4.2. Scenarios
Twenty scenarios are used to represent a variety of clinically feasible true underlying combination-toxicity relationships (
Table 1). We focus on finding one MTC at the end of the trial, regardless of if there are multiple possible correct combinations within the scenario.
In Scenarios 1–7, the MTC is located along each diagonal of the combination space, moving from the lower (1,1) corner of the combination space in scenario 1 to the upper (5,3) corner in Scenario 7. As it is unknown at the planning stage what the true combination-toxicity relationship is, it is important that all these scenarios are used to ensure good operating characteristics across all these scenarios. Scenarios 2 and 6 each have two possible MTC dose combinations. Two more variants of each of these scenarios were added.
Scenarios 2 and 6 were altered by replacing, in turn, one of the two MTCs with a dose combination with a toxicity probability different to 0.3 whilst ensuring that the monotonicity assumption still holds to form the Scenarios 2.1, 2.2, 6.1 and 6.2. The Scenarios 1, 2.1, 2.2, 6.1, 6.2 and 7 represent extreme examples of the dose-toxicity relationship. For Scenarios 1, 2.1 and 2.2, they represent a steep combination-toxicity relationship with many of the doses higher in the combination space having toxicity probabilities far above the MTC. Comparatively, for Scenarios 6.1, 6.2 and 7, they show a flat combination-toxicity relationship where many of the doses lower in the combination have toxicity probabilities space are far below the MTC. Note that Scenarios 2.1, 2.2, 6.1, 6.2 also correspond to cases when one compound increases the toxicity of the combination more than the other—when increasing the dose of one compound leads just to the target toxicity of 30% but increase in another corresponds to an overly toxic (40%) dose combination.
Scenarios 8–12 were proposed by Riviere et al. [
6]. In Scenarios 8, 10, 11 and 12 there are multiple MTCs which are not located along the same diagonal but throughout the combination space and in Scenario 9 there is one MTC located in the centre of the combination space. Furthermore, under Scenarios 8, 10, and 12, it is assumed that one of the compounds is more toxic than the other (i.e., the combination toxicity relationship is steeper in one compound). Scenarios 13 and 14 represent the scenarios with only one MTC located for high doses of one of the agents.
Under Scenario 15, all combinations are too toxic as the lowest combination already has the toxicity rate of 45%. In contrast, under scenario 16, all combinations are safe as the highest combination has a toxicity probability of 20%. Note that as the design does not include stopping rules, it is expected that a design with desirable properties would recommend the lowest and the highest dose in Scenarios 15 and 16, respectively.
Finally, Scenarios 11 and 14 were found to be well-approximated by the original 4-parameter logistic model but with the slope parameters for each agent being equal to . This implies that the underlying combination-toxicity relationships are fully determined by the interaction parameter and these are the scenarios where one could expect to see the most gain in benefit in including the interaction term. Therefore, we will be using these 2 scenarios to assess further potential losses of not including the interaction parameter.
4.3. Calibration
The sets of values for the grid search for the hyper-parameters have been selected as
for each of the considered models. Note that the values of
,
,
and
correspond to the prior distributions specified by Riviere et al. [
6]. Therefore, these grids were chosen to explore lower and higher variance around the mean of the parameters compared to the originally considered prior distribution. Due to the computational costs, the hyper-parameters to be tried were chosen to be noticeably different from each other, e.g., at least increasing the variance of the parameter twofold. This aims at locating an approximately optimal (in terms of PCS) values. We will use these values to compare whether the proposed calibration procedure can provide benefits in terms of the operating characteristics. For each set of hyper-parameters combinations, we simulate 500 trials to evaluate dual-agent drug combinations.
As discussed above, the choice of scenarios for the calibration is crucial. As the calibration over all 20 scenarios would have been to computationally demanding, we specify a subset of four scenarios to reduce the compuational costs while still adequetly exploring the properties of the design specification under extreme scenarios. Specifically, Scenarios 2.1, 2.2, 6.1 and 6.2 from
Table 1 are used for the calibration process. Scenarios 2.1 and 2.2 represent a steep combination-toxicity relationship with many of the doses higher in the combination space far above the MTC and Scenarios 6.1 and 6.2 show a flat combination-toxicity relationship where many of the doses lower in the combination space are far below the MTC. Importantly, we have selected scenarios with one MTC only as it was noted previously that model-based designs can strongly favour one of the MTC in scenarios with several of them. This undesirable favouring of particular combinations cannot be picked up via summary characteristics such as the PCS, and the inclusion of scenarios with one MTC only would mitigate this risk.
The results of the hyper-parameter calibration are given in
Table 2.
For completeness, we also include the prior distribution originally proposed for the 4-parameter model (to which we refer as M0
) that will be further included in the simulation study. Note that the values of the hyper-parameters for M1 yielding the highest PCS were found to be on the bound of the selected grids of
and
. We have further extended these grids to include
and
and it was found that indeed the hyper-parameters in
Table 2 result in the highest PCS among the considered combinations of values (see
Table 3).
Finally, the calibrated hyper-parameters choices imply various prior combination toxicity relationships, all of which could be plausible. For example, under Model M2, the prior point estimate is around 0.05% for the lowest combination (i.e., the starting combination is very safe) and the highest is nearly 40%. Such prior beliefs correspond to a sharp increase in toxicity on the 5th dose on one of the compounds. Then, for example, starting escalation at the lowest combination, if the earlier data would suggest that the highest dose is not as toxic as expected, the escalation to higher doses would be allowed.
4.5. Results
A summary of operating characteristics using 4000 replications for all 4 models under Scenarios 1–14 with at least one MTC is given in
Table 4. As some designs are expected to outperform others in some scenarios and perform worse in others, we also provide the geometric mean (GM) of the PCS and its variance (Var) across scenarios.
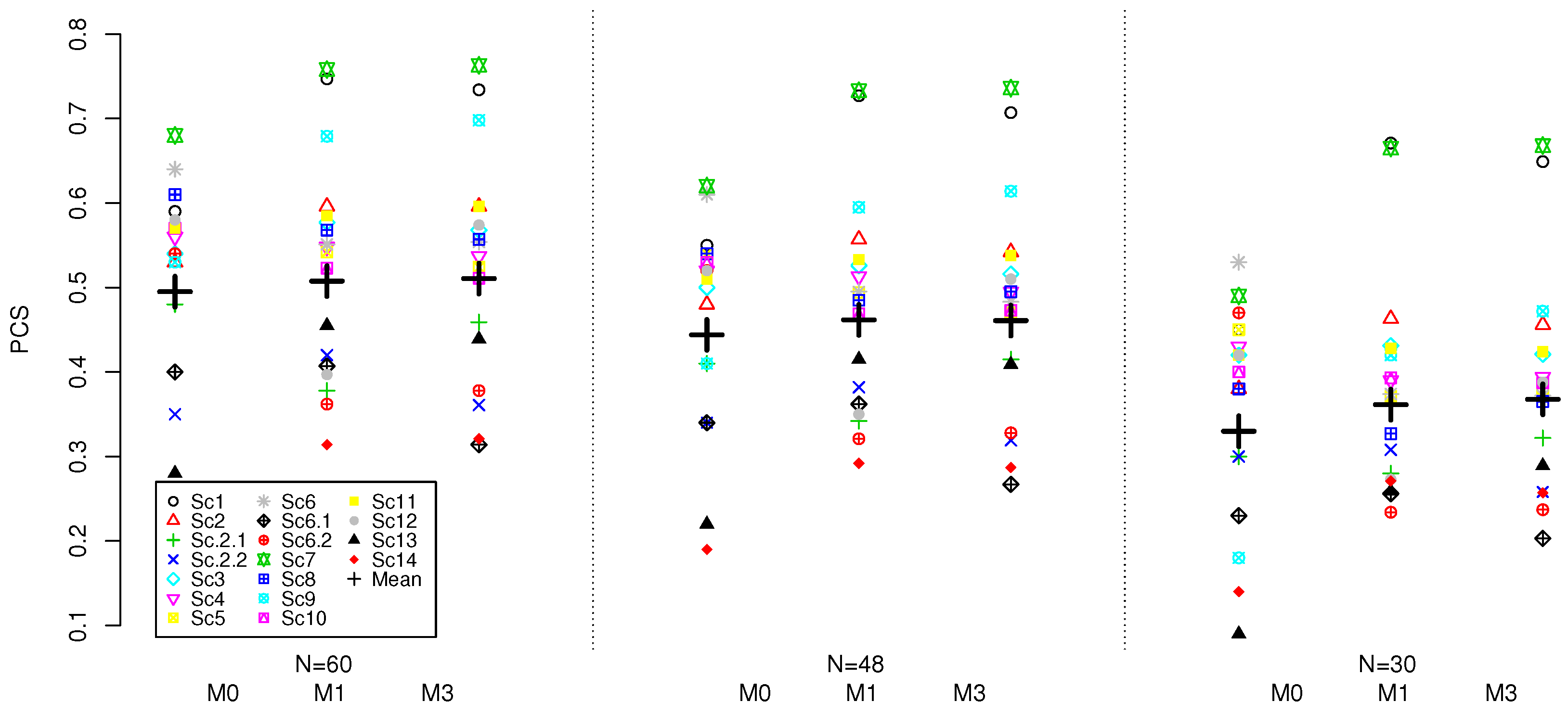
The calibrated model M2, the model with no intercept parameter, has a significantly lower average PCS of 37.0% compared to all other models. In 10 scenarios it has a PCS below 40% and has the lowest PCS amongst all models with a PCS of 11% in Scenario 13. The model M2 also has the lowest average percentage of patients allocated to a true MTC during the trial and the average percentage of DLTs throughout the trial is 32.8% which is the furthest from 30% compared to all other models. As this model is not performing comparably to all other models, it will not be considered further.
For model M0, which is the four-parameter model, two variations, with different set of prior parameters, are considered. The original model M0 and the calibrated M0 have the mean PCS of 48.7% and 49.6%, respectively. Therefore, the use of a calibrated prior allowed to increase the average PCS by nearly 1% on average under all considered scenarios. Comparing the average performance across scenarios that were not included in the calibration (i.e., excluding 2.1, 2.2, 6.1, 6.2), the models perform comparably—within 0.4% for the average PCS. At the same time, the calibrated model results in a noticeably more consistent performance in terms of the PCS across the scenarios—the variance of the PCS is 232.6 for the original prior and 152.0 for the calibrated one. Furthermore, the calibrated model results in nearly the same proportion of patients allocated to the true MTC (difference of 0.3%) but with noticeably lower variance across scenarios—62.0 for the calibrated model against 105.6 for the original prior. The costs for a better and more consistent performance for the calibrated model M0 is having an average percentage of observed DLTs slightly above the target rate, 30.5%, but still close to the target toxicity. Therefore, the model with the calibrated prior results in a more consistent performance of the design, and therefore the model under this prior is taken for further evaluation with the competing models.
Model M1, the model with no interaction parameter, had the highest average PCS of 50.8% compared to the other models 1.2% higher that for the calibrated model M0. At the same time, M0 has slightly lower variance in the PCS across the scenarios of 152.0 compared to 165.2 for M1. In eleven scenarios the model M1 has either higher PCS than the model M0 or is within 3% of it (for 9 scenarios the same can be said for M0). Additionally, M1 allocated the highest average percentage of patients to a true MTC throughout the trial at 31.4% and has 1% lower average percentage of observed DLTs. We will now focus on comparing scenario-by-scenario performance as the two models M0 and M1 seem to perform somewhat comparably.
In scenarios with only one MTC, the two models show uneven performance depending on the location of the MTC. When the true MTC was located in either the lower (1,1) or higher (5,3) extremity or the centre (3,2) of the combination space, as in scenarios 1, 7 and 9 respectively, model M1 showed its best performances with PCS of 75%, 76% and 68% respectively. In all three of these scenarios the model M0 had a PCS at least 8% lower. Importantly, in scenario 1, the difference in PCS of 16% is observed between the two models in favour of the model M1. Scenario 1 shows a steep combination-toxicity relationship with all the doses higher in the combination space having a toxicity probability far above the MTC. Therefore, the significantly higher PCS for the model M1 is of particular preference in this scenario. The model M1 also allocates 56.1% of patients to the true MTC in scenario 1 which is the highest allocation across all scenarios compared to 41% for M0. The higher percentage of patients allocated to the true MTC for the model M1 suggests it is more conservative and less aggressive in its approach at allocating patients compared to the model M0. This is preferable, in particular in scenarios such as scenario 1 which shows such a steep combination-toxicity relationship. The most noticeable costs for this advantage of the model M1 is a loss of 18% PCS in scenario 12 with the target combination lying on various diagonals. The model M0 has a PCS of 58% compared to 40% for M1 that suggests that having a more flexible model (under the calibrated parameters) might be more beneficial under this scenario. Comparatively, in scenarios 2.1, 2.2, 6.1, 6.2, 13 and 14, both models show some of their poorest performances as these are the most challenging scenarios with a single MTC. It also confirmed by the benchmark that these scenarios are the most difficult—the benchmark results in its lowest PCS under these scenarios as well. In all these scenarios, both models have a PCS of 45% or lower.
Comparing the PCS in scenarios 11 and 14 which are approximately generated using the model with the intercept and interaction parameter model, , one can find that the calibrated 4-parameter and 3-parameter with no interaction models perform within 3% of each other under scenario 11, and M1 outperforms M0 by 9% under scenario 14. Therefore, in the scenarios determined by the interaction only, the 4-parameter model does not provide any tangiable benefit and the 3-parameter model can approximate the combination-toxicity relationship well enough (or even better) to locate the true MTC.
Finally, comparing the performance of the models to the benchmark, as expected the benchmark results in the highest average PCS. Specifically, the ratio of the PCS compared to the benchmark, is 90% for M0
and are 92 and 94% for M0 and M1, respectively. Furthermore, the benchmark results in the highest PCS under the majority of scenarios, in 11 out of 18 scenarious, the benchmark results in either higher PCS or within the simulation error. The lowest ratio of the PCS compared to the benchmark is nearly 44% for M0
and around 71–72% for M0 and M1. In some of the scenarios, the models have shown to lead to super-efficiency [
18], the phenomenon when the benchmark is outperformed. This can be explained by a design favouring particular combinations. The highest ratio of PCS compared to the benchmark is also achieved for M1 under Scenario 13–32% PCS for the benchmark versus 46% for M1 resulting in the ratio of 144%. This suggests that the design favours this combination under the calibrated hyper-parameters.
The model M1 assigned at least 30% of patients to a true MTC in more scenarios than M0. Of the two models, M0 was the only one in which for two scenarios—Scenario 9 and 13—the allocation was below 20%. For the model M0, the highest percentage of DLTs observed for all the scenarios was 38% in scenario 1 whereas for M1 this value is lower at 36%, also in scenario 1. This once again highlights that the model M0 is more aggressive in patients allocations. For the model M1, in six scenarios the percentage of observed DLTs lay in the interval [29%, 31%] compared to three scenarios for M0.
The results for scenarios 15 and 16 with no MTC are given in
Table 5.
Under the overly toxic scenario 15, all of the models select the lowest combination with at least 92% with the minimum value of 92.7% for M2 and the highest of 98.9% for M1 (nearly 3% higher than for the calibrated model M0). Similarly, M1 allocated nearly 90% of patients to the lowest combination, which is the highest proportion among all models. Concerning, the safe scenario 16, M2 correspond to the poorest performance and selects the highest combination in nearly 80% compared to nearly 88% for both M0 models and nearly 94% for M1. The proportion of allocation is again the lowest for M2 and the highest for M1. Therefore, under both scenarios, Model 1 selects the closest to the target level combination with the highest probability and allocated more patients to the right dose.
As it was noted above, under the scenarios with several target combinations, model-based designs can favour particular combinations that will be reflected in uneven selection proportion of the target combinations. To explore this aspect of the considered models, we study the variance of each MTC selection within the scenario.
Table 6 shows the variance between the percentage of selections of each possible correct MTC within the scenario for the models M
, M0, and M1.
All of the models show poor performance in evenly selecting between multiple MTC combinations across the scenarios. Comparing calibrated models, Model M1 has a lower average variance across these scenarios of 163.5 compared to 196.4 for M0. Despite having a lower average variance, the model M1 shows a greater range of values across the scenarios. In Scenario 8, M1 shows its highest variance of 1365.0. The model M0 has nearly the same range of variance across these scenarios, where its highest variance is 1336.4 in Scenario 11.
Overall, under the operational prior distributions calibrated to achieve the highest PCS under each parameter model, the model M1 without interaction parameter was found to have the best performance in the set of considered scenarios. The model M1 has the highest average PCS and has the greatest lowest ratio of the PCS compared to the benchmark across scenarios. The model M1 allocates the highest average percentage of patients to a true MTC throughout the trial. It has the closest average percentage of observed DLTs throughout the trial to the target value of 30%. The model M1 also has the highest proportion of MTC selections in the interval [0.2, 0.4] so is, on average, selecting combinations with toxicities around the target value of more often. It has also demonstrated the most accurate performance in scenarios without the MTC. Furthermore, it containts one fewer parameter that can reduce the computations complexity of the proposed calibration procedure noticeably. One of the main drawbacks of model M1 is that it shows high variability when selecting the MTC when there are multiple possible MTCs in the combination space (e.g., Scenario 8).




{kind=link}