
Connective Tissue Disease-Associated Pulmonary Arterial Hypertension in Southern Taiwan: A Single-Center 10-Year Longitudinal Observation Cohort


Abstract
:1. Introduction
2. Methods
2.1. Data Source and Study Subjects
2.2. Covariates and Comorbidities
2.3. Autoantibody Detection
2.4. Treatment
2.5. Statistical Analyses
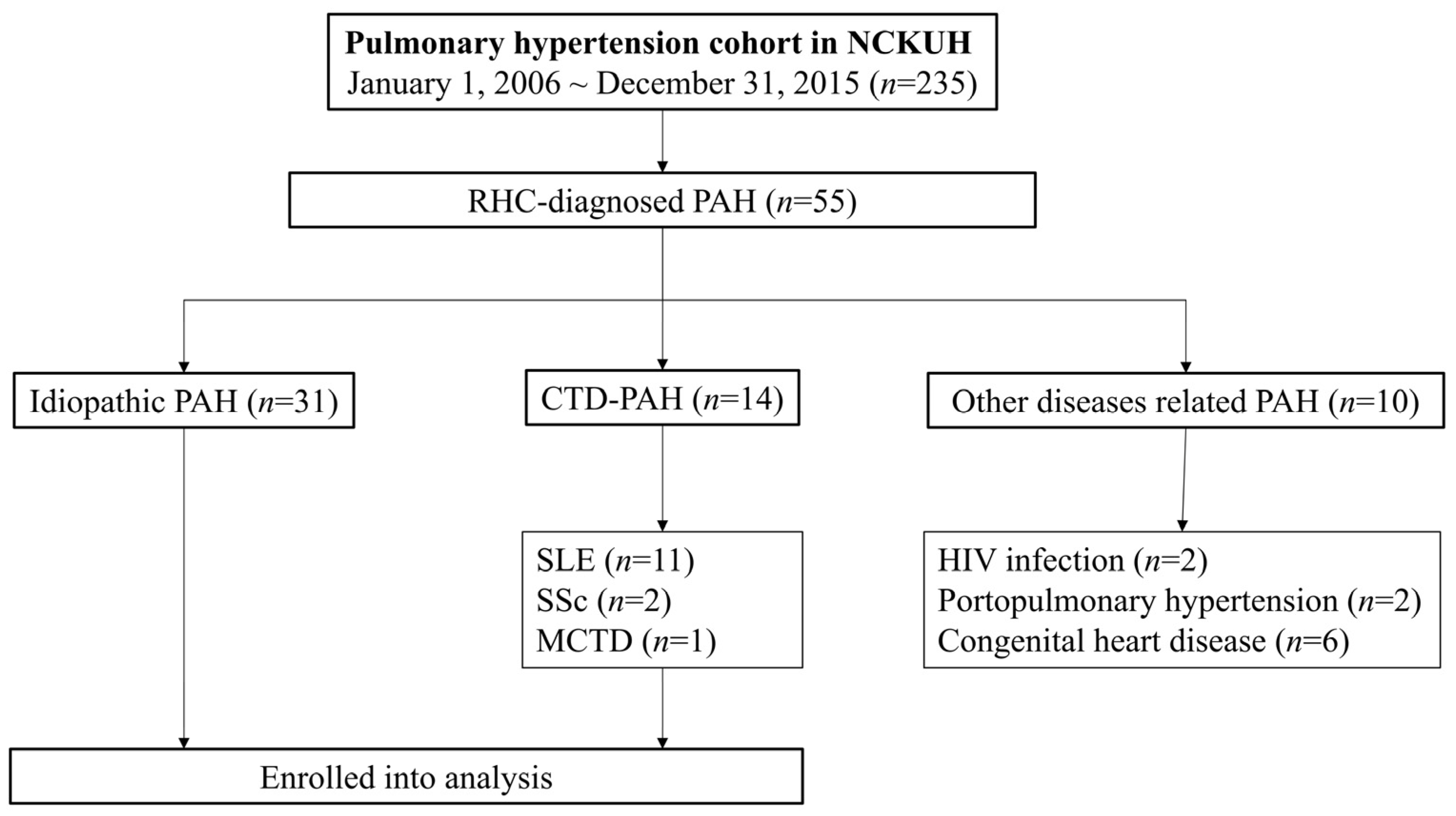
3. Results
3.1. Demographic Features of Patient Characteristics
3.2. Characterizations of Patients with SLE and PAH
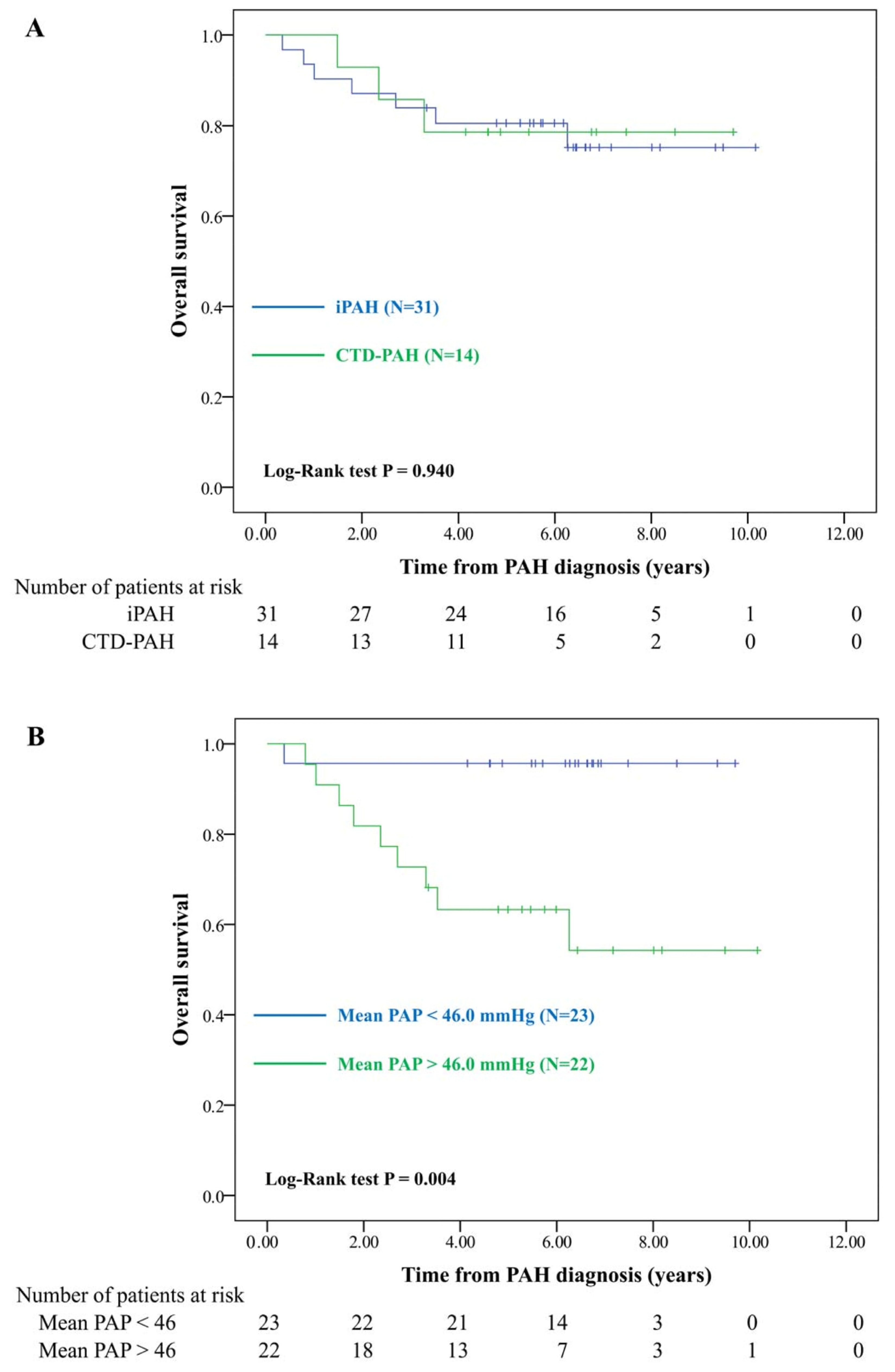
3.3. Survival Analysis of Patients with PAH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Sanchez, M.A.G.; Kumar, R.K.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGoon, M.D.; Miller, D.P. REVEAL: A contemporary US pulmonary arterial hypertension registry. Eur. Respir. Rev. 2012, 21, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.H.; Chai, J.Y.; Seo, Y.I.; Jun, J.B.; Koh, E.M.; Lee, S.K.; Pulmonary Hypertension Study Group of Korean College of Rheumatology. Pulmonary hypertension associated with rheumatic diseases: Baseline characteristics from the Korean registry. Int. J. Rheum. Dis. 2012, 15, e80–e89. [Google Scholar] [CrossRef]
- Shirai, Y.; Yasuoka, H.; Okano, Y.; Takeuchi, T.; Satoh, T.; Kuwana, M. Clinical characteristics and survival of Japanese patients with connective tissue disease and pulmonary arterial hypertension: A single-centre cohort. Rheumatology 2012, 51, 1846–1854. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.-J.; Jiang, X.; Zhou, W.; Wang, Y.; Gao, L.; Wang, Y.; Li, G.-T.; Hong, T.; Huo, Y.; Jing, Z.-C.; et al. Connective tissue disease-associated pulmonary arterial hypertension in Chinese patients. Eur. Respir. J. 2014, 44, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Blyth, K.G.; Syyed, R.; Chalmers, J.; Foster, J.E.; Saba, T.; Naeije, R.; Mélot, C.; Peacock, A.J. Pulmonary arterial pulse pressure and mortality in pulmonary arterial hypertension. Respir. Med. 2007, 101, 2495–2501. [Google Scholar] [CrossRef]
- Ruth, B.K.; Bilchick, K.C.; Mysore, M.M.; Mwansa, H.; Harding, W.C.; Kwon, Y.; Kennedy, J.L.; Mazurek, J.A.; Mihalek, A.D.; Smith, L.A.; et al. Increased pulmonary-systemic pulse pressure ratio is associated with increased mortality in group 1 pulmonary hypertension. Heart Lung Circ. 2019, 28, 1059–1066. [Google Scholar] [CrossRef]
- McGoon, M.D.; Benza, R.L.; Escribano-Subias, P.; Jiang, X.; Miller, D.P.; Peacock, A.J.; Pepke-Zaba, J.; Pulido, T.; Rich, S.; Rosenkranz, S.; et al. Pulmonary arterial hypertension: Epidemiology and registries. J. Am. Coll. Cardiol. 2013, 62, D51–D59. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.W.; Thenappan, T. World Health Organization Group I Pulmonary Hypertension. Cardiol. Clin. 2016, 34, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.K.; Chung, L. Connective tissue disease–associated pulmonary arterial hypertension. Rheum. Dis. Clin. N. Am. 2015, 41, 295–313. [Google Scholar] [CrossRef]
- Chang, W.; Weng, S.; Hsu, C.; Shih, J.; Wang, J.; Wu, C.; Chen, Z. Prognostic factors in patients with pulmonary hypertension—A nationwide cohort study. J. Am. Heart Assoc. 2016, 5, e003579. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.H.; See, L.C.; Kuo, C.F.; Chou, I.J.; Chou, M.J. Prevalence and incidence in patients with autoimmune rheumatic diseases: A nationwide population-based study in Taiwan. Arthritis Care Res. 2013, 65, 244–250. [Google Scholar] [CrossRef]
- Chung, L.; Kawut, S.M. Connective tissue disease-associated pulmonary arterial hypertension: “Beijing style”. Eur. Respir. J. 2014, 44, 839–841. [Google Scholar] [CrossRef] [Green Version]
- Mukerjee, D.; George, D.S.; Coleiro, B.; Knight, C.; Denton, C.P.; Davar, J.; Black, C.M.; Coghlan, J.G. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: Application of a registry approach. Ann. Rheum. Dis. 2003, 62, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Phung, S.; Strange, G.; Chung, L.P.; Leong, J.; Dalton, B.; Roddy, J.; Deague, J.; Playford, D.; Musk, M.; Gabbay, E. Prevalence of pulmonary arterial hypertension in an Australian scleroderma population: Screening allows for earlier diagnosis. Intern. Med. J. 2009, 39, 682–691. [Google Scholar] [CrossRef]
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; E Pope, J.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 73, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Chen, H.A.; Wang, H.P.; Liao, H.T.; Chou, C.T.; Huang, D.F. Pulmonary arterial hypertension in autoimmune diseases: An analysis of 19 cases from a medical center in northern Taiwan. J. Microbiol. Immunol. Infect. 2006, 39, 162–168. [Google Scholar]
- Li, M.; Wang, Q.; Zhao, J.; Li, Z.; Ye, Z.; Li, C.; Zhu, P.; Wang, Z.; Zheng, Y.; Li, X.; et al. Chinese SLE Treatment and Research group (CSTAR) registry: II. Prevalence and risk factors of pulmonary arterial hypertension in Chinese patients with systemic lupus erythematosus. Lupus 2014, 23, 1085–1091. [Google Scholar]
- Chen, H.-A.; Hsu, T.-C.; Yang, S.-C.; Weng, C.-T.; Wu, C.-H.; Sun, C.-Y.; Lin, C.-Y. Incidence and survival impact of pulmonary arterial hypertension among patients with systemic lupus erythematosus: A nationwide cohort study. Arthritis Res. Ther. 2019, 21, 82. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Irastorza, G.; Garmendia, M.; Villar, I.; Egurbide, M.V.; Aguirre, C. Pulmonary hypertension in systemic lupus erythematosus: Prevalence, predictors and diagnostic strategy. Autoimmun. Rev. 2013, 12, 410–415. [Google Scholar] [CrossRef]
- Pérez-Peñate, G.M.; Rúa-Figueroa, I.; Juliá-Serdá, G.; León-Marrero, F.; García-Quintana, A.; Ortega-Trujillo, J.R.; Erausquin-Arruabarrena, C.; Rodríguez-Lozano, C.; Cabrera-Navarro, P.; Ojeda-Betancor, N.; et al. Pulmonary arterial hypertension in systemic lupus erythematosus: Prevalence and predictors. J. Rheumatol. 2016, 43, 323–329. [Google Scholar] [CrossRef]
- Gunnarsson, R.; Andreassen, A.K.; Molberg, Ø.; Lexberg, Å.S.; Time, K.; Dhainaut, A.S.S.; Bertelsen, L.; Palm, Ø.; Irgens, K.; Becker-Merok, A.; et al. Prevalence of pulmonary hypertension in an unselected, mixed connective tissue disease cohort: Results of a nationwide, Norwegian cross-sectional multicentre study and review of current literature. Rheumatology 2013, 52, 1208–1213. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Li, M.; Qian, J.; Wang, Q.; Zhao, J.; Yang, Z.; Tian, Z.; Zhang, X.; Zuo, X.; Zhang, M.; et al. Pulmonary arterial hypertension in systemic lupus erythematosus based on a CSTAR-PAH study: Baseline characteristics and risk factors. Int. J. Rheum. Dis. 2019, 22, 921–928. [Google Scholar] [CrossRef]
- Rhee, R.L.; Gabler, N.B.; Sangani, S.; Praestgaard, A.; Merkel, P.A.; Kawut, S.M. Comparison of treatment response in idiopathic and connective tissue disease-associated pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Acharya, N.; Sharma, S.K.; Mishra, D.; Dhooria, S.; Dhir, V.; Jain, S. Efficacy and safety of pirfenidone in systemic sclerosis-related interstitial lung disease-a randomised controlled trial. Rheumatol. Int. 2020, 40, 703–710. [Google Scholar] [CrossRef]
- Stern, E.P.; Denton, C.P. The pathogenesis of systemic sclerosis. Rheum. Dis. Clin. N. Am. 2015, 41, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Liu, J.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Badesch, D.B.; Miller, D.P.; Nicolls, M.R.; Zamanian, R.T. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: Identifying systemic sclerosis as a unique phenotype. Chest 2010, 138, 1383–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, G.M.Y.; Tay, E.L.; Tai, B.C.; Yip, J.W.L. Idiopathic pulmonary arterial hypertension in Asians: A long-term study on clinical outcomes. Chest 2015, 147, e160–e163. [Google Scholar] [CrossRef]
- Ogawa, A.; Satoh, T.; Tamura, Y.; Fukuda, K.; Matsubara, H. Survival of Japanese patients with idiopathic/heritable pulmonary arterial hypertension. Am. J. Cardiol. 2017, 119, 1479–1484. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, D.; Joo, Y.B.; Won, S.; Lee, J.; Shin, J.; Bae, S.-C. Factors associated with development and mortality of pulmonary hypertension in systemic lupus erythematosus patients. Lupus 2018, 27, 1769–1777. [Google Scholar] [CrossRef]
- Qian, J.; Li, M.; Zhang, X.; Wang, Q.; Zhao, J.; Tian, Z.; Wei, W.; Zuo, X.; Zhang, M.; Zhu, P.; et al. Long-term prognosis of patients with systemic lupus erythematosus-associated pulmonary arterial hypertension: CSTAR-PAH cohort study. Eur. Respir. J. 2019, 53, 1800081. [Google Scholar] [CrossRef]
- Morrisroe, K.; Stevens, W.; Huq, M.; Prior, D.; Sahhar, J.; Ngian, G.-S.; Celermajer, D.; Zochling, J.; Proudman, S.; Nikpour, M.; et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res. Ther. 2017, 19, 122. [Google Scholar] [CrossRef] [Green Version]
- Kolstad, K.D.; Li, S.; Steen, V.; Chung, L.; PHAROS Investigators. Long-term outcomes in systemic sclerosis-associated pulmonary arterial hypertension: From the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS). Chest 2018, 154, 862–871. [Google Scholar] [CrossRef]
- Douschan, P.; Kovacs, G.; Avian, A.; Foris, V.; Gruber, F.; Olschewski, A.; Olschewski, H. Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am. J. Respir. Crit. Care Med. 2018, 197, 509–516. [Google Scholar] [CrossRef]
- Chin, K.M.; Rubin, L.J.; Channick, R.; Di Scala, L.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; McLaughlin, V.V.; et al. Association of N-terminal pro brain natriuretic peptide and long-term outcome in patients with pulmonary arterial hypertension. Circulation 2019, 139, 2440–2450. [Google Scholar] [CrossRef]
- Hsu, V.M.; Chung, L.; Hummers, L.K.; Shah, A.; Simms, R.; Bolster, M.; Hant, F.N.; Silver, R.M.; Fischer, A.; Hinchcliff, M.E.; et al. Risk factors for mortality and cardiopulmonary hospitalization in systemic sclerosis patients at risk for pulmonary hypertension, in the PHAROS Registry. J. Rheumatol. 2019, 46, 176–183. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristics | CTD-PAH | iPAH | p Value: CTD-PAH vs. iPAH | |
|---|---|---|---|---|
| All (N = 14) | SLE (N = 11) | (N = 31) | ||
| Female | 13 (92.9) | 11 (100) | 17 (54.8) | 0.016 |
| Age at PAH diagnosis, years | 38.6 (10.6) | 38.7 (9.7) | 47.7 (14.4) | 0.040 |
| NT-proBNP a (pg/mL) (25–75% IQR) | 1089.3 (636.4–3644.0) | 1264.0 (684.1–3638.0) | 441.5 (213.1–2125.0) | 0.039 |
| ePASP b (mmHg) | 67.0 (22.5) | 71.1 (23.3) | 81.0 (31.2) | 0.141 |
| mPAP (mmHg) | 42.8 (9.5) | 43.2 (9.5) | 46.4 (15.2) | 0.322 |
| DLCO c (% of predicted) | 55.3 (11.8) | 53.5 (11.6) | 75.5 (25.4) | 0.014 |
| ANA positive | 13 (92.9) | 11 (100) | 2 (6.7) | <0.001 |
| ENA positive | 13 (92.9) | 11 (100) | 1 (3.3) | <0.001 |
| PAH-induced mortality | 3 (21.4) | 3 (27.3) | 7 (22.6) | 0.99 |
| Mean follow-up years (min–max) | 5.3 (1.5–9.7) | 4.8 (1.5–9.7) | 5.6 (0.4–10.2) | 0.715 |
| Comorbidity | ||||
| CKD | 2 (14.3) | 2 (18.2) | 9 (29.0) | 0.458 |
| Hypertension | 4 (28.6) | 4 (36.4) | 7 (22.6) | 0.717 |
| Diabetes mellitus | 0 (0.0) | 0 (0.0) | 5 (16.1) | 0.305 |
| Dyslipidemia | 2 (14.3) | 2 (18.2) | 4 (12.9) | 1.000 |
| PAH-specific therapy | ||||
| Prostacyclin agonists | 1 (7.1) | 0 (0) | 6 (19.4) | 0.407 |
| ERA | 1 (7.1) | 1 (9.1) | 10 (32.3) | 0.132 |
| PDE5 inhibitor | 14 (100) | 11 (100) | 20 (64.5) | 0.010 |
| No. | Sex | Age at SLE | Age at PAH | Time to PAH Onset (year) | LN | Anti-RNP | APL | NT-proBNP (pg/mL) | mPAP (mmHg) | DLCO (%) | Outcome | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LAC | aCL | β2GP1 | |||||||||||
| 1 | F | 16 | 17 | 1 | - | + | - | - | - | 3638 | 58 | 51 | Died at age 19 years |
| 2 | F | 41 | 46 | 5 | + | - | - | - | - | 684.1 | 36 | 54 | Survival |
| 3 | F | 24 | 42 | 18 | - | - | - | - | - | 2029 | 45 | 63 | Survival |
| 4 | F | 30 | 34 | 4 | + | + | - | - | - | 109.8 | 47 | 65 | Died at age 35 years |
| 5 | F | 43 | 53 | 10 | - | - | - | - | - | 3424 | 52 | 38 | Died at age 56 years |
| 6 | F | 29 | 29 | 0.3 | + | + | - | - | - | 422.1 | 55 | 67 | Survival |
| 7 | F | 19 | 37 | 18 | - | + | + | - | - | 1264 | 32 | 41 | Survival |
| 8 | F | 26 | 38 | 12 | + | + | + | 8346 | 38 | N/A | Survival | ||
| 9 | F | 30 | 46 | 16 | - | + | - | - | - | 914.5 | 44 | 63 | Survival |
| 10 | F | 33 | 44 | 11 | - | + | - | - | - | 887.9 | 40 | 57 | Survival |
| 11 | F | 29 | 40 | 11 | + | + | - | - | - | 7048 | 28 | 36 | Survival |
| Mortality (N = 10) | Survival (N = 35) | p Value | |
|---|---|---|---|
| Female | 7 (70) | 23 (65.7) | 1.000 |
| Age at PAH diagnosis | 43.2 (16.6) | 45.3 (13.2) | 0.676 |
| CTD | 3 (30) | 8 (22.9) | 0.687 |
| NT-proBNP a (pg/mL) (25–75% IQR) | 1867.5 (535.3–3147.5) | 546.5 (257.9–2077.0) | 0.235 |
| ePASP b (mmHg) | 81.9 (16.6) | 74.9 (32.0) | 0.516 |
| mPAP (mmHg) | 51.9 (7.8) | 43.0 (14.9) | 0.069 |
| DLCO c (% of predicted) | 55.9 (22.1) | 71.7 (23.2) | 0.113 |
| Comorbidity | |||
| CKD | 4 (40.0) | 7 (20.0) | 0.228 |
| Hypertension | 3 (30) | 8 (22.9) | 0.687 |
| Diabetes mellitus | 1 (10) | 4 (11.4) | 1.000 |
| Dyslipidemia | 1 (10) | 5 (14.3) | 1.000 |
| PAH-specific therapy | |||
| Prostacyclin agonists | 3 (30) | 4 (11.4) | 0.172 |
| ERA | 2 (20) | 9 (25.7) | 1.000 |
| PDE5 inhibitor | 8 (80) | 26 (74.3) | 1.000 |
| Crude HR (95% CI) | p Value | Adjusted HR (95% CI) a | p Value | |
|---|---|---|---|---|
| CTD | 0.95 (0.24–3.68) | 0.940 | 3.29 (0.66–16.35) | 0.144 |
| Mean PAP > 46.0 mmHg | 11.36 (1.43–89.98) | 0.021 | 21.81 (2.32–204.88) | 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-H.; Lin, C.-Y.; Hsu, C.-H.; Lin, S.-H.; Weng, C.-T. Connective Tissue Disease-Associated Pulmonary Arterial Hypertension in Southern Taiwan: A Single-Center 10-Year Longitudinal Observation Cohort. Healthcare 2021, 9, 615. https://0-doi-org.brum.beds.ac.uk/10.3390/healthcare9050615
Wu C-H, Lin C-Y, Hsu C-H, Lin S-H, Weng C-T. Connective Tissue Disease-Associated Pulmonary Arterial Hypertension in Southern Taiwan: A Single-Center 10-Year Longitudinal Observation Cohort. Healthcare. 2021; 9(5):615. https://0-doi-org.brum.beds.ac.uk/10.3390/healthcare9050615
Chicago/Turabian StyleWu, Chun-Hsin, Chun-Yu Lin, Chih-Hsin Hsu, Sheng-Hsiang Lin, and Chia-Tse Weng. 2021. "Connective Tissue Disease-Associated Pulmonary Arterial Hypertension in Southern Taiwan: A Single-Center 10-Year Longitudinal Observation Cohort" Healthcare 9, no. 5: 615. https://0-doi-org.brum.beds.ac.uk/10.3390/healthcare9050615