DRAM1 Protects Neuroblastoma Cells from Oxygen-Glucose Deprivation/Reperfusion-Induced Injury via Autophagy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
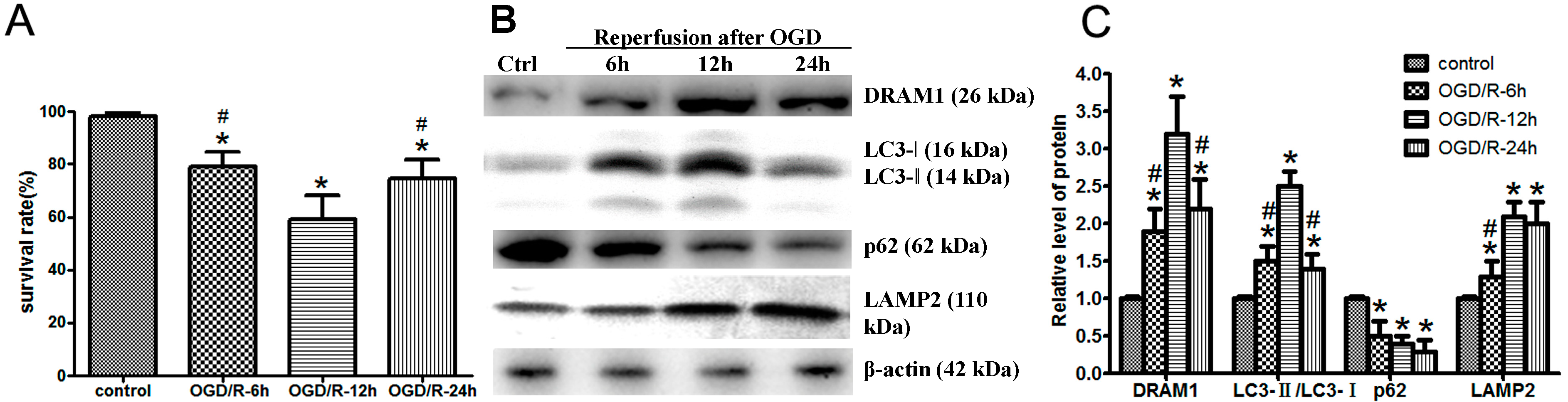
2.1. Oxygen Glucose Deprivation and Reperfusion (OGD/R) Treatment Increases DNA Damage-Regulated Autophagy Modulator Protein 1 (DRAM1) Expression and Induces Autophagy Activation

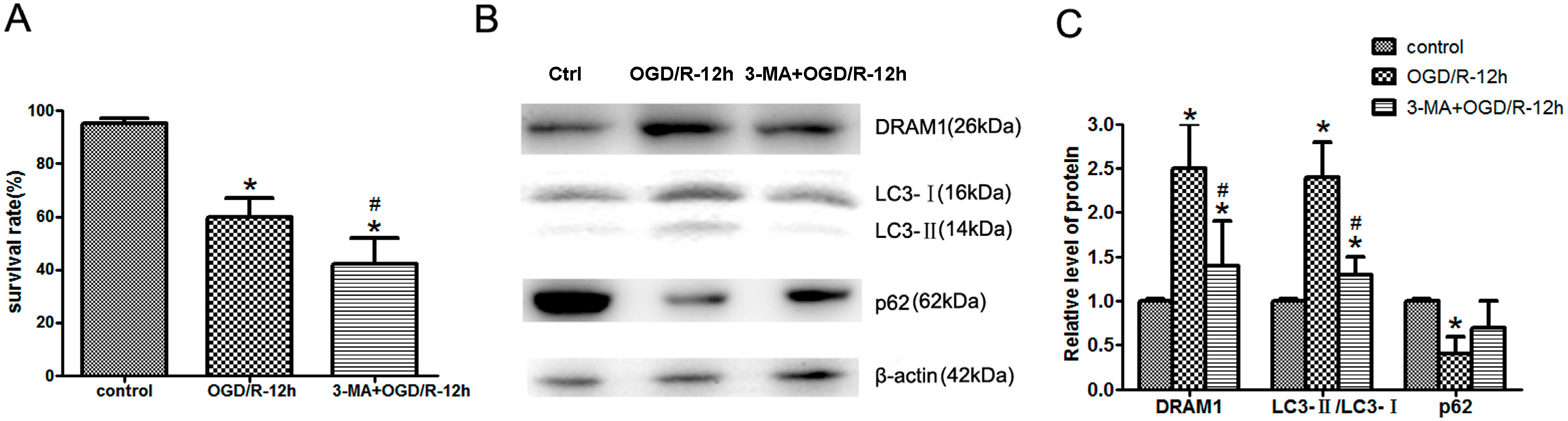
2.2. Inhibition of Autophagy Exacerbates OGD/R-Induced Neuro-2a Cell Injury

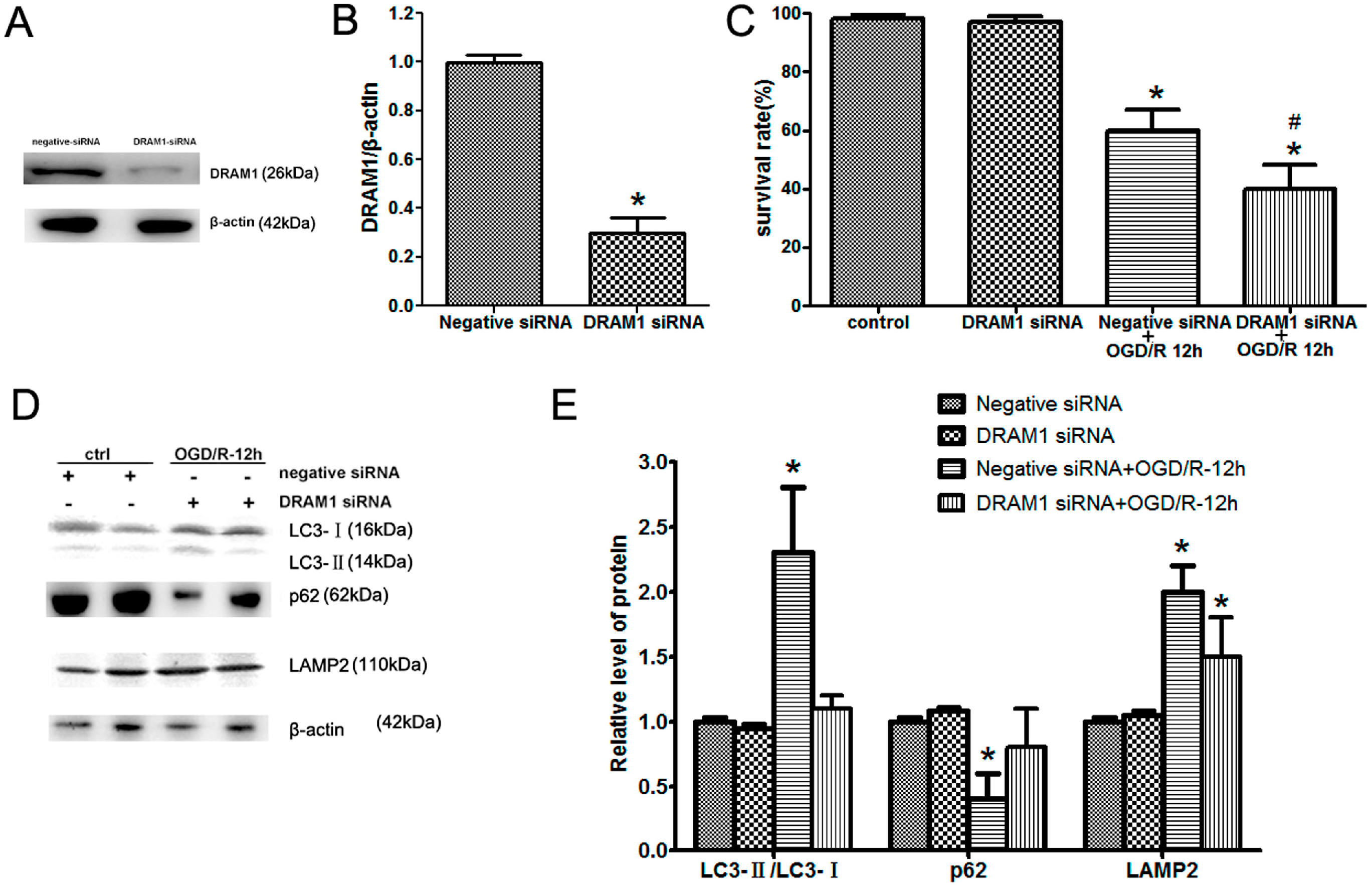
2.3. DRAM1 Knockdown Inhibits Autophagy Activation and Aggravates OGD/R-Induced Neuro-2a Cell Injury

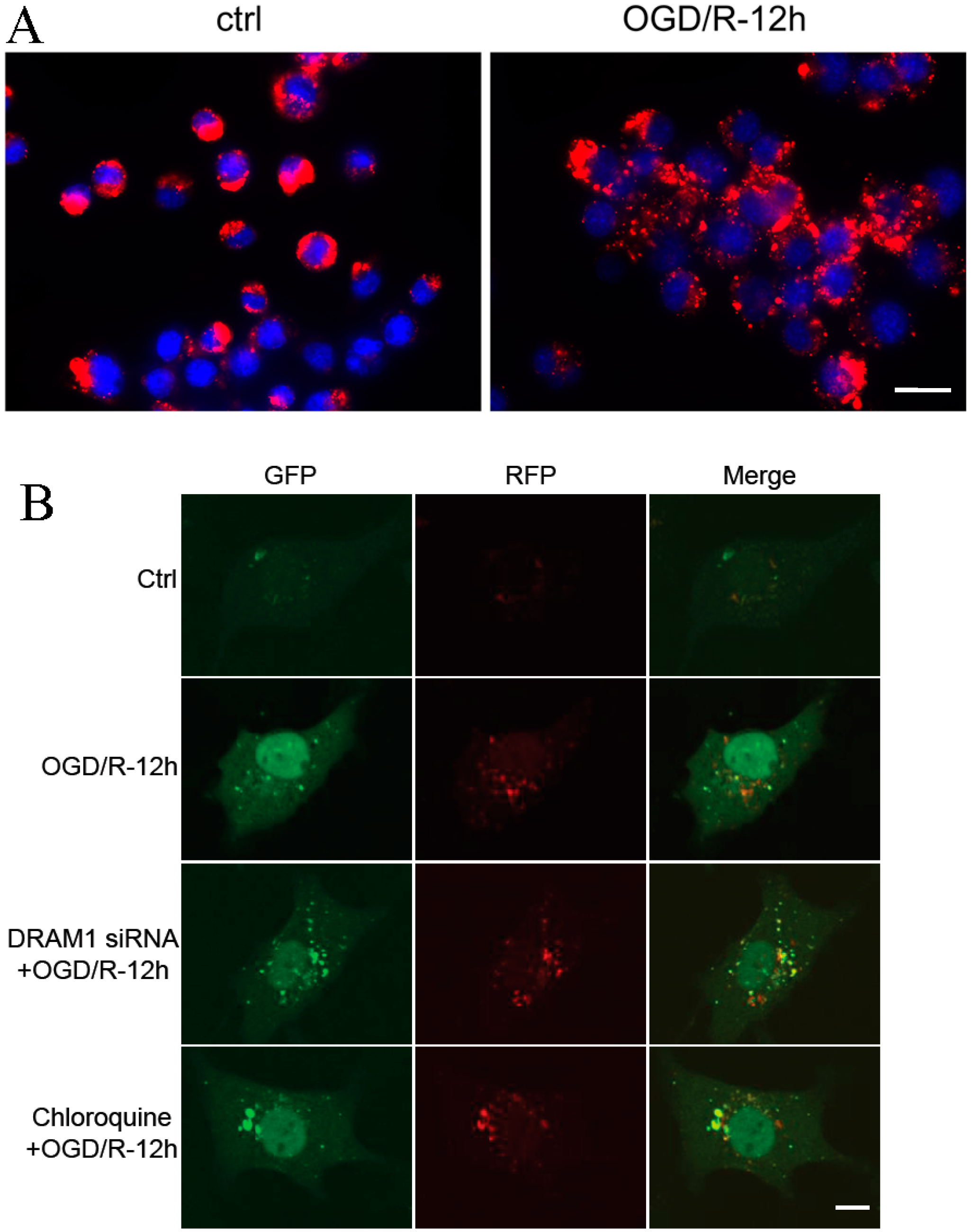
2.4. DRAM1 Knockdown Blocks Autophagy by Decreasing Autophagosome-Lysosome Fusion

3. Experimental Section
3.1. Cell Culture
3.2. Transfection with the Red Fluorescent Protein–Green Fluorescent Protein–Microtubule Associated Protein 1 Light Chain 3 (RFP–GFP–LC3) Expression Vector and DRAM1 Knockdown
3.3. Oxygen-Glucose Deprivation (OGD) and Reperfusion
3.4. Cell Viability Assay
3.5. Lysosome Staining and GFP–RFP–Fluorescence Observation
3.6. Western Blot Analyzes
3.7. Statistical Analyzes
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Pan, J.; Konstas, A.A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar]
- Uchiyama, Y.; Koike, M.; Shibata, M. Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy 2008, 4, 404–408. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Cui, D.; Wang, L.; Qi, A.; Zhou, Q.; Zhang, X.; Jiang, W. Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One 2012, 7, e35324. [Google Scholar] [CrossRef]
- Xu, F.; Li, J.; Ni, W.; Shen, Y.W.; Zhang, X.P. Peroxisome proliferator-activated receptor-γ agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS One 2013, 8, e55080. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Tadakoshi, M.; Gotoh, K.; Komatsu, M.; Waguri, S.; Kawahara, N.; Kuida, K.; Nagata, S.; Kominami, E.; et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am. J. Pathol. 2008, 172, 454–469. [Google Scholar]
- Wen, Y.D.; Sheng, R.; Zhang, LS.; Han, R.; Zhang, X.; Zhang, X.D.; Han, F.; Fukunaga, K.; Qin, Z.H. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008, 4, 762–769. [Google Scholar] [CrossRef]
- Liu, K.; Sun, Y.; Gu, Z.; Shi, N.; Zhang, T.; Sun, X. Mitophagy in ischaemia/reperfusion induced cerebral injury. Neurochem. Res. 2013, 38, 1295–1300. [Google Scholar] [CrossRef]
- Su, F.; Zhang, P.; Jiang, Z.W.; Peng, D.Q.; Gao, L.Y.; Liu, S.Q.; Qian, L.B.; Ye, Z.G.; Xia, Q. Expression and function of autophagy after ischemia/reperfusion in rats hippocampus neuron. Chin. J. Appl. Physiol. 2011, 27, 187–191. [Google Scholar]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia–ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef]
- Wang, P.; Guan, Y.F.; Du, H.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemic stroke. Autophagy 2012, 8, 77–87. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.R.; Wang, X.; Hu, W.W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar]
- Wei, K.; Wang, P.; Miao, C.Y. A double-edged sword with therapeutic potential: An updated role of autophagy in ischemic cerebral injury. CNS Neurosci. Ther. 2012, 18, 879–886. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Tyagi, N.; Qipshidze, N.; Munjal, C.; Vacek, J.C.; Metreveli, N.; Givvimani, S.; Tyagi, S.C. Tetrahydrocurcumin ameliorates homocysteinylated cytochrome-c mediated autophagy in hyperhomocysteinemia mice after cerebral ischemia. J. Mol. Neurosci. 2012, 47, 128–138. [Google Scholar] [CrossRef]
- Contartese, A.; Valoti, M.; Corelli, F.; Pasquini, S.; Mugnaini, C.; Pessina, F.; Aldinucci, C.; Sgaragli, G.; Frosini, M. A novel CB2 agonist, COR167, potently protects rat brain cortical slices against OGD and reperfusion injury. Pharmacol. Res. 2012, 66, 555–563. [Google Scholar] [CrossRef]
- Zhao, L.P.; Ji, C.; Lu, P.H.; Li, C.; Xu, B.; Gao, H. Oxygen glucose deprivation (OGD)/re-oxygenation-induced in vitro neuronal cell death involves mitochondrial cyclophilin-D/P53 signaling axis. Neurochem. Res. 2013, 38, 705–713. [Google Scholar]
- Rami, A.; Langhagen, A.; Steiger, S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol. Dis. 2008, 29, 132–141. [Google Scholar] [CrossRef]
- Adhami, F.; Schloemer, A.; Kuan, C.Y. The roles of autophagy in cerebral ischemia. Autophagy 2007, 3, 42–44. [Google Scholar]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; von, Figura.K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar]
- Shvets, E.; Elazar, Z. Autophagy-independent incorporation of GFP–LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 2008, 4, 1054–1056. [Google Scholar] [CrossRef]
- Xing, S.; Zhang, Y.; Li, J.; Zhang, J.; Li, Y.; Dang, C.; Li, C.; Fan, Y.; Yu, J.; Pei, Z.; et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012, 8, 63–76. [Google Scholar]
- Kerley-Hamilton, J.S.; Pike, A.M.; Hutchinson, J.A.; Freemantle, S.J.; Spinella, M.J. The direct p53 target gene, FLJ11259/DRAM, is a member of a novel family of transmembrane proteins. Biochim. Biophys. Acta 2007, 1769, 209–219. [Google Scholar]
- O’Prey, J.; Skommer, J.; Wilkinson, S.; Ryan, K.M. Analysis of DRAM-related proteins reveals evolutionarily conserved and divergent roles in the control of autophagy. Cell Cycle 2009, 8, 2260–2265. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Cuervo, A.M.; Taylor, M.R.; Nishino, I.; Blum, J.S.; Dice, J.F.; Sandoval, I.V.; Lippincott-Schwartz, J.; August, J.T.; Saftig, P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 2005, 6, 1058–1061. [Google Scholar] [CrossRef]
- Hariharan, N.; Zhai, P.; Sadoshima, J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011, 14, 2179–2190. [Google Scholar]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Ni, H.M.; Bockus, A.; Wozniak, A.L.; Jones, K.; Weinman, S.; Yin, X.M.; Ding, W.X. Dissecting the dynamic turnover of GFP–LC3 in the autolysosome. Autophagy 2011, 7, 188–204. [Google Scholar]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010, 6, 438–448. [Google Scholar]
- Laforge, M.; Limou, S.; Harper, F.; Casartelli, N.; Rodrigues, V.; Silvestre, R.; Haloui, H.; Zagury, J.F.; Senik, A.; Estaquier, J. DRAM triggers lysosomal membrane permeabilization and cell death in CD4+ T cells infected with HIV. PLoS Pathog. 2013, 9, e1003328. [Google Scholar]
- Zhang, X.D.; Qi, L.; Wu, J.C.; Qin, Z.H. DRAM1 regulates autophagy flux through lysosomes. PLoS One 2013, 8, e63245. [Google Scholar]
- Xie, X.; Le, L.; Fan, Y.; Lv, L.; Zhang, J. Autophagy is induced through the ROS–TP53–DRAM1 pathway in response to mitochondrial protein synthesis inhibition. Autophagy 2012, 8, 1071–1084. [Google Scholar] [CrossRef]
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, 5, 339–350. [Google Scholar]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef]
- Chi, Z.; Ma, X.; Cui, G.; Li, M.; Li, F. Cinnamtannin B-1 regulates cell proliferation of spinal cord astrocytes and protects the cell fromoxygen-glucose-serum deprivation/reoxygenation-induced apoptosis. Int. J. Mol. Sci. 2013, 14, 15827–15837. [Google Scholar] [CrossRef]
- Bendix, I.; Schulze, C.; von Haefen, C.; Gellhaus, A.; Endesfelder, S.; Heumann, R.; Felderhoff-Mueser, U.; Sifringer, M. Erythropoietin modulates autophagy signaling in the developing rat brain in an in vivo model of oxygen-toxicity. Int. J. Mol. Sci. 2012, 13, 12939–12951. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, M.; Jiang, Y.; Feng, Q.; Ouyang, Y.; Gan, J. DRAM1 Protects Neuroblastoma Cells from Oxygen-Glucose Deprivation/Reperfusion-Induced Injury via Autophagy. Int. J. Mol. Sci. 2014, 15, 19253-19264. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151019253
Yu M, Jiang Y, Feng Q, Ouyang Y, Gan J. DRAM1 Protects Neuroblastoma Cells from Oxygen-Glucose Deprivation/Reperfusion-Induced Injury via Autophagy. International Journal of Molecular Sciences. 2014; 15(10):19253-19264. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151019253
Chicago/Turabian StyleYu, Mengqiang, Yugang Jiang, Qingliang Feng, Yi'an Ouyang, and Jie Gan. 2014. "DRAM1 Protects Neuroblastoma Cells from Oxygen-Glucose Deprivation/Reperfusion-Induced Injury via Autophagy" International Journal of Molecular Sciences 15, no. 10: 19253-19264. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151019253