DNA Hydroxymethylation at the Interface of the Environment and Nonalcoholic Fatty Liver Disease



Abstract
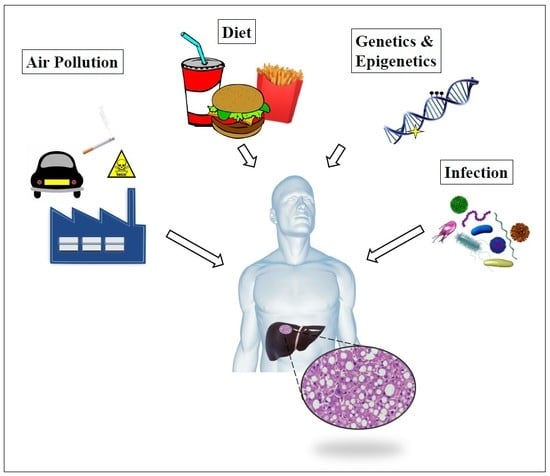
:
{kind=link}
{kind=link}
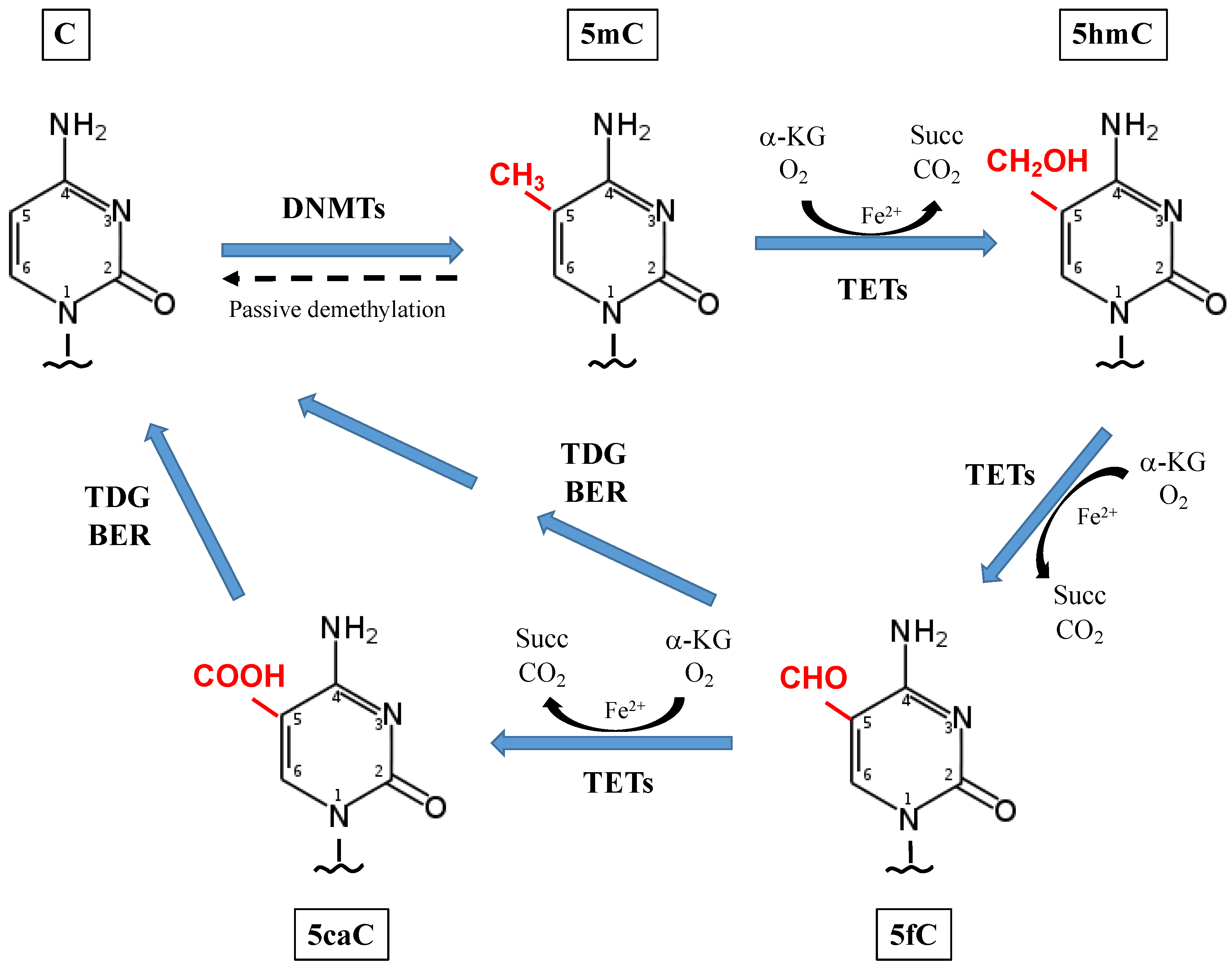{kind=link}
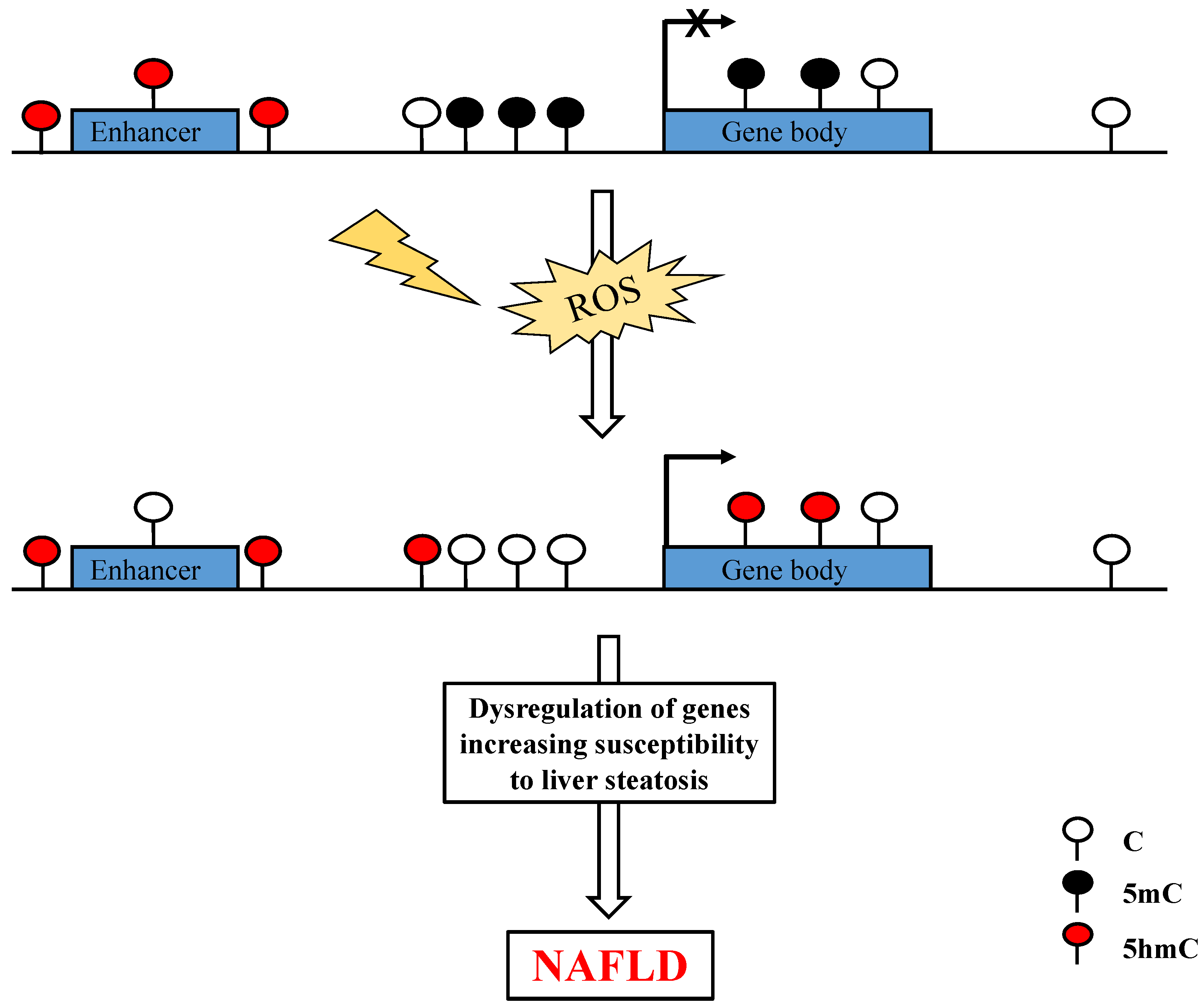{kind=link}
1. Introduction
2. The Contribution of Xenobiotics in the Pathogenesis of NAFLD
3. Modulation of DNA Oxidation and TET Activity by Environmental Exposures
4. The Role of 5hmC and TET Proteins in the Development of NAFLD
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62 (Suppl. 1), S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Ofosu, A.; Ramai, D.; Reddy, M. Non-alcoholic fatty liver disease: Controlling an emerging epidemic, challenges, and future directions. Ann. Gastroenterol. 2018, 31, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Adams, L.A.; de Lédinghen, V.; Wong, G.L.-H.; Sookoian, S. Noninvasive biomarkers in NAFLD and NASH—Current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Fatty liver and the metabolic syndrome. Curr. Opin. Gastroenterol. 2007, 23, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukusato, T. Pediatric nonalcoholic fatty liver disease: Overview with emphasis on histology. World J. Gastroenterol. 2010, 16, 5280–5285. [Google Scholar] [CrossRef]
- Brunt, E.M. Histopathology of non-alcoholic fatty liver disease. Clin. Liver Dis. 2009, 13, 533–544. [Google Scholar] [CrossRef]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpai, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.E.; Guo, G.L. Understanding environmental contaminants’ direct effects on non-alcoholic fatty liver disease progression. Curr. Environ. Health Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Li, X.; Wang, Z. Role of xenobiotics in the induction and progression of fatty liver disease. Toxicol. Res. 2018, 7, 664–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VoPham, T. Environmental risk factors for liver cancer and nonalcoholic fatty liver disease. Curr. Epidemiol. Rep. 2019, 6, 50–66. [Google Scholar] [CrossRef]
- Milic, S.; Lulic, D.; Stimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J. Gastroenterol. 2014, 20, 9330–9337. [Google Scholar]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug metabolism in the liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of environmental chemicals associated with the development of toxicant-associated fatty liver disease in rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Rabinowich, L.; Shibolet, O. Drug induced steatohepatitis: An uncommon culprit of a common disease. Biomed. Res. Int. 2015, 2015, 168905. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative mechanisms of environmental chemical-induced steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Trevino, L.S.; Katz, T.A. Endocrine disruptors and developmental origins of nonalcoholic fatty liver disease. Endocrinology 2018, 159, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Yao, H.; Rahman, I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid. Redox Signal. 2013, 18, 1956–1971. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Pulliero, A. Molecular damage and lung tumors in cigarette smoke-exposed mice. Ann. N. Y. Acad. Sci. 2015, 1340, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Lai, T.; Li, M.; Zhou, H.; Lv, D.; Deng, Z.; Ying, S.; Chen, Z.; Li, W.; Shen, H. Smoking-promoted oxidative DNA damage response is highly correlated to lung carcinogenesis. Oncotarget 2016, 7, 18919–18926. [Google Scholar] [CrossRef] [PubMed]
- Azzalini, L.; Ferrer, E.; Ramalho, L.N.; Moreno, M.; Domínguez, M.; Colmenero, J.; Peinado, V.I.; Barberà, J.A.; Arroyo, V.; Ginès, P.; et al. Cigarette smoking exacerbates nonalcoholic fatty liver disease in obese rats. Hepatology 2010, 51, 1567–1576. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Tong, M.; Agarwa, A.R.; Cadenas, E. Tobacco smoke-induced hepatic injury with steatosis, inflammation, and impairments in insulin and insulin-like growth factor signaling. J. Clin. Exp. Pathol. 2016, 6, 269. [Google Scholar] [CrossRef]
- Yuan, H.; Shyy, J.Y.; Martins-Green, M. Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP-1. J. Hepatol. 2009, 51, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumber, Y.; Issa, J.P. Epigenetics in cancer: what’s the future? Oncology 2011, 25, 220–226, 228. [Google Scholar] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.R.; O’Donnell, A.H.; Rollins, R.A.; Peckham, H.E.; Lee, C.; Milekic, M.H.; Chanrion, B.; Fu, Y.; Su, T.; Hibshoosh, H.; et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010, 20, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Branco, M.R.; Ficz, G.; Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 2011, 13, 7–13. [Google Scholar] [CrossRef]
- Laird, A.; Thomson, J.P.; Harrison, D.J.; Meehan, R.R. 5-hydroxymethylcytosine profiling as an indicator of cellular state. Epigenomics 2013, 5, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef]
- Xu, G.L.; Walsh, C.P. Enzymatic DNA oxidation: Mechanisms and biological significance. BMB Rep. 2014, 47, 609–618. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. TET enzymatic oxidation of 5-methylcytosine, 5-hydroxymethylcytosine and 5-formylcytosine. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 764–765, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.Q.; Ali, I.; Tang, J.; Yang, W.C. New insights into 5hmC DNA modification: Generation, distribution and function. Front. Genet. 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA demethylation dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011, 25, 2436–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Scian, R.; Gianotti, T.F.; Dopazo, H.; Rohr, C.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modifications in the biology of nonalcoholic fatty liver disease: The role of DNA hydroxymethylation and TET proteins. Medicine 2015, 94, e1480. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, Y. 5-Hydroxymethylcytosine: Generation, fate, and genomic distribution. Curr. Opin. Cell Biol. 2013, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; Yi, C.; He, C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat. Biotechnol. 2012, 30, 1107–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Salz, T.; Hansen, K.D.; Feinberg, A. Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver. Genome Res. 2016, 26, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jiang, J.; Mo, J.; Liu, D.; Cao, D.; Wang, H.; He, Y.; Wang, H. Global DNA 5-Hydroxymethylcytosine and 5-Formylcytosine Contents Are Decreased in the Early Stage of Hepatocellular Carcinoma. Hepatology 2019, 69, 196–208. [Google Scholar] [CrossRef]
- Nestor, C.E.; Ottaviano, R.; Reddington, J.; Sproul, D.; Reinhardt, D.; Dunican, D.; Katz, E.; Dixon, J.M.; Harrison, D.J.; Meehan, R.R. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012, 22, 467–477. [Google Scholar] [CrossRef]
- Thomson, J.P.; Hunter, J.M.; Lempiäinen, H.; Müller, A.; Terranova, R.; Moggs, J.G.; Meehan, R.R. Dynamic changes in 5-hydroxymethylation signatures underpin early and late events in drug exposed liver. Nucleic Acids Res. 2013, 41, 5639–5654. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Chia, N.; Wang, L.; Lu, X.; Senut, M.C.; Brenner, C.; Ruden, D.M. Hypothesis: Environmental regulation of 5-hydroxymethylcytosine by oxidative stress. Epigenetics 2011, 6, 853–856. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.; Cheng, R.Y.; Revelo, M.P.; Mitzner, W.; Tang, W. Hydroxymethylation as a novel environmental biosensor. Curr. Environ. Health Rep. 2014, 1, 1–10. [Google Scholar] [CrossRef]
- Ruiz-Hernandez, A.; Kuo, C.C.; Rentero-Garrido, P.; Tang, W.Y.; Redon, J.; Ordovas, J.M.; Navas-Acien, A.; Tellez-Plaza, M. Environmental chemicals and DNA methylation in adults: A systematic review of the epidemiologic evidence. Clin. Epigenet. 2015, 7, 55. [Google Scholar] [CrossRef]
- Coulter, J.B.; O’Driscoll, C.M.; Bressler, J.P. Hydroquinone increases 5-hydroxymethylcytosine formation through ten eleven translocation 1 (TET1) 5-methylcytosine dioxygenase. J. Biol. Chem. 2013, 288, 28792–28800. [Google Scholar] [CrossRef]
- Zhao, B.; Yang, Y.; Wang, X.; Chong, Z.; Yin, R.; Song, S.H.; Zhao, C.; Li, C.; Huang, H.; Sun, B.F.; et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and Tet-dependent mechanism. Nucleic Acids Res. 2014, 42, 1593–1605. [Google Scholar] [CrossRef]
- Delatte, B.; Jeschke, J.; Defrance, M.; Bachman, M.; Creppe, C.; Calonne, E.; Bizet, M.; Deplus, R.; Marroquí, L.; Libin, M. Genome-wide hydroxymethylcytosine pattern changes in response to oxidative stress. Sci. Rep. 2015, 5, 12714. [Google Scholar] [CrossRef]
- Thomson, J.P.; Lempiäinen, H.; Hackett, J.A.; Nestor, C.E.; Müller, A.; Bolognani, F.; Oakeley, E.J.; Schübeler, D.; Terranova, R.; Reinhardt, D.; et al. Non-genotoxic carcinogen exposure induces defined changes in the 5-hydroxymethylome. Genome Biol. 2012, 13, R93. [Google Scholar] [CrossRef]
- Tellez-Plaza, M.; Tang, W.Y.; Shang, Y.; Umans, J.G.; Francesconi, K.A.; Goessler, W.; Ledesma, M.; Leon, M.; Laclaustra, M.; Pollak, J.; et al. Association of global DNA methylation and global DNA hydroxymethylation with metals and other exposures in human blood DNA samples. Environ. Health Perspect. 2014, 122, 946–954. [Google Scholar] [CrossRef]
- Meehan, R.R.; Thomson, J.P.; Lentini, A.; Nestor, C.E.; Pennings, S. DNA methylation as a genomic marker of exposure to chemical and environmental agents. Curr. Opin. Chem. Biol. 2018, 45, 48–56. [Google Scholar] [CrossRef]
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef]
- Nano, J.; Ghanbari, M.; Wang, W.; de Vries, P.S.; Dhana, K.; Muka, T.; Uitterlinden, A.G.; van Meurs, J.B.J.; Hofman, A.; BIOS Consortium; et al. Epigenome-wide association study identifies methylation sites associated with liver enzymes and hepatic steatosis. Gastroenterology 2017, 153, 1096–1106. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and epigenetic regulation in nonalcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef]
- Ashraf, N.U.; Altaf, M. Epigenetics: An emerging field in the pathogenesis of nonalcoholic fatty liver disease. Mutat. Res. 2018, 778, 1–12. [Google Scholar] [CrossRef]
- Kovalic, A.J.; Banerjee, P.; Tran, Q.T.; Singal, A.K.; Satapathy, S.K. Genetic and epigenetic culprits in the pathogenesis of nonalcoholic fatty liver disease. J. Clin. Exp. Hepatol. 2018, 8, 390–402. [Google Scholar] [CrossRef]
- Sinton, M.C.; Hay, D.C.; Drake, A.J. Metabolic control of gene transcription in non-alcoholic fatty liver disease: The role of the epigenome. Clin. Epigenetics 2019, 11, 104. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueño, A.L.; Fernández Gianotti, T.; Castaño, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Tryndyak, V.P.; Han, T.; Fuscoe, J.C.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Status of hepatic DNA methylome predetermines and modulates the severity of non-alcoholic fatty liver injury in mice. BMC Genom. 2016, 17, 298. [Google Scholar] [CrossRef]
- Pirola, C.J.; Gianotti, T.F.; Burgueño, A.L.; Rey-Funes, M.; Loidl, C.F.; Mallardi, P.; Martino, J.S.; Castaño, G.O.; Sookoian, S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013, 62, 1356–1363. [Google Scholar] [CrossRef]
- Mann, D.A. Epigenetics in liver disease. Hepatology 2014, 60, 1418–1425. [Google Scholar] [CrossRef] [Green Version]
- Page, A.; Paoli, P.; Moran Salvador, E.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 2016, 64, 661–673. [Google Scholar] [CrossRef]
- Lyall, M.J.; Cartier, J.; Thomson, J.P.; Cameron, K.; Meseguer-Ripolles, J.; O’Duibhir, E.; Szkolnicka, D.; Villarin, B.L.; Wang, Y.; Blanco, G.R.; et al. Modelling non-alcoholic fatty liver disease in human hepatocyte-like cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170362. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: Novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Pirola, C.J.; Sookoian, S. The modulation of liver methylome in liver diseases. J. Hepatol. 2016, 64, 987–988. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ivanov, M.; Kals, M.; Kacevska, M.; Barragan, I.; Kasuga, K.; Rane, A.; Metspalu, A.; Milani, L.; Ingelman-Sundberg, M. Ontogeny, distribution and potential roles of 5-hydroxymethylcytosine in human liver function. Genome Biol. 2013, 14, R83. [Google Scholar] [CrossRef]
- Mischke, M.; Plosch, T. More than just a gut instinct-the potential interplay between a baby’s nutrition, its gut microbiome, and the epigenome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R1065–R1069. [Google Scholar] [CrossRef]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Mischke, M.; Plosch, T. The gut microbiota and their metabolites: Potential implications for the host epigenome. Adv. Exp. Med. Biol. 2016, 902, 33–44. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Kobayashi, S.; Seo, H.G. Mutual interaction between oxidative stress and endoplasmic reticulum stress in the pathogenesis of diseases specifically focusing on non-alcoholic fatty liver disease. World J. Biol. Chem. 2018, 9, 1–15. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tommasi, S.; Besaratinia, A. DNA Hydroxymethylation at the Interface of the Environment and Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2791. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph16152791
Tommasi S, Besaratinia A. DNA Hydroxymethylation at the Interface of the Environment and Nonalcoholic Fatty Liver Disease. International Journal of Environmental Research and Public Health. 2019; 16(15):2791. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph16152791
Chicago/Turabian StyleTommasi, Stella, and Ahmad Besaratinia. 2019. "DNA Hydroxymethylation at the Interface of the Environment and Nonalcoholic Fatty Liver Disease" International Journal of Environmental Research and Public Health 16, no. 15: 2791. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph16152791