The Environmental Pollutant Cadmium Promotes Influenza Virus Replication in MDCK Cells by Altering Their Redox State


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
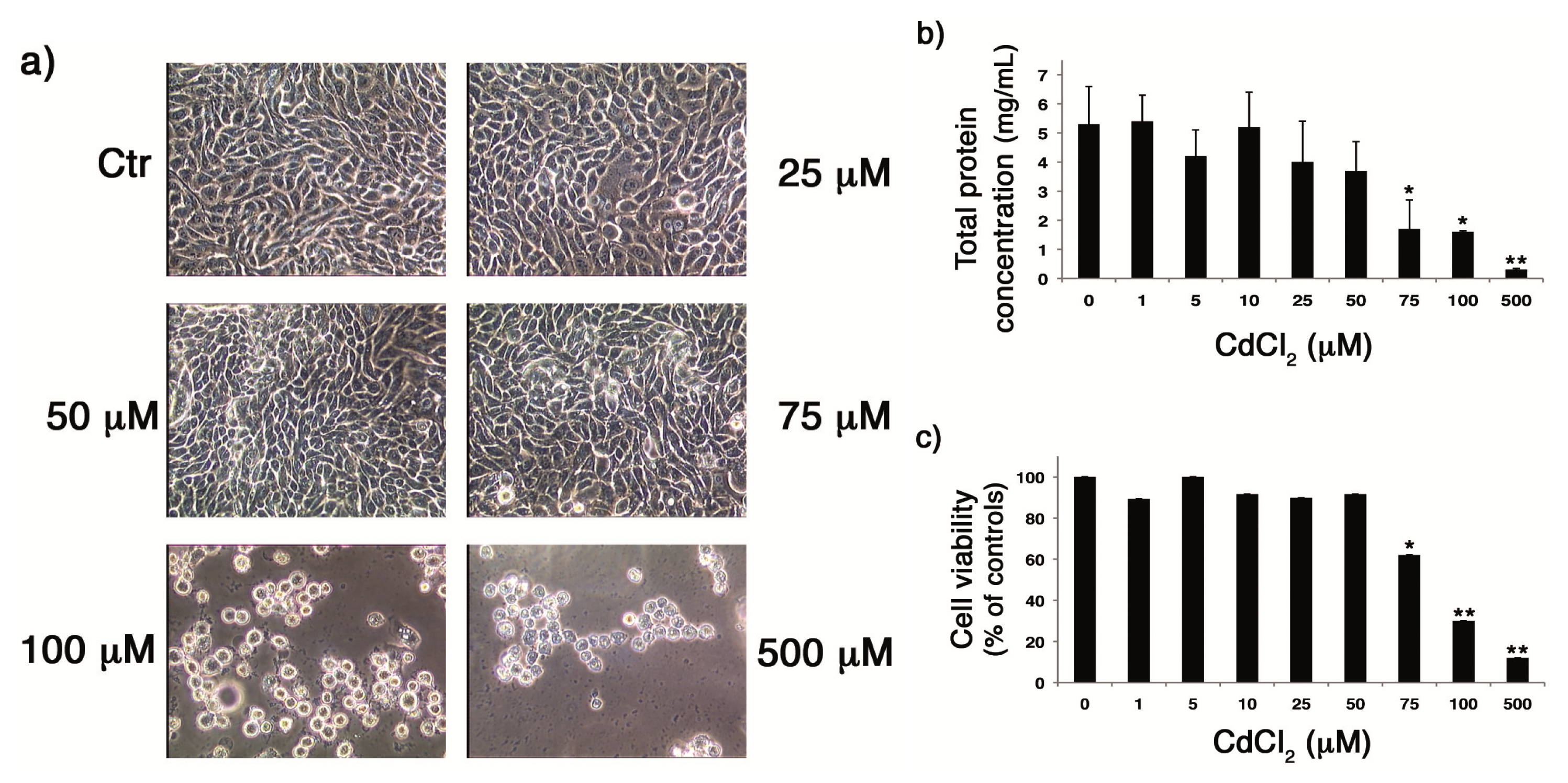
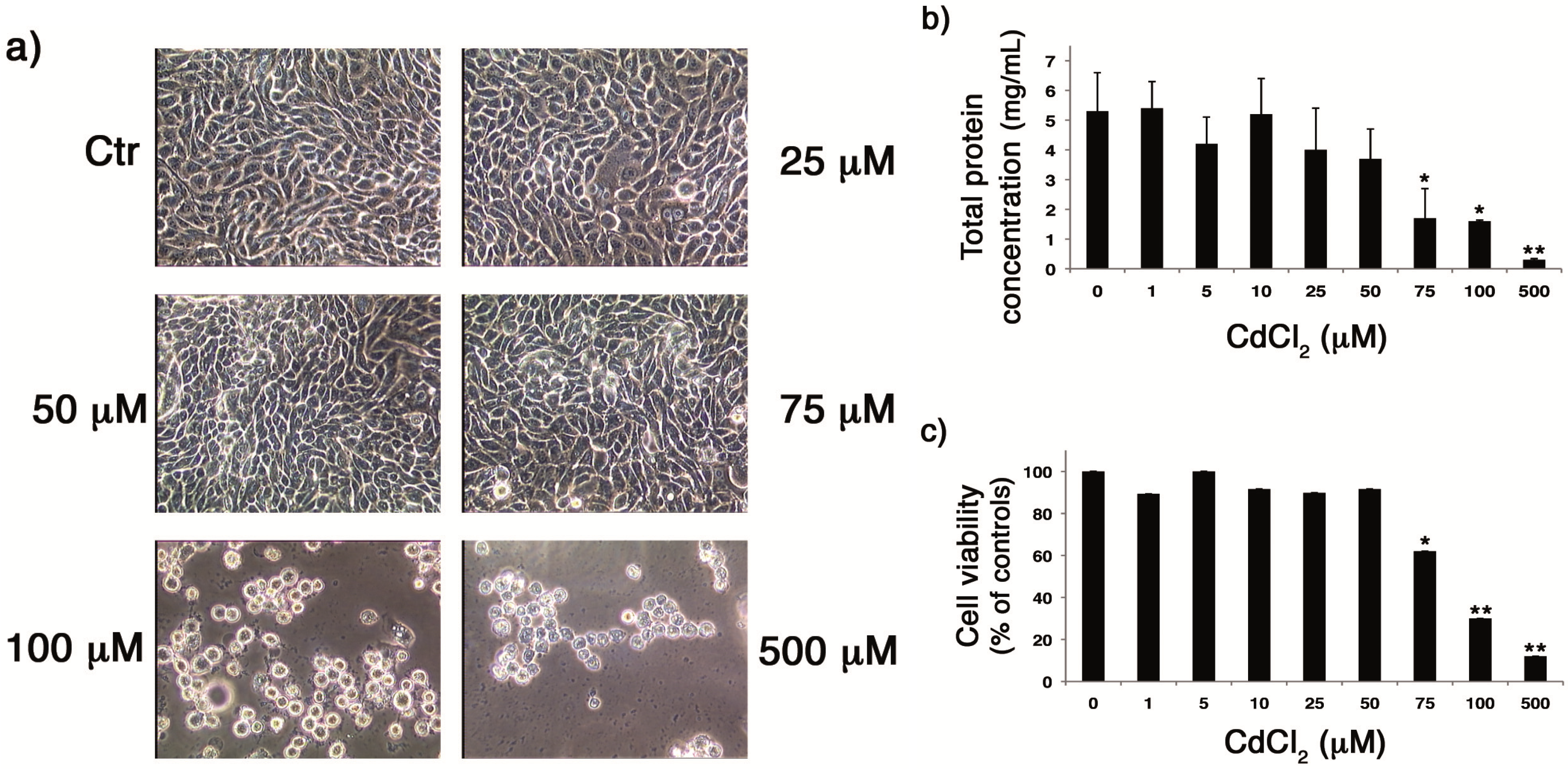
2.1. CdCl2 Was Not Toxic for Cells until the Concentration of 50 µM

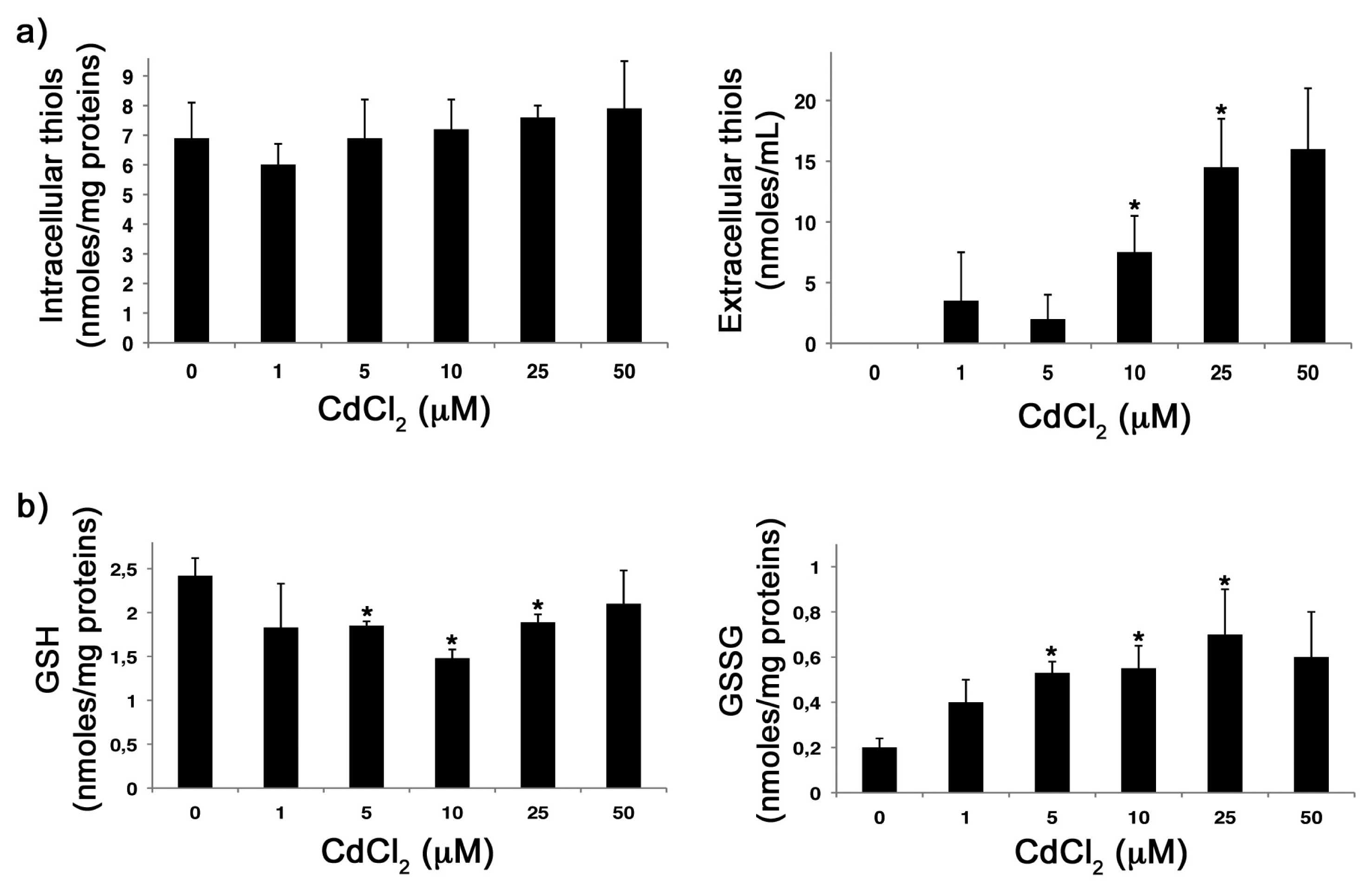
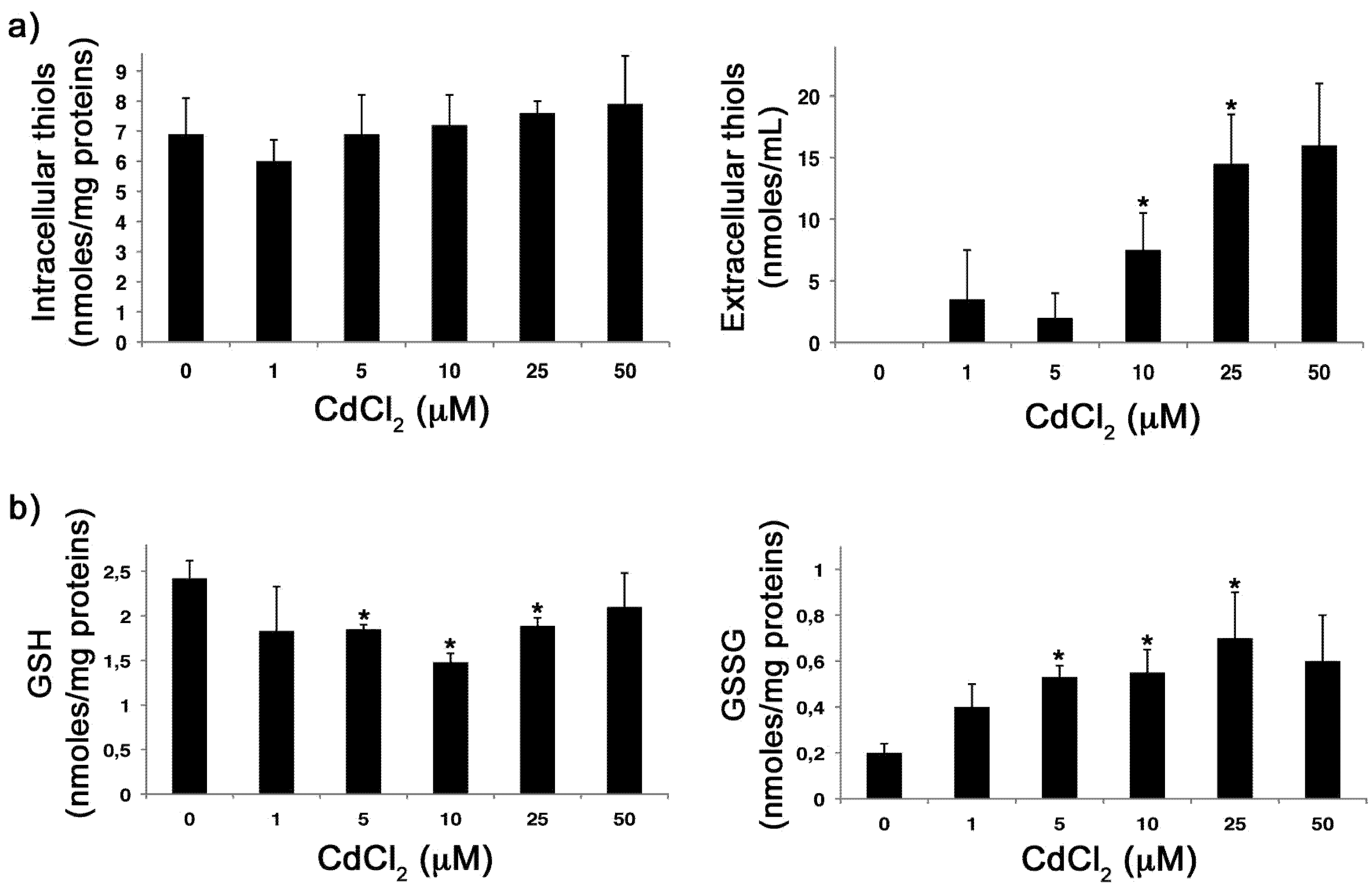
2.2. CdCl2 Alters the Intracellular Redox Balance versus an Oxidized State

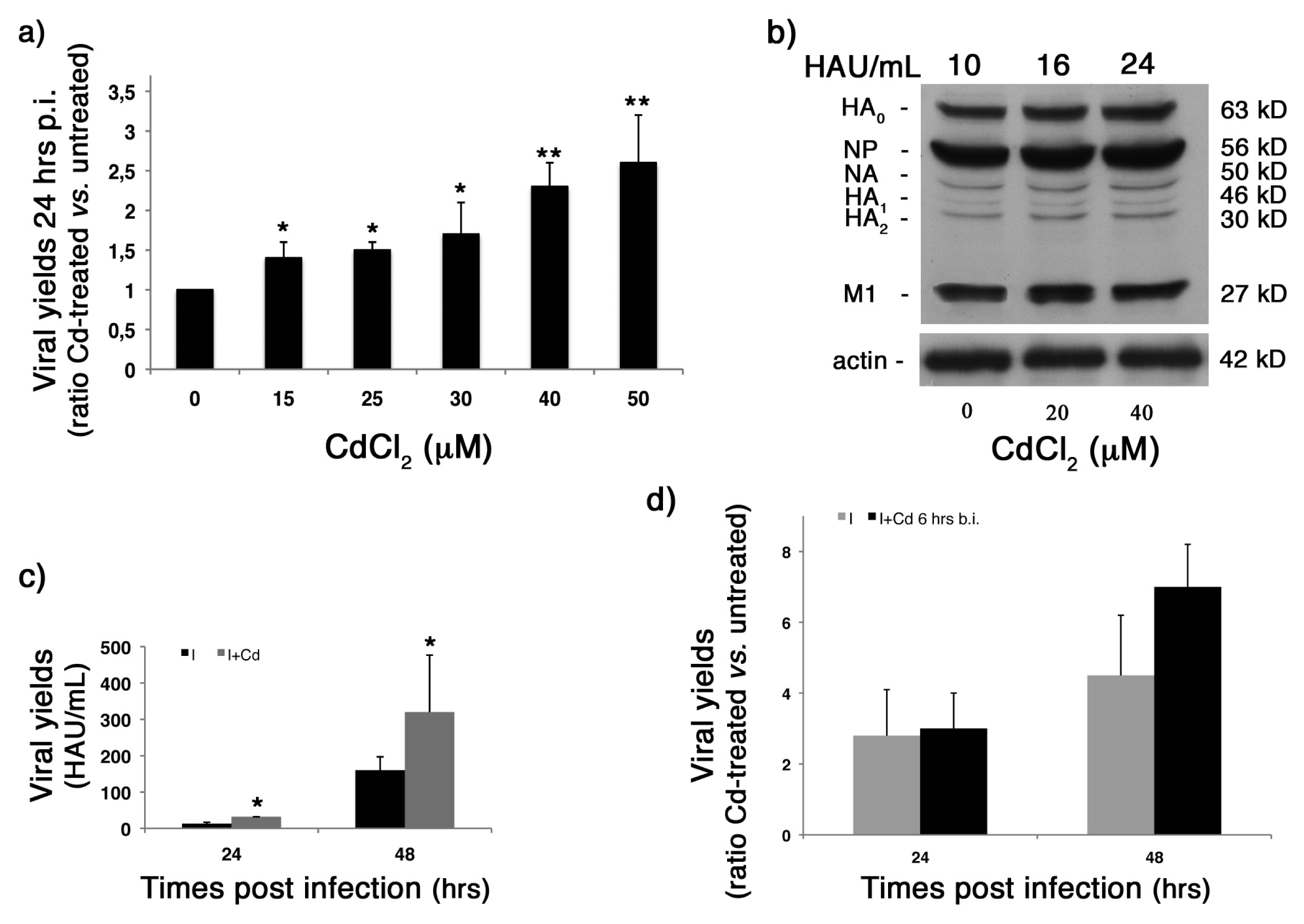
2.3. CdCl2 Induces an Increase in Influenza Virus Replication

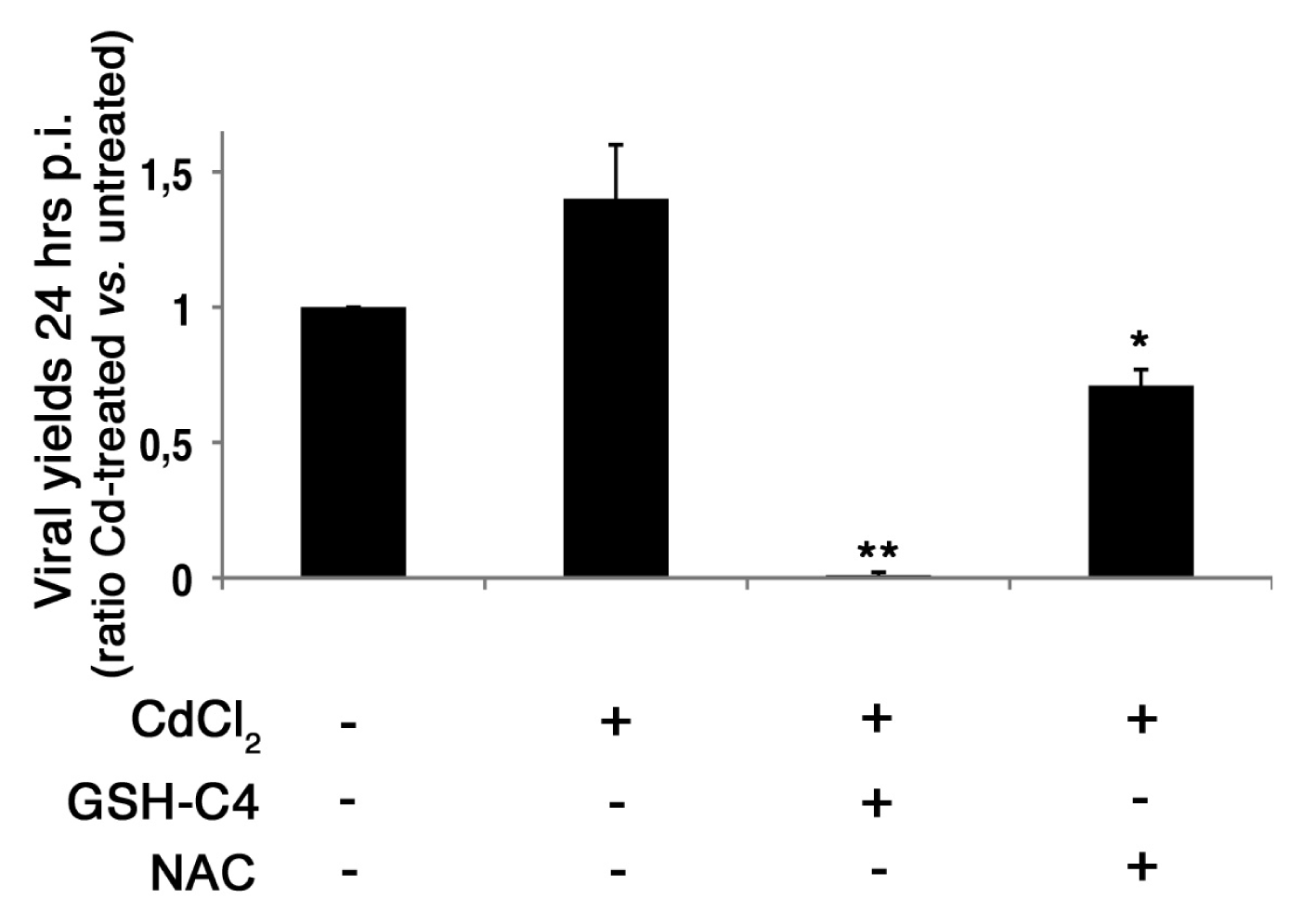
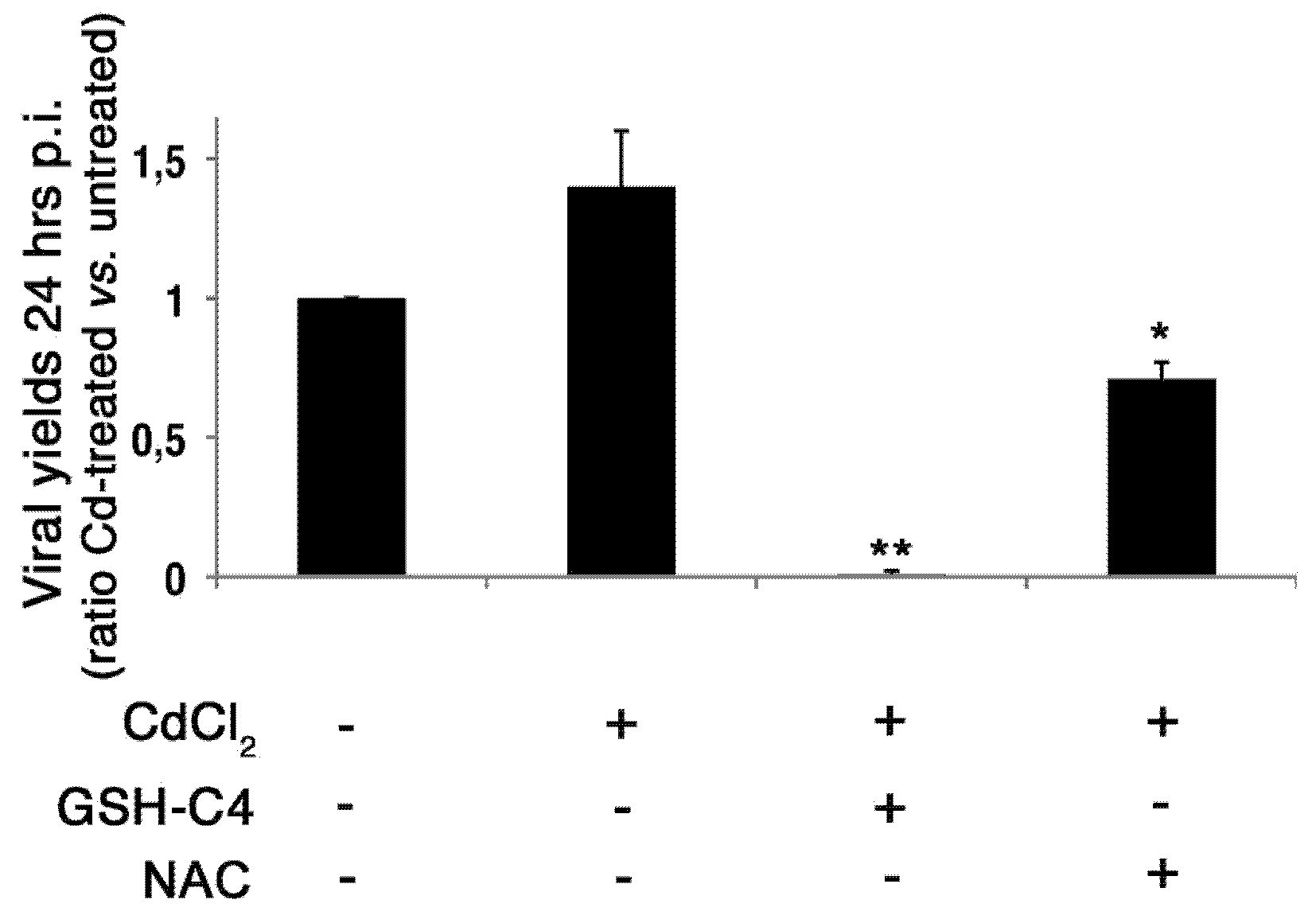
2.4. Treatment with Antioxidants Reverts the CdCl2-Induced Increase of Influenza Virus Replication

2.5. Discussion
3. Experimental Section
3.1. Cell Cultures
3.2. Cytotoxicity Assay
3.3. Virus Production, Infection and Titration
3.4. Immunoblotting
3.5. Quantitative Determination of Thiols
3.6. Glutathione Assay
3.7. Statistical Analyses
4. Conclusions
Acknowledgments
Conflict of Interest
References
- World Health Organization, Cadmium. In Air Quality Guidelines, 2nd ed.; WHO, Regional Office for Europe: Copenhagen, Denmark, 2000; pp. 136–138.
- Rennolds, J.; Butler, S.; Maloney, K.; Boyaka, P.N.; Davis, I.C.; Knoell, D.L.; Parinandi, N.L.; Cormet-Boyaka, E. Cadmium regulates the expression of the CFTR chloride channel in human airway epithelial cells. Toxicol. Sci. 2010, 116, 349–358. [Google Scholar] [CrossRef]
- Lòpez, E.; Arce, C.; Oset-Gasque, M.J.; Canadas, S.; Gonzàlez, M.P. Cadmium induces reactive oxygen species generation and lipid peroxidation in cortical neurons in culture. Free Radic. Biol. Med. 2006, 40, 940–951. [Google Scholar] [CrossRef]
- Manca, D.; Richard, A.C.; van Tra, H.; Chevalier, G. Relation between lipid peroxidation and inflammation in the pulmonary toxicity of cadmium. Arch. Toxicol. 1994, 68, 364–369. [Google Scholar] [CrossRef]
- Bagchi, D.; Vuchetich, P.J.; Bagchi, M.; Hassoun, E.A.; Tran, M.X.; Tang, L.; Stohs, S.J. Induction of oxidative stress by chronic administration of sodium dichromate and cadmium chloride to rats. Free Radic. Biol. Med. 1997, 22, 471–478. [Google Scholar] [CrossRef]
- Liu, J.; Qu, W.; Kadiiska, M.B. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol. Appl. Pharmacol. 2009, 238, 209–214. [Google Scholar] [CrossRef]
- Magder, S. Reactive oxygen species: Toxic molecules or spark of life? Crit. Care 2006, 10, 208. [Google Scholar] [CrossRef]
- Mates, M. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Nencioni, L.; Sgarbanti, R.; De Chiara, G.; Garaci, E.; Palamara, A.T. Influenza virus and redox mediated cell signaling: A complex network of virus/host interaction. New Microbiol. 2007, 30, 367–375. [Google Scholar]
- Palamara, A.T.; Perno, C.F.; Ciriolo, M.R.; Dini, L.; Balestra, E.; D’Agostini, C.; Di Francesco, P.; Favalli, C.; Rotilio, G.; Garaci, E. Evidence for antiviral activity of glutathione: In vitro inhibition of herpes simplex virus type 1 replication. Antiviral Res. 1995, 27, 237–253. [Google Scholar] [CrossRef]
- Garaci, E.; Palamara, A.T.; Di Francesco, P.; Favalli, C.; Ciriolo, M.R.; Rotilio, G. Glutathione inhibits replication and expression of viral proteins in cultured cells infected with Sendai virus. Biochem. Biophys. Res. Commun. 1992, 188, 1090–1096. [Google Scholar] [CrossRef]
- Garaci, E.; Palamara, A.T.; Ciriolo, M.R.; D’Agostini, C.; Abdel-Latif, M.S.; Aquaro, S.; Lafavia, E.; Rotilio, G. Intracellular GSH content and HIV replication in human macrophages. J. Leukoc. Biol. 1997, 62, 54–59. [Google Scholar]
- Palamara, A.T.; Garaci, E.; Rotilio, G.; Ciriolo, M.R.; Casabianca, A.; Fraternale, A.; Rossi, L.; Schiavano, G.F.; Chiarantini, L.; Magnani, M. Inhibition of murine AIDS by reduced glutathione. AIDS Res. Hum. Retrovir. 1996a, 12, 1373–1381. [Google Scholar] [CrossRef]
- Nucci, C.; Palamara, A.T.; Ciriolo, M.R.; Nencioni, L.; Savini, P.; D’Agostini, C.; Rotilio, G.; Cerulli, L.; Garaci, E. Imbalance in corneal redox state during herpes simplex virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp. Eye Res. 2000, 70, 215–220. [Google Scholar] [CrossRef]
- Hennet, T.; Peterhans, E.; Stocker, R. Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J. Gen. Virol. 1992, 73, 39–46. [Google Scholar] [CrossRef]
- Mileva, M.; Tancheva, L.; Bakalova, R.; Galabov, A.; Savov, V.; Ribarov, S. Effect of vitamin E on lipid peroxidation and liver monooxigenase activity in experimental influenza virus infection. Toxicol. Lett. 2000, 114, 39–45. [Google Scholar] [CrossRef]
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003, 17, 758–760. [Google Scholar]
- Cai, J.; Chen, Y.; Seth, S.; Furukawa, S.; Compans, R.W.; Jones, D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003, 34, 928–936. [Google Scholar] [CrossRef]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox-regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox Signal. 2011, 15, 593–606. [Google Scholar] [CrossRef]
- Buffinton, G.D.; Christen, S.; Peterhans, E.; Stocker, R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic. Res. Commun. 1992, 16, 99–110. [Google Scholar] [CrossRef]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radical in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar]
- Suliman, H.B.; Ryan, L.K.; Bishop, L.; Folz, R.J. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am. J. Physiol. 2001, 280, L69–L78. [Google Scholar]
- Savarino, A.; Mai, A.; Norelli, S.; El Daker, S.; Valente, S.; Rotili, D.; Altucci, L.; Palamara, A.T.; Garaci, E. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology 2009, 6, 52. [Google Scholar] [CrossRef]
- Lewis, M.G.; DaFonseca, S.; Chomont, N.; Palamara, A.T.; Tardugno, M.; Mai, A.; Collins, M.; Wagner, W.L.; Yalley-Ogunro, J.; Greenhouse, J.; et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 2011, 25, 1347–1356. [Google Scholar] [CrossRef]
- Palamara, A.T.; Di Francesco, P.; Ciriolo, M.R.; Buè, C.; Lafavia, E.; Rotilio, G.; Garaci, E. Cocaine increases Sendai virus replication in cultured epithelial cells: Critical role of the intracellular redox status. Biochem. Biophys. Res. Commun. 1996b, 228, 579–585. [Google Scholar] [CrossRef]
- Macchia, I.; Palamara, A.T.; Buè, C.; Savini, P.; Ciriolo, M.R.; Gaziano, R.; Di Francesco, P. Increased replication of Sendai virus in morphine-treated epithelial cells: Evidence for the involvement of the intracellular levels of glutathione. Int. J. Immunopharmacol. 1999, 21, 185–193. [Google Scholar] [CrossRef]
- Jaspers, I.; Ciencewicki, J.M.; Zhang, W.; Brighton, L.E.; Carson, J.L.; Beck, M.A.; Madden, M.C. Diesel exhaust enhances influenza virus infections in respiratory epithelial cells. Toxicol. Sci. 2005, 85, 990–1002. [Google Scholar] [CrossRef]
- Nencioni, L.; Sgarbanti, R.; Amatore, D.; Checconi, P.; Celestino, I.; Limongi, D.; Anticoli, S.; Palamara, A.T.; Garaci, E. Intracellular redox signaling as therapeutic target for novel antiviral strategy. Curr. Pharm. Des. 2011, 17, 3898–3904. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef]
- Thévenod, F. Cadmium and cellular signaling cascades: To be or not to be? Toxicol. Appl. Pharmacol. 2009, 238, 221–239. [Google Scholar] [CrossRef]
- Cuypers, A.; Plusquin, M.; Remans, T.; Jozefczak, M.; Keunen, E.; Gielen, H.; Opdenakker, K.; Nair, A.R.; Munters, E.; Artois, T.J.; et al. Cadmium stress: an oxidative challenge. Biometals 2010, 23, 927–940. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Müller, L. Consequences of cadmium toxicity in rat hepatocytes: Effects of cadmium on the glutathione-peroxidase system. Toxicol. Lett. 1986, 30, 259–265. [Google Scholar] [CrossRef]
- Sidhu, M.; Bhatia, M.; Awasthi, Y.C.; Nath, R. Effect of chronic cadmium exposure on glutathione S-transferase and glutathione peroxidase activities in rhesus monkey: The role of selenium. Toxicology 1993, 83, 203–213. [Google Scholar] [CrossRef]
- Chrestensen, C.A.; Starke, D.W.; Mieyal, J.J. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000, 275, 26556–26565. [Google Scholar] [CrossRef]
- Chin, T.A.; Templeton, D.M. Protective elevations of glutathione and metallothionein in cadmium-exposed mesangial cells. Toxicology 1993, 77, 145–156. [Google Scholar] [CrossRef]
- Hart, B.A.; Lee, C.H.; Shukla, G.S.; Shukla, A.; Osier, M.; Eneman, J.D.; Chiu, J.-F. Characterization of cadmium-induced apoptosis in rat lung epithelial cells: Evidence for the participation of oxidant stress. Toxicology 1999, 133, 43–58. [Google Scholar] [CrossRef]
- Hart, B.A.; Potts, R.J.; Watkin, R.D. Cadmium adaptation in the lung—A double-edged sword? Toxicology 2001, 160, 65–70. [Google Scholar] [CrossRef]
- Amatore, D.; Aquilano, K.; Sgarbanti, R.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. NOX4-dependent ROS burst controls the replication of influenza virus in lung epithelial cells. Cell. Microbiol. 2013. submitted for publication. [Google Scholar]
- Nencioni, L.; De Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and p38MAPK activity in cells infected with influenza A virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem. 2009, 284, 16004–16015. [Google Scholar]
- Palamara, A.T.; Nencioni, L.; Aquilano, K.; De Chiara, G.; Hernandez, L.; Cozzolino, F.; Ciriolo, M.R.; Garaci, E. Resveratrol inhibits Influenza A virus replication in vitro and in vivo. J. Infect. Dis. 2005, 191, 1719–1729. [Google Scholar] [CrossRef]
- Fioravanti, R.; Celestino, I.; Costi, R.; Cuzzucoli Crucitti, G.; Pescatori, L.; Mattiello, L.; Novellino, E.; Checconi, P.; Palamara, A.T.; Nencioni, L.; et al. Effects of polyphenol compounds on influenza A virus replication and definition of their mechanism of action. Bioorg.Med.Chem. 2012, 20, 5046–5052. [Google Scholar] [CrossRef]
- Lag, M.; Refsnes, M.; Lilleaas, E.M.; Holme, J.A.; Becher, R.; Schwarze, P.E. Role of mitogen activated protein kinases and protein kinase C in cadmium-induced apoptosis of primary epithelial lung cells. Toxicology 2005, 211, 253–264. [Google Scholar] [CrossRef]
- Chuang, S.M.; Wang, I.C.; Yang, J.L. Roles of JNK, p38 and ERK mitogen-activated protein kinasesin the growth inhibition and apoptosis induced by cadmium. Carcinogenesis 2000, 21, 1423–1432. [Google Scholar] [CrossRef]
- Galan, A.; Garcia-Bermejo, M.L.; Troyano, A.; Vilaboa, N.E.; de Blas, E.; Kazanietz, M.G.; Aller, P. Stimulation of p38 mitogen-activated protein kinase is an early regulatory event for the cadmium-induced apoptosis in human promonocytic cells. J. Biol. Chem. 2000, 275, 11418–11424. [Google Scholar]
- Hung, J.J.; Cheng, T.J.; Lai, Y.K.; Chang, M.D. Differential activation of p38 mitogen-activated protein kinase and extracellular signal-regulated protein kinases confers cadmium-induced HSP70 expression in 9L rat brain tumor cells. J. Biol. Chem. 1998, 273, 31924–31931. [Google Scholar]
- Iryo, Y.; Matsuoka, M.; Wispriyono, B.; Sugiura, T.; Igisu, H. Involvement of the extracellular signal-regulated protein kinase (ERK) pathway in the induction of apoptosis by cadmium chloride in CCRF–CEM cells. Biochem. Pharmacol. 2000, 60, 1875–1882. [Google Scholar] [CrossRef]
- Palamara, A.T.; Brandi, G.; Rossi, L.; Millo, E.; Benatti, U.; Nencioni, L.; Iuvara, A.; Garaci, E.; Magnani, M. New synthetic glutathione derivatives with increased antiviral activities. Antivir. Chem. Chemother. 2004, 15, 83–91. [Google Scholar]
- Fraternale, A.; Paoletti, M.F.; Casabianca, A.; Nencioni, L.; Garaci, E.; Palamara, A.T.; Magnani, M. GSH and analogs in antiviral therapy. Mol. Aspects Med. 2009, 30, 99–110. [Google Scholar] [CrossRef]
- Pope, C.A. Air pollution and health—Good news and bad. N. Engl. J. Med. 2004, 351, 1132–1134. [Google Scholar] [CrossRef]
- Gowdy, K.M.; Krantz, Q.T.; King, C.; Boykin, E.; Jaspers, I.; Linak, W.P.; Gilmour, M.I. Role of oxidative stress on diesel-enhanced influenza infection in mice. Part. Fibre Toxicol. 2010, 7, 34. [Google Scholar]
- Lin, C.C.; Chen, S.J.; Huang, K.L.; Hwang, W.I.; Chang-Chien, G.P.; Lin, W.Y. Characteristics of metals in nano/ultrafine/fine/coarse particles collected beside a heavily trafficked road. Environ. Sci. Technol. 2005, 39, 8113–8122. [Google Scholar]
- May, B.V.J. Virology: A practical Approach; IRL Press: Oxford, UK, 1991; pp. 119–150. [Google Scholar]
- Baranovich, T.; Wong, S.S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (Favipiravir) Induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013. [Google Scholar] [CrossRef]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Checconi, P.; Sgarbanti, R.; Celestino, I.; Limongi, D.; Amatore, D.; Iuvara, A.; Alimonti, A.; Garaci, E.; Palamara, A.T.; Nencioni, L. The Environmental Pollutant Cadmium Promotes Influenza Virus Replication in MDCK Cells by Altering Their Redox State. Int. J. Mol. Sci. 2013, 14, 4148-4162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14024148
Checconi P, Sgarbanti R, Celestino I, Limongi D, Amatore D, Iuvara A, Alimonti A, Garaci E, Palamara AT, Nencioni L. The Environmental Pollutant Cadmium Promotes Influenza Virus Replication in MDCK Cells by Altering Their Redox State. International Journal of Molecular Sciences. 2013; 14(2):4148-4162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14024148
Chicago/Turabian StyleChecconi, Paola, Rossella Sgarbanti, Ignacio Celestino, Dolores Limongi, Donatella Amatore, Alessandra Iuvara, Alessandro Alimonti, Enrico Garaci, Anna Teresa Palamara, and Lucia Nencioni. 2013. "The Environmental Pollutant Cadmium Promotes Influenza Virus Replication in MDCK Cells by Altering Their Redox State" International Journal of Molecular Sciences 14, no. 2: 4148-4162. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms14024148