL-6 [130]. Transient repression of ATF6 alters macrophages expressing scavenging receptor CD36, leading to reduced uptake of oxidized low-density lipoprotein (ox-LDL) [131]. Finally, ATF6 activation in epithelial and myeloid cells transactivates factors to maintain ER homeostasis.
Altogether, the ER-UPR branches, IRE1, PERK, and ATF6, are the critical components of epithelial and myeloid cells to restore ER homeostasis, thereby attenuating an aberrant inflammatory response in the GI tract (Figure 1). Further studies with genetically engineered mice may enrich our understanding and find potential therapeutic targets.
3. Mitochondria-Associated UPR Activation and IBD
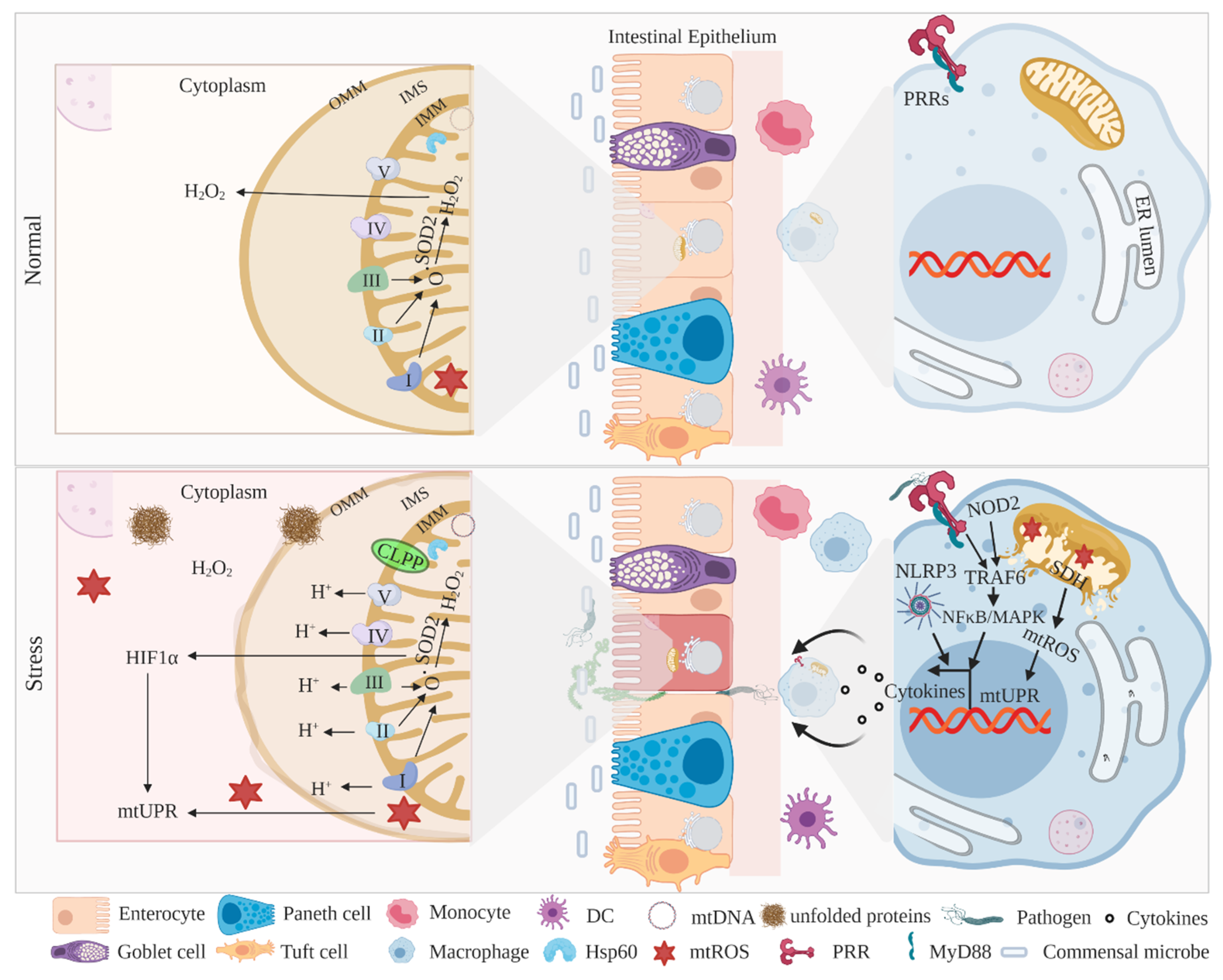
The mitochondrial matrix expresses a set of proteins to coordinate various mechanisms, including ATP synthesis, oxidative imbalance, calcium transport, and apoptosis [64,132,133]. The mitochondrial matrix also consistently responds to abnormal intracellular changes and pathogenic intrusions via a specialized group of proteins to restore cellular homeostasis [58,134,135]. Indeed, mitochondrial DNA (mtDNA) encodes electron transport chain (ETC) complex subunits, complex I (NADH dehydrogenase), complex IV (cytochrome c oxidase), complex V (ATP synthase), and complex III, with its own sets of translation machinery and chaperons capable of the proper folding of proteins in the matrix [136] (Figure 2). Particularly, mitochondrial chaperones such as heat shock protein 60 (HSP60) and caseinolytic mitochondrial matrix peptidase (CLPP) regulate the accumulation of unfolded proteins in the matrix to maintain homeostasis [58,62,137]. Consistent with ER-UPR signaling, mitochondrial UPR (mitoUPR) is critical to maintaining homeostasis in IECs [138,139]. Interestingly, the ER-UPR and mitoUPR share some common signaling axis, such as phosphorylation of eIF2α and activation of CHOP, suggesting a high degree of cooperation between these two organelles in regulating intestinal homeostasis [140,141].
Considering the fact that mitochondrial ETC complexes are the major source of ROS and superoxide molecules (Figure 2), impaired ETC signaling imposes an oxidative burden to cells, leading to various idiopathic diseases [142,143,144]. Earlier studies have examined a deregulated ETC complex activity in the mucosal biopsies of patients with IBD, which was substantially restored by anti-TNFα treatment [145,146]. Furthermore, the ultrastructure analysis of mitochondrion revealed altered cristae in the epithelial cells of patients with CD [147]. In this context, a robust expression of mitochondrial chaperone chaperonin 60 (CPN60) was examined in IECs from patients with IBD and in murine colitis models [65], suggesting a direct role of mitoUPR in intestinal inflammation. Indeed, an in-depth analysis of GWAS data sets identified ~5% of IBD-associated genes that are involved in mitochondrial health, highlighting that IBD susceptibility genes and loci may prospectively modulate the expression of mitochondrial-encrypted proteins and, in turn, elevate the severity of disease [148]. The underlying mechanism of mitoUPR in intestinal cell homeostasis is still enigmatic, and future studies with IEC-specific genetic ablation will enhance our understanding.
3.1. MitoUPR Signaling in IECs and Myeloid Cells
The rapidly proliferating and differentiating ISCs need sufficient building blocks and have energy demands, including ATP production, thereby possess an abundance of mitochondria to meet that requirement [56]. In this context, IEC- or ISC-specific Hsp60 knockout mice exhibited loss of Lgr5+ intestinal stem cells with perturbed IEC proliferation [56], suggesting the importance of mitoUPR signaling in maintaining intestinal homeostasis. An interesting study showed that a mutant of mitochondrial-localized ornithine transcarbamylase (involved in urea synthesis) accumulates its own misfolded protein in the matrix, leading to the activation of mitoUPR downstream targets such as mitochondrial HSP60 and the matrix-localized AAA protease CLPP [58] (Figure 2). Another study in small IEC line Mode-K cells showed that the induced expression of ornithine transcarbamylase activates double-stranded RNA-activated protein kinase (PKR) expression and eIF2α phosphorylation, which, in turn, induce mitoUPR signaling and lead to intestinal inflammation [65]. Conditional IEC-specific CHOP transgenic mice have consistently showed an overexpression of CHOP-conferred DSS-induced susceptibility to intestinal inflammation and attenuated epithelial proliferation [149]. In line with the contribution of the PERK–CHOP axis in ER-UPR signaling, the activation of CHOP and its cofactor C/EBPβ, in turn, activates mitoUPR target genes such as HSP60, HSP10, MPPβ, and proteases like ATP-dependent caseinolytic peptidase to restore intestinal homeostasis [58,150].
In the colon, a high expression of multidrug-resistance-1 (MDR1; encodes an ATP-dependent efflux transporter) protects mitochondria against aberrant stress, and loss of MDR1 expression (MDR1−/− mice) leads to an accumulation of damaged mitochondria and exacerbates mitochondrial ROS (mtROS), leading to colitis, as observed in IBD [148]. Importantly, the physiological oxygen concentrations across the GI tract are critical to intestinal inflammation; reduced oxygen levels within intestinal tissues generate hypoxia that leads to a low-grade inflammatory response from IECs, and to overcome this hypoxic condition, IECs induce the expression of hypoxia-inducible factor (HIF)-1α to maintain homeostasis [151,152]. The transcription factor HIF-1α in the inflamed mucosa incites an immune response and activates antimicrobial responses to protect mucosal integrity [152].
Notably, the mitochondrial enzyme SOD2 (superoxide dismutase-2), required to turnover superoxide into hydrogen peroxide, in turn, restores mitochondrial oxidative imbalance [153] and is implicated as a sentinel of mitoUPR [64] (Figure 2). HIF1α coordinates with mitochondrial SOD2 and activates mitoUPR signaling to maintain intestinal homeostasis [140]. Interestingly, genetic ablation of SOD2 in mice confers susceptibility to colitis; in contrast, mitochondria protecting MDR1 is negatively associated with SOD2 [148]. Prohibitin (PHB), which is predominantly expressed in the inner mitochondrial membrane (IMM), serves as a chaperon to stabilize mitochondrial proteins and is also required for the optimal functioning of complexes I and IV of ETC [154,155]. A recent study examined the conditional deletion of PHB1 in mice, which exhibited dysregulated mitochondrial ETC function and activation of mitoUPR. In addition, IEC Phb1−/− mice spontaneously develop ileitis, and loss of Phb1 in intestinal organoids leads to ISC abnormalities and Paneth cell dysfunction [156], suggesting that mitochondrial chaperons, apart from maintaining protein folding machinery, simultaneously regulate inflammatory responses.
Given the importance of mitochondrial ETC complexes in ROS production, some of the complex protein interacts with innate immune mediators and regulates intestinal inflammation [157,158]. In myeloid cells, the mitochondrial mechanisms, including mtROS, mitochondrial membrane potential (MMP), oxidized mtDNA, and ATP efflux, activates NLRP3 inflammasome signaling, suggesting that inflammatory outcomes are under metabolic control [159]. NLRP3−/− mice consistently exhibit low production of proinflammatory cytokines, leading to reduced severity of experimental colitis [160]. An interesting study with a mouse colitis model and patients with IBD has demonstrated that IL-10 deficiency results in impaired mitochondrial health and NLRP3 inflammasome activation in macrophages [161]. These studies highlight the potential involvement of innate signaling components in the regulation of mitochondrial outcomes during the pathogenesis of IBD. Furthermore, the PRR adaptor protein TRAF6 (Tumor necrosis factor receptor-associated factor 6) interacts with ECSIT (evolutionarily conserved signaling intermediate in Toll pathway) in mouse macrophages [162] and human monocyte THP-1 cells to enhance the ROS response and TLR4-induced innate immune responses [163]. Notably, mitochondrial metabolic processes, such as glutamine metabolism, activate macrophage polarization and differentiation in response to TLR stimulation [164]. In addition, PRR NOD2 (nucleotide-binding oligomerization domain containing 2) stimulation in human macrophages induces NFκB and MAPK (mitogen-activated protein kinase) activation, mitochondrial succinate dehydrogenase (SDH) (located in the IMM) activity, and mtROS for optimal cytokine secretion and microbial clearance [165] as NOD2 polymorphisms are associated with CD [166,167], suggesting that innate immune regulators also shape mitochondrial homeostasis (Figure 2). Altogether, these studies highlight the importance of mitoUPR signaling in epithelial and myeloid cells to regulate metabolic as well as inflammatory signaling to restore intestinal homeostasis.
3.2. Mitochondria in the Pathogenesis of IBD
Altered mitochondrial health and an imbalanced oxidative metabolism in the colonic epithelium confer pathogenesis of IBD [168]. GWAS have identified several IBD risk genes and locus implicated in mitochondrial dysfunction [32,135]. Notably, in European cohorts, mitochondrial-encrypted polymorphisms have been identified in patients with UC [169,170]. Furthermore, genetic variants of mitochondrial-signaling protein SLC22A5 (encodes OCTN2, carnitine transporter) were associated with susceptibility to IBD [171,172]. In addition, genetic ablation or pharmacological inhibition of OCTN2 caused spontaneous colitis in mice [173]. The IBD-associated polymorphism in autophagy-related proteins ATG16L1 is implicated in mitochondrial dysfunction, leading to impaired intestinal inflammation [34,174]. Furthermore, a polymorphism associated with an additional autophagy protein, IRGM, confers susceptibility to DSS-induced colitis, similar to the pathogenesis of CD [37,38,175]. A polymorphism in mitochondrial E3 ubiquitin ligase SMURF1 (smad ubiquitin regulatory factor 1) has been associated with the pathogenesis of IBD [33]. An interesting study showed that reduced mitochondrial complex I activity in patients with UC [176] leads to loss of epithelial permeability and microbial intrusion [177]. In addition, mitochondrial dysfunction may damage intestinal barrier permeability in patients with UC, with active and emission stages [178,179]. A reduced expression of mitochondrial chaperone PHB1 in the mucosa intensifies the disease severity of patients with IBD [154,180]. In the context of mitochondria’s role in metabolic regulation, a high turnover of lipid peroxidation in the colonic mucosa of patients with IBD highlights a feedback mechanism to protect the oxidative breakdown of lipids in maintaining intestinal homeostasis [181]. The hyperexpression of transcription factor HIF1α in the epithelium of patients with IBD further demonstrate a protective mechanism played by hypoxia to maintain the integrity of mucosal barriers [151]. The examination of mtDNA in the serum in patients with IBD consistently provides evidence of robust innate immune activation, which may intensify disease severity [182].
Altogether, mitochondrial health and resident mitoUPR machinery are interconnected to maintain homeostasis (Figure 2), and identifying signaling molecules that are associated with disease could be a therapeutic target for IBD treatment.
4. Therapeutic Implications Targeting UPR Restoration
Given the importance of UPR signaling within ER and mitochondria in the regulation of IEC outcomes, mitigating stress in these organelles is imperative to maintaining GI homeostasis. In recent years, an emerging body of experimental data has proposed several molecules implicated in UPR homeostasis, which can be prospectively used as therapeutic candidates for the treatment of IBD.
A recent study showed that azathioprine, an immunosuppressant drug used for IBD, modulates mTORC1 (mammalian target of rapamycin complex 1) signaling and induces the expression of PERK and associated downstream genes ATF4, CHOP, and ER stress chaperone protein disulfide isomerase (PDI) in THP-1-derived macrophages, suggesting that azathioprine induces a parallel mechanism of PERK–eIF2α-mediated global translation inhibition with simultaneous mTORC1 inhibition, leading to the activation of autophagy genes [183]. An earlier study demonstrated that chemical chaperones, tauroursodeoxycholate (TUDCA) and 4-phenylbutyrate (PBA), enhance protein refolding to impede ER stress and were shown to reduce DSS-induced colitis in mice with diminished ER stress in colonic epithelial cells [106]. Furthermore, ursodeoxycholic acid (the unconjugated form of TUDCA) was shown to be more effective in the inhibition of ER stress, compared to TUDAC, and it represents a potential therapeutic drug to resolve ER stress [184].
Food supplemented with glutamine reduces TNBS (2, 4, 6-trinitrobenzenesulfonic acid)-induced colitis in rats by alleviating ER stress in colonic epithelial cells [185]. In mice, administration of fexofenadine significantly reduces the severity of DSS-induced colitis by activation of NF-κB signaling and phosphorylation of eIF2α in IECs, representing a potential therapeutic agent for the treatment of IBD [186]. Another study in mice showed that salubrinal, an inhibitor of eIF2α dephosphorylation, enhances the expression of BiP, ATF4, and HSP70 (heat-shock protein 70) in colonic IECs and protects against DSS-induced colitis [187,188]. In the context of mitoUPR signaling, a recent study showed that in SOD2, mimetic antioxidant Mito-Tempo ameliorated Paneth cell abnormalities and ileitis in Prohibitin 1 (critical for ETC signaling) impaired mice, and are implicated as therapeutic options in a subset of patients with CD [156]. In another study, treatment by a tetracycline antibiotic, methacycline hydrochloride, induces the expression of mitochondrial chaperons in mouse 3T3 cells; however, it failed to induce mitoUPR signaling in mice [189]. These findings suggest that restoration of UPR homeostasis in ER and mitochondria through pharmacological intervention might be a promising therapeutic option in patients with IBD.
5. Conclusion and Future Perspectives
The integrity of the GI tract is critical to protect the host from a challenging environment, and perturbation in the barrier results in gastrointestinal disorders, including IBD. Several studies have established that IECs and myeloid cells induce several response mechanisms to maintain intestinal homeostasis. A continuous course of action imposes stress on these cells, including the accumulation of unfolded proteins, oxidative imbalance, and apoptosis. Notably, the intracellular organelles such as ER and mitochondria are well-equipped with conserved sets of molecules, which help in resolving the intracellular stress. UPR signaling is a sequential activation of an organized intracellular response mechanism encrypted by ER (ER-UPR) and mitochondria (mitoUPR) to regain cellular homeostasis. Many studies have shown that a coordinated response from all three branches of ER-UPR signaling (IRE1, PERK, and ATF6) are critical to resolving cellular stress and, in turn, protecting GI integrity. Moreover, the innate immune components in coordination with UPR signaling mediators provide an additional arm to mitigate cellular stress. Indeed, IBD-associated polymorphism-encompassing genes, which are vital for the cellular defense mechanism, further intensify the severity of the disease. Therefore, in the light of this evidence, further studies for the underlying mechanisms associated with pathophysiological alterations of UPR within the intestines may allow us to decipher the intricate etiology and pathogenesis of IBD.
Funding
This research received no external funding.
Acknowledgments
I am thankful to Clara Abraham, Yale University, for her guidance and support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Mowat, A.; Agace, W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Ji, M.; Liang, H.-E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 2015, 529, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchenough, G.M.; Johansson, M.E.V.; Gustafsson, J.K.; Bergström, J.H.; Hansson, G.C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015, 8, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoop, K.A.; Newberry, R.D. Goblet cells: Multifaceted players in immunity at mucosal surfaces. Mucosal Immunol. 2018, 11, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Genet. 2011, 9, 265–278. [Google Scholar] [CrossRef]
- Van Der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That MUC2 Is Critical for Colonic Protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Enteroendocrine Cells: Chemosensors in the Intestinal Epithelium. Annu. Rev. Physiol. 2016, 78, 277–299. [Google Scholar] [CrossRef]
- Birchenough, G.M.H.; Nyström, E.E.L.; Johansson, M.E.V.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Oshima, S.; Nakamura, T.; Namiki, S.; Okada, E.; Tsuchiya, K.; Okamoto, R.; Yamazaki, M.; Yokota, T.; Aida, M.; Yamaguchi, Y.; et al. Interferon Regulatory Factor 1 (IRF-1) and IRF-2 Distinctively Up-Regulate Gene Expression and Production of Interleukin-7 in Human Intestinal Epithelial Cells. Mol. Cell. Biol. 2004, 24, 6298–6310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunliffe, R.N.; Rose, F.R.A.J.; Keyte, J.; Abberley, L.; Chan, W.; Mahida, Y.R. Human defensin 5 is stored in precursor form in normal Paneth cells and is expressed by some villous epithelial cells and by metaplastic Paneth cells in the colon in inflammatory bowel disease. Gut 2001, 48, 176–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevalainen, T.J.; Grönroos, J.M.; Kallajoki, M. Expression of group II phospholipase A2 in the human gastrointestinal tract. Lab. Investig. 1995, 72, 201–208. [Google Scholar] [PubMed]
- Hooper, L.V.; Stappenbeck, T.S.; Hong, C.V.; Gordon, J.I. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 2003, 4, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin regiiigamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Bevins, C.L. Paneth Cells: Maestros of the Small Intestinal Crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- Ayabe, T.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of microbicidal α-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar] [CrossRef]
- Ganz, T. Paneth cells—Guardians of the gut cell hatchery. Nat. Immunol. 2000, 1, 99–100. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Schmitt, M.; Schewe, M.; Sacchetti, A.; Feijtel, D.; Van De Geer, W.S.; Teeuwssen, M.; Sleddens, H.F.; Joosten, R.; Van Royen, M.E.; Van De Werken, H.J.; et al. Paneth Cells Respond to Inflammation and Contribute to Tissue Regeneration by Acquiring Stem-like Features through SCF/c-Kit Signaling. Cell Rep. 2018, 24, 2312–2328.e7. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Everard, A.; Duparc, T. Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 2013, 13, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Van Der Flier, L.G.; Clevers, H. Stem Cells, Self-Renewal, and Differentiation in the Intestinal Epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; Van Der Flier, L.G.; Sato, T.; Van Es, J.H.; Born, M.V.D.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; Van Rheenen, J.; Simons, B.D.; et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Born, M.V.D.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, R.; Watanabe, M. Role of epithelial cells in the pathogenesis and treatment of inflammatory bowel disease. J. Gastroenterol. 2015, 51, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Surawicz, C.M.; Haggitt, R.C.; Husseman, M.; McFarland, L.V. Mucosal biopsy diagnosis of colitis: Acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology 1994, 107, 755–763. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Johansson, M.E.V.; Schütte, A.; Hansson, G.C.; Sjövall, H. An ex vivo method for studying mucus formation, properties, and thickness in human colonic biopsies and mouse small and large intestinal explants. Am. J. Physiol. Liver Physiol. 2012, 302, G430–G438. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.V.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2013, 63, 281–291. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J. IL-23 and Autoimmunity: New Insights into the Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Med. 2009, 60, 97–110. [Google Scholar] [CrossRef] [Green Version]
- Barmada, M.M.; Brant, S.R.; Nicolae, D.L.; Achkar, J.-P.; Panhuysen, C.I.; Bayless, T.M.; Cho, J.; Duerr, R.H. A genome scan in 260 inflammatory bowel disease-affected relative pairs. Inflamm. Bowel Dis. 2004, 10, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; The NIDDK IBD Genetics Consortium; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, A.; And the IBSEN Study Group; Balschun, T.; Sina, C.; Ellinghaus, E.; Häsler, R.; Mayr, G.; Albrecht, M.; Wittig, M.; Buchert, E.; et al. Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat. Genet. 2010, 42, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2006, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; Devoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [Green Version]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef]
- van der Sloot, K.W.J.; Amini, M.; Peters, V.; Dijkstra, G.; Alizadeh, B.Z. Inflammatory bowel diseases: Review of known environmental protective and risk factors involved. Inflamm. Bowel Dis. 2017, 23, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Adolph, T.E.; Blumberg, R.S. The unfolded protein response and gastrointestinal disease. Semin. Immunopathol. 2013, 35, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Martinez-Naves, E.; Blumberg, R.S. Endoplasmic reticulum stress: Implications for inflammatory bowel disease pathogenesis. Curr. Opin. Gastroenterol. 2010, 26, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, E.; Hironiwa, N.; Kitagawa, M.; Futamura, Y.; Suzuki, S.-I.; Nishio, M.; Imoto, M. ChemInform Abstract: Trierixin, a Novel Inhibitor of ER Stress-Induced XBP1 Activation from Streptomyces sp. Part 1. Taxonomy, Fermentation, Isolation, and Biological Activities. ChemInfrom 2008, 39, 547–553. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kaplan, G.G. Environmental Risk Factors for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2010, 6, 339–346. [Google Scholar]
- Rogler, G.; Zeitz, J.; Biedermann, L. The Search for Causative Environmental Factors in Inflammatory Bowel Disease. Dig. Dis. 2016, 34, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosnes, J. What is the link between the use of tobacco and ibd? Inflamm. Bowel Dis. 2008, 14, S14–S15. [Google Scholar] [CrossRef] [PubMed]
- Cosnes, J.; Gower-Rousseau, C.; Seksik, P.; Cortot, A. Epidemiology and Natural History of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1785–1794.e4. [Google Scholar] [CrossRef]
- Birrenbach, T.; Böcker, U. Inflammatory bowel disease and smoking: A review of epidemiology, pathophysiology, and therapeutic implications. Inflamm. Bowel Dis. 2004, 10, 848–859. [Google Scholar] [CrossRef]
- Verschuere, S.; Bracke, K.R.; Demoor, T.; Plantinga, M.; Verbrugghe, P.L.; Ferdinande, L.; Lambrecht, B.N.; Brusselle, G.G.G.; Cuvelier, C.A. Cigarette smoking alters epithelial apoptosis and immune composition in murine GALT. Lab. Investig. 2011, 91, 1056–1067. [Google Scholar] [CrossRef]
- Berg, D.J.; Zhang, J.; Weinstock, J.V.; Ismail, H.F.; Earle, K.A.; Alila, H.; Pamukcu, R.; Moore, S.A.; Lynch, R.G. Rapid development of colitis in NSAID-treated IL-10–deficient mice. Gastroenterology 2002, 123, 1527–1542. [Google Scholar] [CrossRef]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2014, 64, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Author Correction: Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 898. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; MacPherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva Medica 2019, 110, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Rosso, C.; Ribaldone, D.G.; Dughera, F.; Fagoonee, S.; Astegiano, M.; Pellicano, R. Physiopathology of intestinal barrier and the role of zonulin. Minerva Biotecnol. 2019, 31, 83–92. [Google Scholar] [CrossRef]
- Berger, E.; Rath, E.; Yuan, D.; Waldschmitt, N.; Khaloian, S.; Allgäuer, M.; Staszewski, O.; Lobner, E.M.; Schöttl, T.; Giesbertz, P.; et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016, 7, 13171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-S.; Chen, Y.; Fan, L.; Xi, Q.-L.; Wu, G.-H.; Li, X.-X.; Yuan, T.-L.; He, S.-Q.; Yu, Y.; Shao, M.; et al. The Endoplasmic Reticulum Stress Sensor IRE1α in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J. Biol. Chem. 2015, 290, 15327–15336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.-M.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Todd, D.J.; Lee, A.-H.; Glimcher, L.H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 2008, 8, 663–674. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Broadley, S.A.; Hartl, F.U. Mitochondrial stress signaling: A pathway unfolds. Trends Cell Biol. 2008, 18, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.; Moschetta, A.; Haller, D. Mitochondrial function—Gatekeeper of intestinal epithelial cell homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 497–516. [Google Scholar] [CrossRef]
- Rath, E.; Berger, E.; Messlik, A.; Nunes, T.; Liu, B.; Kim, S.C.; Hoogenraad, N.; Sans, M.; Sartor, R.B.; Haller, D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 2011, 61, 1269–1278. [Google Scholar] [CrossRef]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.D.; Price, G.R.; Tauro, S.B.; Taupin, U.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant Mucin Assembly in Mice Causes Endoplasmic Reticulum Stress and Spontaneous Inflammation Resembling Ulcerative Colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Bertolotti, A.; Wang, X.; Novoa, I.; Jungreis, R.; Schlessinger, K.; Cho, J.H.; West, A.B.; Ron, D. Increased sensitivity to dextran sodium sulfate colitis in ire1beta-deficient mice. J. Clin. Investg. 2001, 107, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Bakunts, A.; Orsi, A.; Vitale, M.; Cattaneo, A.; Lari, F.; Tadè, L.; Sitia, R.; Raimondi, A.; Bachi, A.; Van Anken, E. Ratiometric sensing of BiP-client versus BiP levels by the unfolded protein response determines its signaling amplitude. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Hetz, C.; Glimcher, L.H. Fine-Tuning of the Unfolded Protein Response: Assembling the IRE1α Interactome. Mol. Cell 2009, 35, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.C.; Larburu, N.; Durairaj, V.; Adams, C.J.; Ali, M.M.U. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol. 2019, 26, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.-H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Yu, X.; Wang, H.; Zuo, D.; Guo, C.; Yi, H.; Tirosh, B.; Subjeck, J.R.; Qiu, X.; Wang, X.-Y. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur. J. Immunol. 2011, 41, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic ifn-beta induction via x-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Núñez, G.; Grethe, J.S.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Hedl, M.; Ranjan, K.; Abraham, C. LACC1 Required for NOD2-Induced, ER Stress-Mediated Innate Immune Outcomes in Human Macrophages and LACC1 Risk Variants Modulate These Outcomes. Cell Rep. 2019, 29, 4525–4539.e4. [Google Scholar] [CrossRef] [Green Version]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Kopp, M.C.; Nowak, P.R.; Larburu, N.; Adams, C.J.; Ali, M.M.U. In vitro FRET analysis of IRE1 and BiP association and dissociation upon endoplasmic reticulum stress. eLife 2018, 7, e30257. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. Ire1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, B.; Iwakoshi, N.N.; Glimcher, L.H.; Ploegh, H.L. Rapid Turnover of Unspliced Xbp-1 as a Factor That Modulates the Unfolded Protein Response. J. Biol. Chem. 2005, 281, 5852–5860. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nature 2001, 3, 158–164. [Google Scholar] [CrossRef]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M. The ER stress transducer ire1beta is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, Y.; Hosoda, A.; Sasaka, S.-I.; Tsuru, A.; Kohno, K. RNase domains determine the functional difference between IRE1α and IRE1β. FEBS Lett. 2008, 582, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Grey, M.J.; Cloots, E.; Simpson, M.S.; LeDuc, N.; Serebrenik, Y.V.; De Luca, H.; De Sutter, D.; Luong, P.; Thiagarajah, J.R.; Paton, A.W.; et al. IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. J. Cell Biol. 2020, 219, e201904048. [Google Scholar] [CrossRef] [Green Version]
- Tsuru, A.; Fujimoto, N.; Takahashi, S.; Saito, M.; Nakamura, D.; Iwano, M.; Iwawaki, T.; Kadokura, H.; Ron, D.; Kohno, K. Negative feedback by ire1beta optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2864–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillich, H.; Loose, M.; Zimmer, K.-P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell. Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Kim, M.-J.; Blumenthal, A.; Koo, J.; Ehrt, S.; Wainwright, H.; Bekker, L.-G.; Kaplan, G.; Nathan, C.; Tabas, I.; et al. Induction of ER Stress in Macrophages of Tuberculosis Granulomas. PLoS ONE 2010, 5, e12772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Joe, Y.; Kim, H.J.; Kim, Y.S.; Jeong, S.O.; Pae, H.O.; Ryter, S.W.; Surh, Y.J.; Chung, H.T. Endoplasmic reticulum stress-induced ire1alpha activation mediates cross-talk of gsk-3beta and xbp-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, S.; Rutgeerts, P.; Van Steen, K.; Joossens, S.; Claessens, G.; Pierik, M.; Peeters, M.; Vlietinck, R. Genome wide scan in a Flemish inflammatory bowel disease population: Support for the IBD4 locus, population heterogeneity, and epistasis. Gut 2004, 53, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Hampe, J.; Schreiber, S.; Shaw, S.H.; Lau, K.F.; Bridger, S.; MacPherson, A.J.; Cardon, L.R.; Sakul, H.; Harris, T.J.; Buckler, A.; et al. A Genomewide Analysis Provides Evidence for Novel Linkages in Inflammatory Bowel Disease in a Large European Cohort. Am. J. Hum. Genet. 1999, 64, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, C.Y.; Back, S.H.; Clark, R.L.; Peisach, D.; Xu, Z.; Kaufman, R.J. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 2006, 103, 14343–14348. [Google Scholar] [CrossRef] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M.U. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Cao, S.S.; Harrington, J.C.; Chuang, B.-M.; Kaufman, R.J.; Wang, M.; Eckmann, L. Phosphorylation of eIF2α Is Dispensable for Differentiation but Required at a Posttranscriptional Level for Paneth Cell Function and Intestinal Homeostasis in Mice. Inflamm. Bowel Dis. 2014, 20, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.-J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989–1000.e6. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Namba, T.; Tanaka, K.-I.; Ito, Y.; Ishihara, T.; Hoshino, T.; Gotoh, T.; Endo, M.; Sato, K.; Mizushima, T. Positive Role of CCAAT/Enhancer-Binding Protein Homologous Protein, a Transcription Factor Involved in the Endoplasmic Reticulum Stress Response in the Development of Colitis. Am. J. Pathol. 2009, 174, 1786–1798. [Google Scholar] [CrossRef] [Green Version]
- Goodall, J.C.; Wu, C.; Zhang, Y.; McNeill, L.; Ellis, L.; Saudek, V.; Gaston, J.S.H. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17698–17703. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.W.; Cui, D.; Arellano, J.; Dorweiler, B.; Harding, H.P.; Fitzgerald, K.A.; Ron, D.; Tabas, I. Adaptive suppression of the ATF4–CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 2009, 11, 1473–1480. [Google Scholar] [CrossRef] [Green Version]
- Shkoda, A.; Ruiz, P.A.; Daniel, H.; Kim, S.C.; Rogler, G.; Sartor, R.B.; Haller, D. Interleukin-10 Blocked Endoplasmic Reticulum Stress in Intestinal Epithelial Cells: Impact on Chronic Inflammation. Gastroenterology 2007, 132, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Burczynski, M.E.; Peterson, R.L.; Twine, N.C.; Zuberek, K.A.; Brodeur, B.J.; Casciotti, L.; Maganti, V.; Reddy, P.S.; Strahs, A.; Immermann, F.; et al. Molecular Classification of Crohn’s Disease and Ulcerative Colitis Patients Using Transcriptional Profiles in Peripheral Blood Mononuclear Cells. J. Mol. Diagn. 2006, 8, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaert, S.; De Vos, M.; Olievier, K.; Peeters, H.; Elewaut, D.; Lambrecht, B.; Pouliot, P.; Laukens, D. Involvement of Endoplasmic Reticulum Stress in Inflammatory Bowel Disease: A Different Implication for Colonic and Ileal Disease? PLoS ONE 2011, 6, e25589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treton, X.; Pedruzzi, E.; Cazals–Hatem, D.; Grodet, A.; Panis, Y.; Groyer, A.; Moreau, R.; Bouhnik, Y.; Daniel, F.; Ogier-Denis, E. Altered Endoplasmic Reticulum Stress Affects Translation in Inactive Colon Tissue From Patients With Ulcerative Colitis. Gastroenterology 2011, 141, 1024–1035. [Google Scholar] [CrossRef]
- Hu, X.; Deng, J.; Yu, T.; Chen, S.; Ge, Y.; Zhou, Z.; Guo, Y.; Ying, H.; Zhai, Q.; Chen, Y.; et al. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology 2019, 156, 1098–1111. [Google Scholar] [CrossRef]
- Jorgensen, E.D.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, Y.; Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med. 2008, 45, 50–59. [Google Scholar] [CrossRef]
- Adair-Kirk, T.L.; Atkinson, J.J.; Griffin, G.L.; Watson, M.A.; Kelley, D.G.; Demello, D.; Senior, R.M.; Betsuyaku, T. Distal Airways in Mice Exposed to Cigarette Smoke: Nrf2-regulated genes are increased in clara cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Hackett, N.R.; Heguy, A.; Harvey, B.-G.; O’Connor, T.P.; Luettich, K.; Flieder, D.B.; Kaplan, R.; Crystal, R.G. Variability of Antioxidant-Related Gene Expression in the Airway Epithelium of Cigarette Smokers. Am. J. Respir. Cell Mol. Biol. 2003, 29, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian Transcription Factor ATF6 Is Synthesized as a Transmembrane Protein and Activated by Proteolysis in Response to Endoplasmic Reticulum Stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.; O’Hare, P. Transmembrane bZIP Transcription Factors in ER Stress Signaling and the Unfolded Protein Response. Antioxid. Redox Signal. 2007, 9, 2305–2322. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable Binding of ATF6 to BiP in the Endoplasmic Reticulum Stress Response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandl, K.; Rutschmann, S.; Li, X.; Du, X.; Xiao, N.; Schnabl, B.; Brenner, D.A.; Beutler, B. Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6-driven unfolded protein response. Proc. Natl. Acad. Sci. USA 2009, 106, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Asada, R.; Saito, A.; Kawasaki, N.; Kanemoto, S.; Iwamoto, H.; Oki, M.; Miyagi, H.; Izumi, S.; Imaizumi, K. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J. Biol. Chem. 2012, 287, 8144–8153. [Google Scholar] [CrossRef] [Green Version]
- Hino, K.; Saito, A.; Asada, R.; Kanemoto, S.; Imaizumi, K. Increased Susceptibility to Dextran Sulfate Sodium-Induced Colitis in the Endoplasmic Reticulum Stress Transducer OASIS Deficient Mice. PLoS ONE 2014, 9, e88048. [Google Scholar] [CrossRef] [Green Version]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C.; Zhu, H.; Toso, R.; Camire, R.M. Effects of the Isoform-specific Characteristics of ATF6 α and ATF6 β on Endoplasmic Reticulum Stress Response Gene Expression and Cell Viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [Green Version]
- Thuerauf, D.J.; Morrison, L.; Glembotski, C.C. Opposing roles for atf6alpha and atf6beta in endoplasmic reticulum stress response gene induction. J. Biol. Chem. 2004, 279, 21078–21084. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Zhai, Y. ATF6 Mediates a Pro-Inflammatory Synergy Between ER Stress and TLR Activation in the Pathogenesis of Liver Ischemia-Reperfusion Injury. Arab. Archaeol. Epigr. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Miao, C.; Tian, H.; Sang, H.; Yang, N.; Jiao, P.; Han, J.; Zong, C.; Qin, S. Endoplasmic Reticulum Stress Promotes Macrophage-derived Foam Cell Formation by Up-regulating Cluster of Differentiation 36 (CD36) Expression. J. Biol. Chem. 2013, 289, 4032–4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjan, K.; Pathak, C. Expression of FADD and cFLIPL balances mitochondrial integrity and redox signaling to substantiate apoptotic cell death. Mol. Cell. Biochem. 2016, 422, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.-M.; Rungi, A.A.; Robinson, B.H.; Hood, D.A. Compensatory responses of protein import and transcription factor expression in mitochondrial DNA defects. Am. J. Physiol. Physiol. Cell 2004, 286, C867–C875. [Google Scholar] [CrossRef]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of Genes Activated by the Mitochondrial Unfolded Protein Response (mtUPR) and Cognate Promoter Elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-Nuclear Communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef]
- Fung, K.Y.; Brierley, G.V.; Henderson, S.; Hoffmann, P.; McColl, S.R.; Lockett, T.J.; Head, R.; Cosgrove, L. Butyrate-Induced Apoptosis in HCT116 Colorectal Cancer Cells Includes Induction of a Cell Stress Response. J. Proteome Res. 2011, 10, 1860–1869. [Google Scholar] [CrossRef]
- Kolar, S.; Barhoumi, R.; Jones, C.K.; Wesley, J.; Lupton, J.R.; Fan, Y.-Y.; Chapkin, R.S.; Bs, C.K.J. Interactive effects of fatty acid and butyrate-induced mitochondrial Ca2+ loading and apoptosis in colonocytes. Cancer 2011, 117, 5294–5303. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Hart, P.; Germain, D.; Bonini, M.G. SOD2 and the Mitochondrial UPR: Partners Regulating Cellular Phenotypic Transitions. Trends Biochem. Sci. 2016, 41, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Rath, E.; Haller, D. Mitochondria at the Interface Between Danger Signaling and Metabolism: Role of Unfolded Protein Responses in Chronic Inflammation. Inflamm. Bowel Dis. 2012, 18, 1364–1377. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; Van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Schürmann, G.; Brüwer, M.; Klotz, A.; Schmid, K.W.; Senninger, N.; Zimmer, K.-P. Transepithelial transport processes at the intestinal mucosa in inflammatory bowel disease. Int. J. Color. Dis. 1999, 14, 41–46. [Google Scholar] [CrossRef]
- Heller, S.; Penrose, H.M.; Cable, C.; Biswas, D.; Nakhoul, H.; Baddoo, M.; Flemington, E.; Crawford, S.E.; Savkovic, S.D. Reduced mitochondrial activity in colonocytes facilitates ampkalpha2-dependent inflammation. FASEB J. 2017, 31, 2013–2025. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzen, L.E.; Lindmark, T.; Wiren, M.; Tagesson, C.; Sjodahl, R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of crohn’s disease. Gut 2002, 50, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.T.; Aird, R.E.; Liu, B.; Boyapati, R.K.; Kennedy, N.A.; Dorward, D.A.; Noble, C.L.; Shimizu, T.; Carter, R.N.; Chew, E.T.; et al. MDR1-deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. 2017, 11, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Waldschmitt, N.; Berger, E.; Rath, E.; Sartor, R.B.; Weigmann, B.; Heikenwälder, M.; Gerhard, M.; Janssen, K.-P.; Haller, D. C/EBP homologous protein inhibits tissue repair in response to gut injury and is inversely regulated with chronic inflammation. Mucosal Immunol. 2014, 7, 1452–1466. [Google Scholar] [CrossRef]
- Horibe, T.; Hoogenraad, N.J. The Chop Gene Contains an Element for the Positive Regulation of the Mitochondrial Unfolded Protein Response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [Green Version]
- Giatromanolaki, A.; Sivridis, E.; Maltezos, E.; Papazoglou, D.; Simopoulos, C.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Hypoxia inducible factor 1α and 2α overexpression in inflammatory bowel disease. J. Clin. Pathol. 2003, 56, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitouni, N.E.; Chotikatum, S.; Von Köckritz-Blickwede, M.; Naim, H.Y. The impact of hypoxia on intestinal epithelial cell functions: Consequences for invasion by bacterial pathogens. Mol. Cell. Pediatr. 2016, 3, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Theiss, A.L.; Idell, R.D.; Srinivasan, S.; Klapproth, J.-M.; Jones, D.P.; Merlin, D.; Sitaraman, S.V. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 2006, 21, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; De Jong, L.; Artal-Sanz, M.; Coates, P.J.; Berden, J.A.; Back, J.W.; Muijsers, A.O.; Van Der Spek, H.; Grivell, L.A. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000, 19, 2444–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C.; et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J.C. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 2002, 166, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Próchnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (dss) is mediated by the nlrp3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wi, S.M.; Moon, G.; Kim, J.; Kim, S.T.; Shim, J.H.; Chun, E.; Lee, K.Y. Tak1-ecsit-traf6 complex plays a key role in the tlr4 signal to activate nf-kappab. J. Biol. Chem. 2014, 289, 35205–35214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. Alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Lahiri, A.; Hedl, M.; Yan, J.; Abraham, C. Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nat. Commun. 2017, 8, 15614. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Abraham, C.; Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [Green Version]
- Roediger, W. The colonic epithelium in ulcerative colitis: An energy-deficiency disease? Lancet 1980, 316, 712–715. [Google Scholar] [CrossRef]
- Dankowski, T.; Schröder, T.; Möller, S.; Yu, X.; Ellinghaus, D.; Bär, F.; Fellermann, K.; Lehnert, H.; Stefansson, K.; Franke, A.; et al. Male-specific association between MT-ND4 11719 A/G polymorphism and ulcerative colitis: A mitochondria-wide genetic association study. BMC Gastroenterol. 2016, 16, 118. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Abrantes, P.; Sousa, I.; Francisco, V.; Santos, P.; Francisco, D.; Xavier, J.M.; Oliveira, S.A. Ulcerative Colitis Is Under Dual (Mitochondrial and Nuclear) Genetic Control. Inflamm. Bowel Dis. 2016, 22, 774–781. [Google Scholar] [CrossRef]
- Lin, Z.; Nelson, L.; Franke, A.; Poritz, L.; Li, T.-Y.; Wu, R.; Wang, Y.; MacNeill, C.; Thomas, N.J.; Stefansson, K.; et al. OCTN1 variant L503F is associated with familial and sporadic inflammatory bowel disease. J. Crohns Coliti 2010, 4, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, S.; Tremelling, M.; Bredin, F.; Godfrey, L.; Howson, J.M.M.; Parkes, M. Evidence for association of OCTN genes and IBD5 with ulcerative colitis. Gut 2006, 55, 809–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekhawat, P.S.; Srinivas, S.R.; Matern, D.; Bennett, M.J.; Boriack, R.; George, V.; Xu, H.; Prasad, P.D.; Roon, P.; Ganapathy, V. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (octn2(-/-)) mice. Mol. Genet. Metab. 2007, 92, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef] [PubMed]
- Rogala, A.R.; Schoenborn, A.A.; Fee, B.E.; Cantillana, V.A.; Joyce, M.J.; Gharaibeh, R.Z.; Roy, S.; Fodor, A.A.; Sartor, R.B.; Taylor, G.A.; et al. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis. Model. Mech. 2017, 11, dmm031070. [Google Scholar] [CrossRef] [Green Version]
- Haberman, Y.; Karns, R.; Dexheimer, P.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Keita, Å.V.; Phan, V.; McKay, C.M.; Schoultz, I.; Lee, J.; Murphy, M.P.; Fernando, M.; Ronaghan, N.; Balce, D.; et al. Targeting Mitochondria-Derived Reactive Oxygen Species to Reduce Epithelial Barrier Dysfunction and Colitis. Am. J. Pathol. 2014, 184, 2516–2527. [Google Scholar] [CrossRef] [Green Version]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.-D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2006, 56, 61–72. [Google Scholar] [CrossRef]
- Büning, C.; Geissler, N.; Prager, M.; Sturm, A.; Baumgart, D.C.; Büttner, J.; Bühner, S.; Haas, V.; Lochs, H. Increased small intestinal permeability in ulcerative colitis: Rather genetic than environmental and a risk factor for extensive disease? Inflamm. Bowel Dis. 2012, 18, 1932–1939. [Google Scholar] [CrossRef]
- Hsieh, S.-Y.; Shih, T.-C.; Yeh, C.-Y.; Lin, C.-J.; Chou, Y.-Y.; Lee, Y.-S. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 2006, 6, 5322–5331. [Google Scholar] [CrossRef]
- Pereira, C.C.; Grácio, D.; Teixeira, J.P.; Magro, F. Oxidative stress and DNA damage: Implications in inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, R.; Dorward, D.A.; Tamborska, A.; Kalla, R.; Ventham, N.T.; Doherty, M.K.; Whitfield, P.D.; Gray, M.; Loane, J.; Rossi, A.G.; et al. Mitochondrial DNA Is a Pro-Inflammatory Damage-Associated Molecular Pattern Released During Active IBD. Inflamm. Bowel Dis. 2018, 24, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.M.; Casanova, V.; Kemp, S.; Staines, K.A.; Satsangi, J.; Barlow, P.G.; Henderson, P.; Stevens, C. The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 2019, 25, 1481–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, E.; Haller, D. Structure–function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2011, 409, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; San-Miguel, B.; Prause, C.; Marroni, N.; Cuevas, M.J.; González-Gallego, J.; Tuñón, M.J. Glutamine Treatment Attenuates Endoplasmic Reticulum Stress and Apoptosis in TNBS-Induced Colitis. PLoS ONE 2012, 7, e50407. [Google Scholar] [CrossRef]
- Koh, S.J.; Kim, J.W.; Kim, B.G.; Lee, K.L.; Chun, J.; Kim, J.S. Fexofenadine regulates nuclear factor-kappab signaling and endoplasmic reticulum stress in intestinal epithelial cells and ameliorates acute and chronic colitis in mice. J. Pharmacol. Exp. Ther. 2015, 352, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, N.; Kaufman, R.J.; Ma, D.; Coen, N.M.; Ron, D.; et al. A Selective Inhibitor of eIF2α Dephosphorylation Protects Cells from ER Stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Okazaki, T.; Nishio, A.; Takeo, M.; Sakaguchi, Y.; Fukui, T.; Uchida, K.; Okazaki, K. Inhibition of the Dephosphorylation of Eukaryotic Initiation Factor 2α Ameliorates Murine Experimental Colitis. Digestion 2014, 90, 147–158. [Google Scholar] [CrossRef]
- Rauthan, M.; Pilon, M. A chemical screen to identify inducers of the mitochondrial unfolded protein response in C. elegans. Worm 2015, 4, e1096490. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Endoplasmic reticulum (ER)-associated unfolded protein response (UPR) signaling in intestinal epithelial cells (IECs) and myeloid cells. The three branches of ER-UPR, IRE1a, PERK, and ATF6, remain inactive and sequestered with chaperon BiP in the resting stage. Intestinal epithelium perturbation activates all three branches of ER-UPR and leads to sequential activation of downstream target gene-encoding chaperons and transcriptional regulators to restore ER homeostasis. Similarly, the macrophages (right), upon encountering pathogenic intrusion, activate PRRs and coordinate with UPR signaling to induce proinflammatory cytokines and UPR-associated transcripts to protect against microbial intrusion. DC, dendritic cell; PRRs, pathogen recognition receptors; S1/2P, site 1/2 proteases; UPR, unfolded protein response; XBP1u/s, XBP1 unspliced/spliced.
Figure 1.
Endoplasmic reticulum (ER)-associated unfolded protein response (UPR) signaling in intestinal epithelial cells (IECs) and myeloid cells. The three branches of ER-UPR, IRE1a, PERK, and ATF6, remain inactive and sequestered with chaperon BiP in the resting stage. Intestinal epithelium perturbation activates all three branches of ER-UPR and leads to sequential activation of downstream target gene-encoding chaperons and transcriptional regulators to restore ER homeostasis. Similarly, the macrophages (right), upon encountering pathogenic intrusion, activate PRRs and coordinate with UPR signaling to induce proinflammatory cytokines and UPR-associated transcripts to protect against microbial intrusion. DC, dendritic cell; PRRs, pathogen recognition receptors; S1/2P, site 1/2 proteases; UPR, unfolded protein response; XBP1u/s, XBP1 unspliced/spliced.

Figure 2.
Mitochondrial-associated UPR signaling in IECs and myeloid cells. Mitochondrial ETC molecules (complex I, II, III, IV, and V) regulate ROS, superoxide, and ATP balance in IMM. The mitochondrial chaperon Hsp60 regulates protein-folding machinery in the mitochondrial matrix and maintains homeostasis; however, accumulation of unfolded proteins in the mitochondrial intermembrane space (IMS) induces mitochondrial ROS (mtROS) and superoxide signaling and coordinates with HIF1α to activate mitoUPR signaling. On the other side, the activated macrophage (right), in response to pathogen recognition, induces SDH activity to produce robust mtROS and restrict invading microbes and further NLRP3 inflammasome-mediated activation of proinflammatory IL-1β to restrict microbial growth. CLPP, caseinolytic mitochondrial matrix peptidase; HSP60, heat shock protein 60; IMM, inner mitochondrial membrane; IMS, inner mitochondrial space; MAPK, mitogen-activated protein kinase; OMM, outer mitochondrial membrane; PRRs, pathogen recognition receptors; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SOD2, superoxide dismutase 2.
Figure 2.
Mitochondrial-associated UPR signaling in IECs and myeloid cells. Mitochondrial ETC molecules (complex I, II, III, IV, and V) regulate ROS, superoxide, and ATP balance in IMM. The mitochondrial chaperon Hsp60 regulates protein-folding machinery in the mitochondrial matrix and maintains homeostasis; however, accumulation of unfolded proteins in the mitochondrial intermembrane space (IMS) induces mitochondrial ROS (mtROS) and superoxide signaling and coordinates with HIF1α to activate mitoUPR signaling. On the other side, the activated macrophage (right), in response to pathogen recognition, induces SDH activity to produce robust mtROS and restrict invading microbes and further NLRP3 inflammasome-mediated activation of proinflammatory IL-1β to restrict microbial growth. CLPP, caseinolytic mitochondrial matrix peptidase; HSP60, heat shock protein 60; IMM, inner mitochondrial membrane; IMS, inner mitochondrial space; MAPK, mitogen-activated protein kinase; OMM, outer mitochondrial membrane; PRRs, pathogen recognition receptors; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SOD2, superoxide dismutase 2.

© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ranjan, K. Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointest. Disord. 2020, 2, 246-266. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2030024
AMA Style
Ranjan K. Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms. Gastrointestinal Disorders. 2020; 2(3):246-266. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2030024
Chicago/Turabian StyleRanjan, Kishu. 2020. "Intestinal Immune Homeostasis and Inflammatory Bowel Disease: A Perspective on Intracellular Response Mechanisms" Gastrointestinal Disorders 2, no. 3: 246-266. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord2030024