
Virulence Profiles of Vibrio vulnificus in German Coastal Waters, a Comparison of North Sea and Baltic Sea Isolates

Abstract
:1. Introduction
1.1. Background
1.2. Aim of the Study
2. Experimental Section
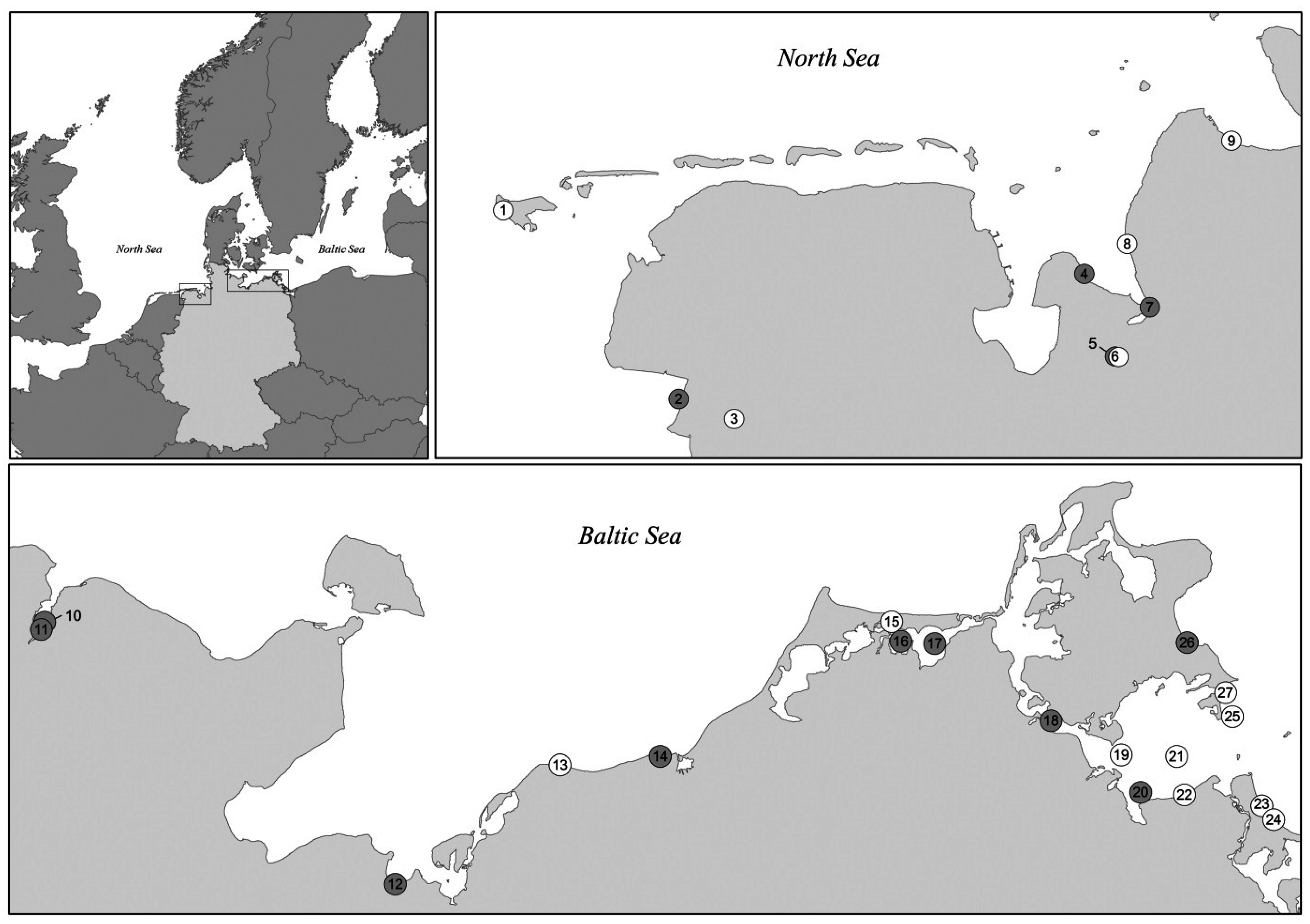
2.1. Bacterial Strains

2.2. Biotyping and Mannitol Fermentation
2.3. Multilocus Sequence Typing
2.4. PCR Analyses
2.5. Serum Resistance
2.6. Statistical Analyses
3. Results and Discussion
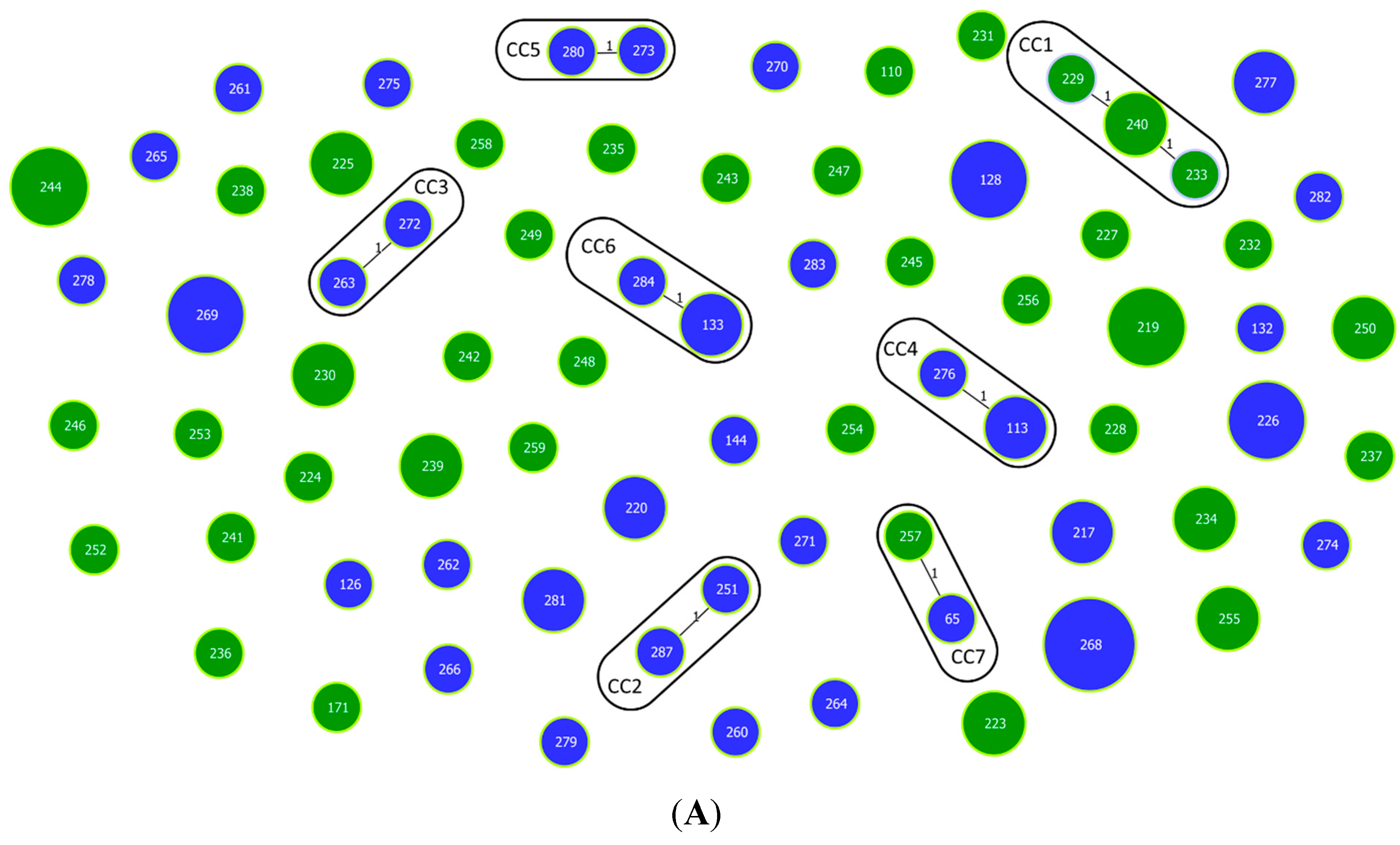
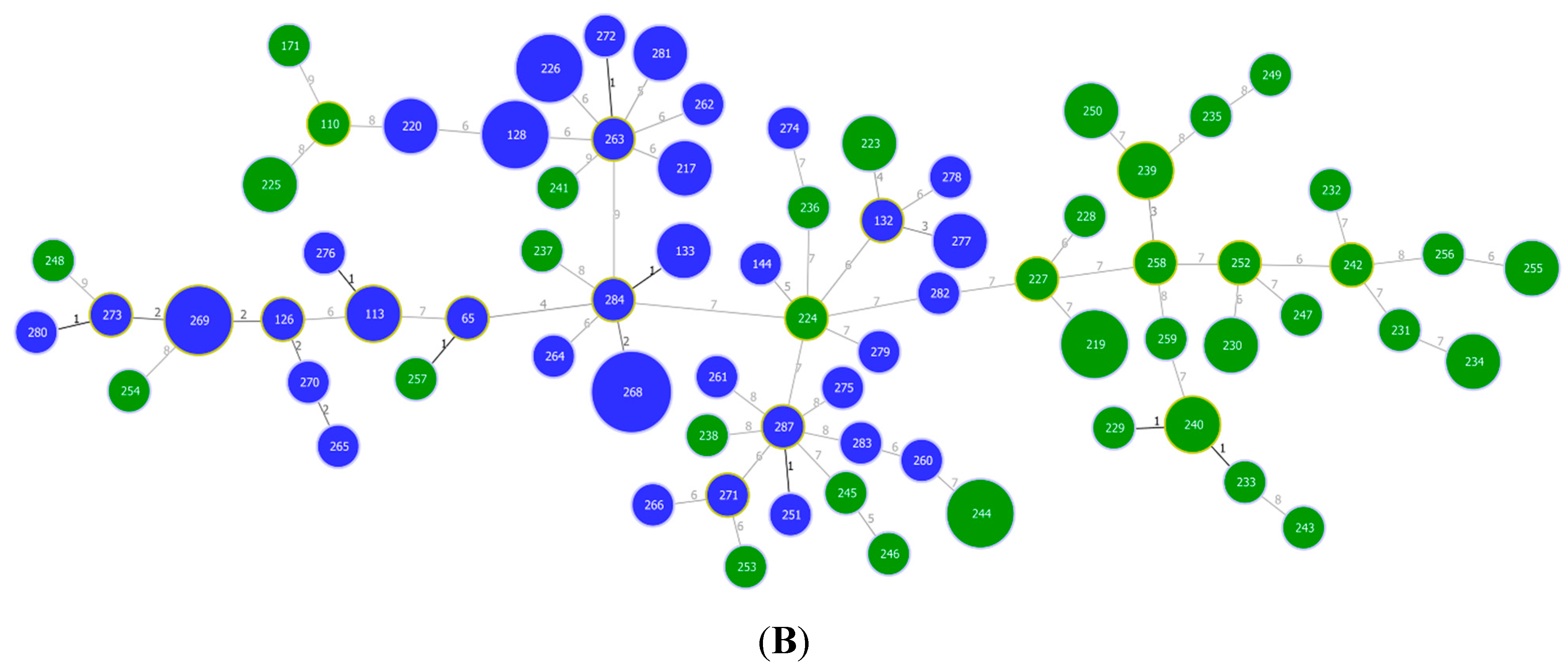
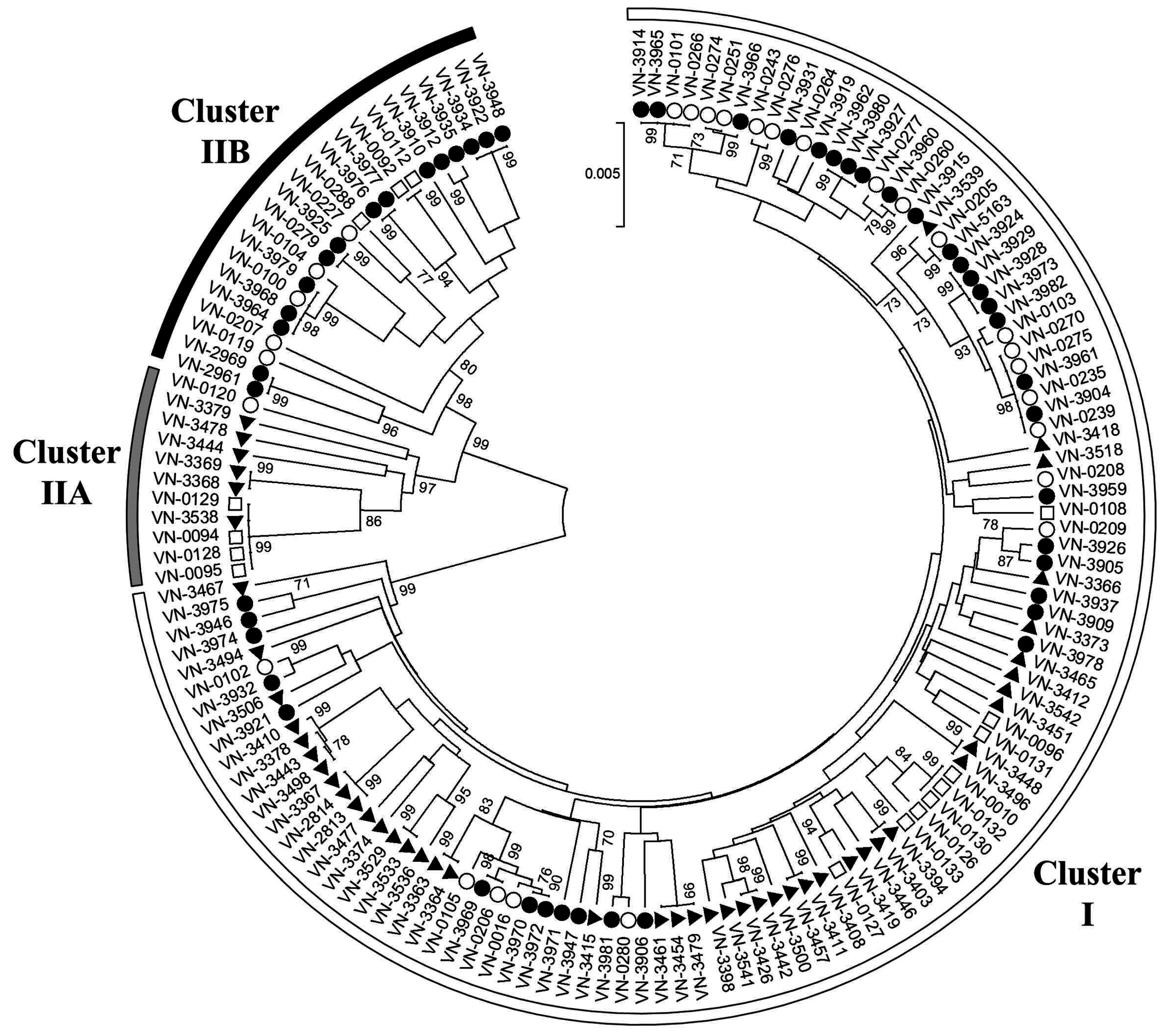
3.1. Multilocus Sequence Typing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geographical Region (Number of Strains) | MLST Cluster (%) | vcg-Type (%) | 16S rRNA-Type (%) | nanA (%) | Region XII (%) | Mannitol Fermentation (%) | Serum Resistance (%) | Risk Group (%) a | No. of Different Virulence Profiles (N) | No. of Strains (N) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| – | I | IIA | IIB | C | E | A | AB | B | – | – | – | R | I | S | 1 | 2 | – | – |
| Total (n = 101) | 79 | 6 | 15 | 6 | 94 | 79 | 14 | 7 | 47 | 37 | 40 | 79 | 14 | 7 | 42 | 59 | 17 | 74 |
| Baltic Sea (n = 51) | 71 | 0 | 29 | 0 | 100 | 71 | 27 | 2 | 24 | 35 | 14 | 76 | 18 | 6 | 61 | 39 | 8 | 36 |
| North Sea (n = 50) | 88 | 12 | 0 | 12 | 88 | 88 | 0 | 12 | 70 | 38 | 66 | 82 | 10 | 8 | 22 | 78 | 14 | 38 |



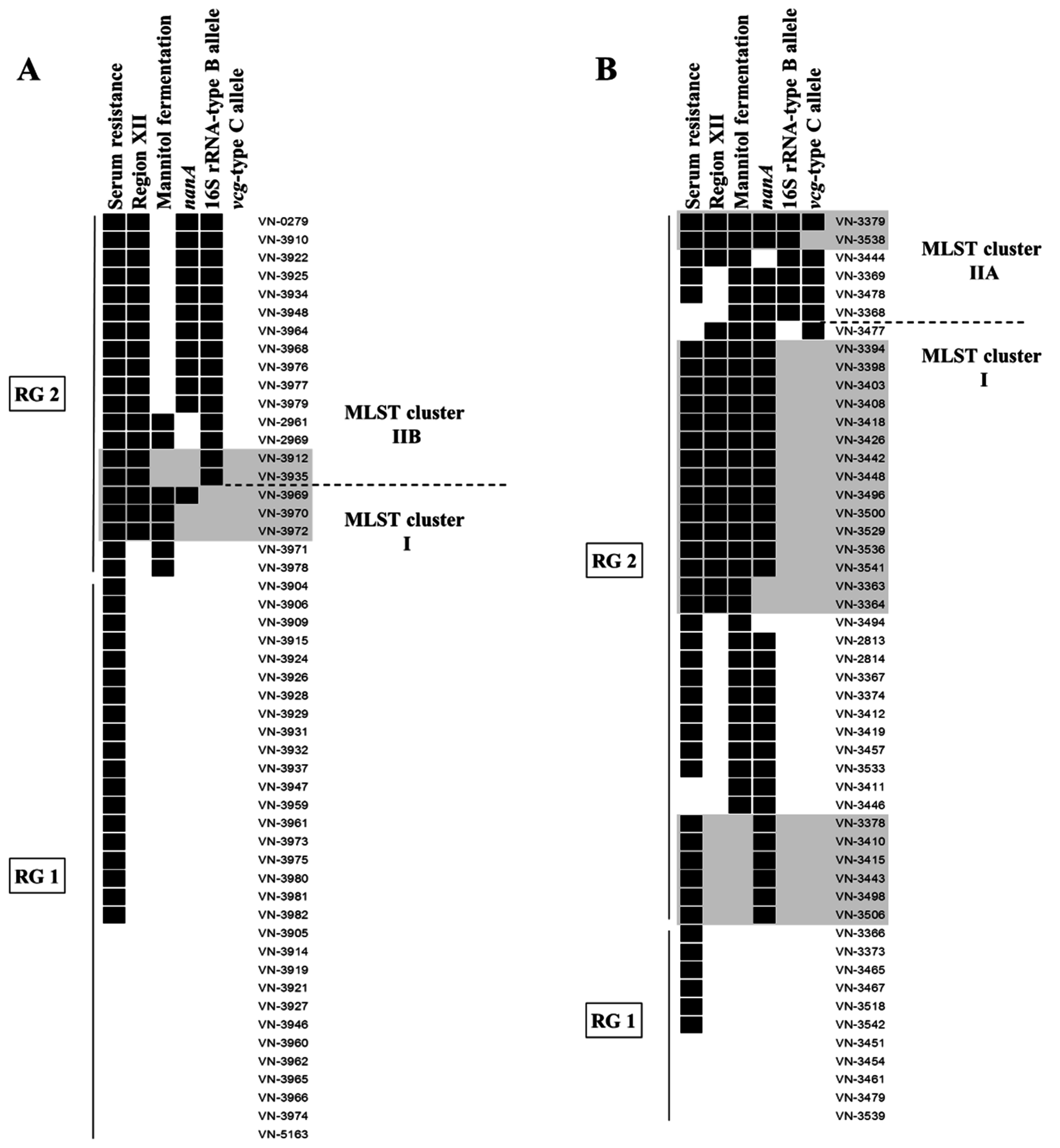
3.2. Distribution of Virulence-Associated Traits and Genotypes in MLST Clusters

3.3. Distribution of Virulence-Associated Traits and Genotypes among Isolates from the North Sea and the Baltic Sea
3.4. Assessment of the Pathogenicity Potential of Environmental Strains
| Risk Group a | Virulence Profile | No. of Isolates (Geographical Origin) | MLST Cluster |
|---|---|---|---|
| Risk Group 1 | – | 17 (12 BS, 5 NS) | I |
| Res | 25 (19 BS, 6 NS) | I | |
| Risk Group 2 | Man-nanA | 2 (2 NS) | I |
| Region XII-Man-nanA-vcgC | 1 (1 NS) | I | |
| Res-Man | 3 (2 BS, 1 NS) | I | |
| Res-Man-nanA | 8 (8 NS) | I | |
| Res-nanA b | 6 (6 NS c) | I | |
| Res-Region XII-Man b | 4 (2 BS, 2 NS) | I | |
| Res-Region XII-Man-nanA b | 14 (1 BS, 13 NS) | I | |
| Man-nanA-16S_B-vcgC | 1 (1 NS) | IIA | |
| Res-Man-nanA-16S_B-vcgC | 2 (2 NS) | IIA | |
| Res-Region XII-16S_B b | 2 (2 BS) | IIB | |
| Res-Region XII-Man-16S_B | 2 (2 BS) | IIB | |
| Res-Region XII-Man-16S_B-vcgC | 1 (1 NS) | IIA | |
| Res-Region XII-Man-nanA-16S_B b | 1 (1 NS c) | IIA | |
| Res-Region XII-Man-nanA-16S_B-vcgC b | 1 (1 NS c) | IIA | |
| Res-Region XII-nanA-16S_B | 11 (11 BS) | IIB |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thompson, J.R.; Polz, M.F. Dynamics of vibrio populations and their role in environmental nutrient cycling. In The Biology of Vibrios; Thompson, F.L., Austin, B., Swings, J., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 190–203. [Google Scholar]
- Oliver, J.D. Vibrio vulnificus . In The Biology of Vibrios; Thompson, F.L., Austin, B., Swings, J., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 349–366. [Google Scholar]
- Shapiro, R.L.; Altekruse, S.; Hutwagner, L.; Bishop, R.; Hammond, R.; Wilson, S.; Ray, B.; Thompson, S.; Tauxe, R.V.; Griffin, P.M. The role of gulf coast oysters harvested in warmer months in Vibrio vulnificus infections in the United States, 1988–1996. Vibrio working group. J. Infect. Dis. 1998, 178, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.D. Vibrio vulnificus . In Oceans and Health: Pathogens in the Marine Environment; Belkin, S., Colwell, R.R., Eds.; Springer: New York, NY, USA, 2005; pp. 253–276. [Google Scholar]
- Oliver, J.D. Wound infections caused by Vibrio vulnificus and other marine bacteria. Epidemiol. Infect. 2005, 133, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Huehn, S.; Eichhorn, C.; Urmersbach, S.; Breidenbach, J.; Bechlars, S.; Bier, N.; Alter, T.; Bartelt, E.; Frank, C.; Oberheitmann, B.; et al. Pathogenic vibrios in environmental, seafood and clinical sources in Germany. Int. J. Med. Microbiol. 2014, 304, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.D. The biology of Vibrio vulnificus. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Bisharat, N.; Cohen, D.I.; Maiden, M.C.; Crook, D.W.; Peto, T.; Harding, R.M. The evolution of genetic structure in the marine pathogen, Vibrio vulnificus. Infect. Genet. Evol. 2007, 7, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Bisharat, N.; Cohen, D.I.; Harding, R.M.; Falush, D.; Crook, D.W.; Peto, T.; Maiden, M.C. Hybrid Vibrio vulnificus. Emerg. Infect. Dis. 2005, 11, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.V.; Oliver, J.D.; DePaola, A.; Feil, E.J.; Boyd, E.F. Emergence of a virulent clade of Vibrio vulnificus and correlation with the presence of a 33-kilobase genomic island. Appl. Environ. Microbiol. 2007, 73, 5553–5565. [Google Scholar] [CrossRef] [PubMed]
- Bier, N.; Bechlars, S.; Diescher, S.; Klein, F.; Hauk, G.; Duty, O.; Strauch, E.; Dieckmann, R. Genotypic diversity and virulence characteristics of clinical and environmental Vibrio vulnificus isolates from the Baltic Sea region. Appl. Environ. Microbiol. 2013, 79, 3570–3581. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, W.B.; Paranjype, R.N.; DePaola, A.; Strom, M.S. Sequence polymorphism of the 16S rRNA gene of Vibrio vulnificus is a possible indicator of strain virulence. J. Clin. Microbiol. 2003, 41, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Rosche, T.M.; Yano, Y.; Oliver, J.D. A Rapid and simple PCR analysis indicates there are two subgroups of Vibrio vulnificus which correlate with clinical or environmental isolation. Microbiol. Immunol. 2005, 49, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Vickery, M.C.L.; Nilsson, W.B.; Strom, M.S.; Nordstrom, J.L.; DePaola, A. A Real-Time PCR assay for the rapid determination of 16S rRNA genotype in Vibrio vulnificus. J. Microbiol. Methods 2007, 68, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Thiaville, P.C.; Bourdage, K.L.; Wright, A.C.; Farrell-Evans, M.; Garvan, C.W.; Gulig, P.A. Genotype is correlated with but does not predict virulence of Vibrio vulnificus biotype 1 in subcutaneously inoculated, iron dextran-treated mice. Infect. Immun. 2011, 79, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, Y.; Pitchford, S.; de Decker, S.; Wikfors, G.H.; Brown, C.L. Molecular typing of environmental and clinical strains of Vibrio vulnificus isolated in the northeastern USA. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Randa, M.A.; Polz, M.F.; Lim, E. Effects of temperature and salinity on Vibrio vulnificus population dynamics as assessed by quantitative PCR. Appl. Environ. Microbiol. 2004, 70, 5469–5476. [Google Scholar] [CrossRef] [PubMed]
- Böer, S.I.; Heinemeyer, E.A.; Luden, K.; Erler, R.; Gerdts, G.; Janssen, F.; Brennholt, N. Temporal and spatial distribution patterns of potentially pathogenic Vibrio spp. at recreational beaches of the German North Sea. Microb. Ecol. 2013, 65, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- National Cholera and Vibriosis Surveillance. Available online: http://www.cdc.gov/nationalsurveillance/cholera-vibrio-surveillance.html (accessed on 8 September 2015).
- Baker-Austin, C.; Trinanes, J.A.; Taylor, N.G.H.; Hartnell, R.; Siitonen, A.; Martinez-Urtaza, J. Emerging Vibrio risk at high latitudes in response to ocean warming. Nat. Clim. Chang. 2012, 3, 73–77. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Stockley, L.; Rangdale, R.; Martinez-Urtaza, J. Environmental occurrence and clinical impact of Vibrio vulnificus and Vibrio parahaemolyticus: A European perspective. Environ. Microbiol. Rep. 2010, 2, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Urtaza, J.; Bowers, J.C.; Trinanes, J.; DePaola, A. Climate anomalies and the increasing risk of Vibrio parahaemolyticus and Vibrio vulnificus illnesses. Food Res. Int. 2010, 43, 1780–1790. [Google Scholar] [CrossRef]
- Brennholt, N.; Böer, S.I.; Heinemeyer, E.-A.; Luden, K.; Hauk, G.; Duty, O.; Baumgarten, A.-L.; Potau Núñez, R.; Rösch, T.; Wehrmann, A.; et al. Klimabedingte Änderungen der Gewässerhygiene und Auswirkungen auf das Baggergutmanagement in den Küstengewässern. Schlussbericht KLIWAS-projekt 3.04. KLIWAS-38/2014. German Federal Institute for Hydrology: Koblenz, Germany. Available online: http://doi.bafg.de/KLIWAS/2014/Kliwas_38_2014_3.04.pdf (accessed on 8 September 2015). (In German)
- Böer, S.; Hauk, G.; Duty, O.; Luden, K.; Heinemeyer, E.-A.; Brennholt, N. Pathogenic Vibrio species in German coastal waters of the North Sea and the Baltic Sea—A comparison. In Pathogenic Vibrio spp. in Northern European Waters, Proceedings of the International Symposium, Koblenz, Germany, 31 May–1 June 2012; German Federal Institute for Hydrology: Koblenz, Germany, 2012; pp. 36–42. (In German)[Google Scholar]
- Vibrionet Europe. Available online: http://www.vibrionet.de/ (accessed on 4 September 2015).
- Kliwas Microbes 3.04. Available online: http://www.kliwas.de/KLIWAS/EN/03_ResearchTasks/03_vh3/04_304/304_node.html (accessed on 4 September 2015).
- Bauer, A.; Roervik, L.M. A novel multiplex PCR for the identification of Vibrio parahaemolyticus, Vibrio cholerae and Vibrio vulnificus. Lett. Appl. Microbiol. 2007, 45, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Schirmeister, F.; Dieckmann, R.; Bechlars, S.; Bier, N.; Faruque, S.M.; Strauch, E. Genetic and phenotypic analysis of Vibrio cholerae non-O1, non-O139 isolated from German and Austrian patients. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, E.; Amaro, C. Multiplex PCR assay for detection of Vibrio vulnificus biotype 2 and simultaneous discrimination of serovar E strains. Appl. Environ. Microbiol. 2007, 73, 2029–2032. [Google Scholar] [CrossRef] [PubMed]
- Pubmlst.Org: MLST Databases and Software. Available online: http://pubmlst.org/vvulnificus/ (accessed on 4 September 2015).
- Phyloviz. Available online: http://www.phyloviz.net/ (accessed on 4 September 2015).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Sneath, P.H.A.; Sokal, R.R. Numerical Taxonomy; William H. Freeman and Company: San Francisco, CA, USA, 1973. [Google Scholar]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for Inferring Very Large Phylogenies By Using the Neighbor-Joining Method. Available online: http://www.pnas.org/content/101/30/11030.short (accessed on 16 July 2004).
- Froelich, B.; Oliver, J. Orientation of mannitol related genes can further differentiate strains of Vibrio vulnificus possessing the vcgC allele. Adv. Stud. Biol. 2011, 3, 151–160. [Google Scholar]
- Lubin, J.B.; Kingston, J.J.; Chowdhury, N.; Boyd, E.F. Sialic acid catabolism and transport gene clusters are lineage specific in Vibrio vulnificus. Appl. Environ. Microbiol. 2012, 78, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, E.; Gonzalez-Candelas, F.; Amaro, C. Polyphyletic origin of Vibrio vulnificus biotype 2 as revealed by sequence-based analysis. Appl. Environ. Microbiol. 2011, 77, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.S.; Williams, T.; Cain, A.; Froelich, B.; Taylor, C.; Baker-Austin, C.; Verner-Jeffreys, D.; Hartnell, R.; Oliver, J.D.; Gibas, C.J. Pyrosequencing-based comparative genome analysis of Vibrio vulnificus environmental isolates. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Yokochi, N.; Tanaka, S.; Matsumoto, K.; Oishi, H.; Tashiro, Y.; Yoshikane, Y.; Nakashima, M.; Kanda, K.; Kobayashi, G. Distribution of virulence markers among Vibrio vulnificus isolates of clinical and environmental origin and regional characteristics in Japan. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Hauk, G.; Duty, O.; Littmann, M. Vibrio vulnificus in der Ostsee—klinische “Ausgangsfälle”. Messstellen und Messdaten. In Pathogene Vibrionen in der marinen Umwelt, Proceedings of the workshop, Pathogene Vibrionen in der marinen Umwelt, Koblenz, Germany, 14–15 April 2010; German Federal Institute for Hydrology: Koblenz, Germany, 2010; pp. 23–30. (In German)[Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bier, N.; Jäckel, C.; Dieckmann, R.; Brennholt, N.; Böer, S.I.; Strauch, E. Virulence Profiles of Vibrio vulnificus in German Coastal Waters, a Comparison of North Sea and Baltic Sea Isolates. Int. J. Environ. Res. Public Health 2015, 12, 15943-15959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph121215031
Bier N, Jäckel C, Dieckmann R, Brennholt N, Böer SI, Strauch E. Virulence Profiles of Vibrio vulnificus in German Coastal Waters, a Comparison of North Sea and Baltic Sea Isolates. International Journal of Environmental Research and Public Health. 2015; 12(12):15943-15959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph121215031
Chicago/Turabian StyleBier, Nadja, Claudia Jäckel, Ralf Dieckmann, Nicole Brennholt, Simone I. Böer, and Eckhard Strauch. 2015. "Virulence Profiles of Vibrio vulnificus in German Coastal Waters, a Comparison of North Sea and Baltic Sea Isolates" International Journal of Environmental Research and Public Health 12, no. 12: 15943-15959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph121215031