Lidocaine Sensitizes the Cytotoxicity of Cisplatin in Breast Cancer Cells via Up-Regulation of RARβ2 and RASSF1A Demethylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
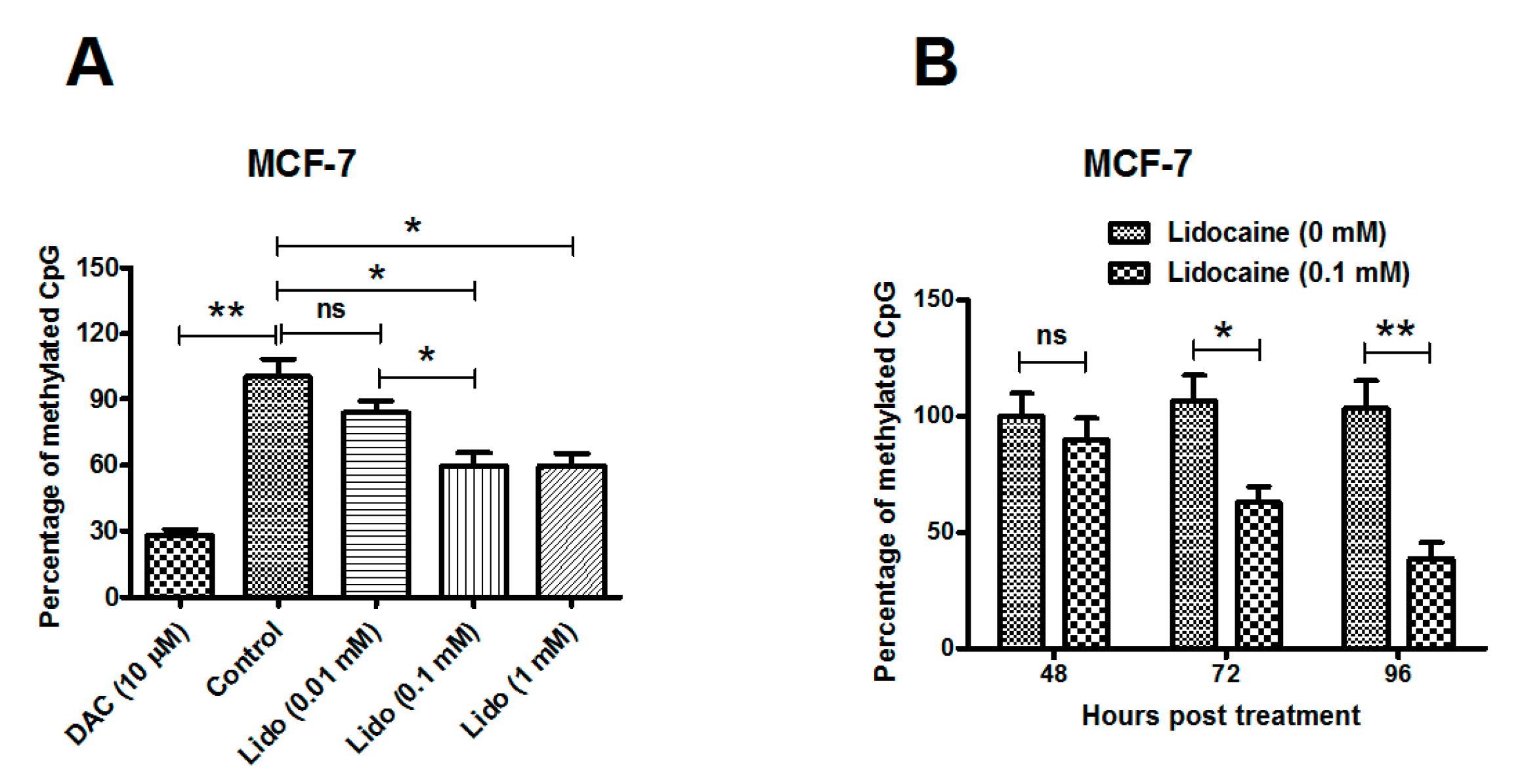
2.1. Lidocaine Promotes Global Genomic Demethylation of the CpG Island in Human Breast Cancer Lines


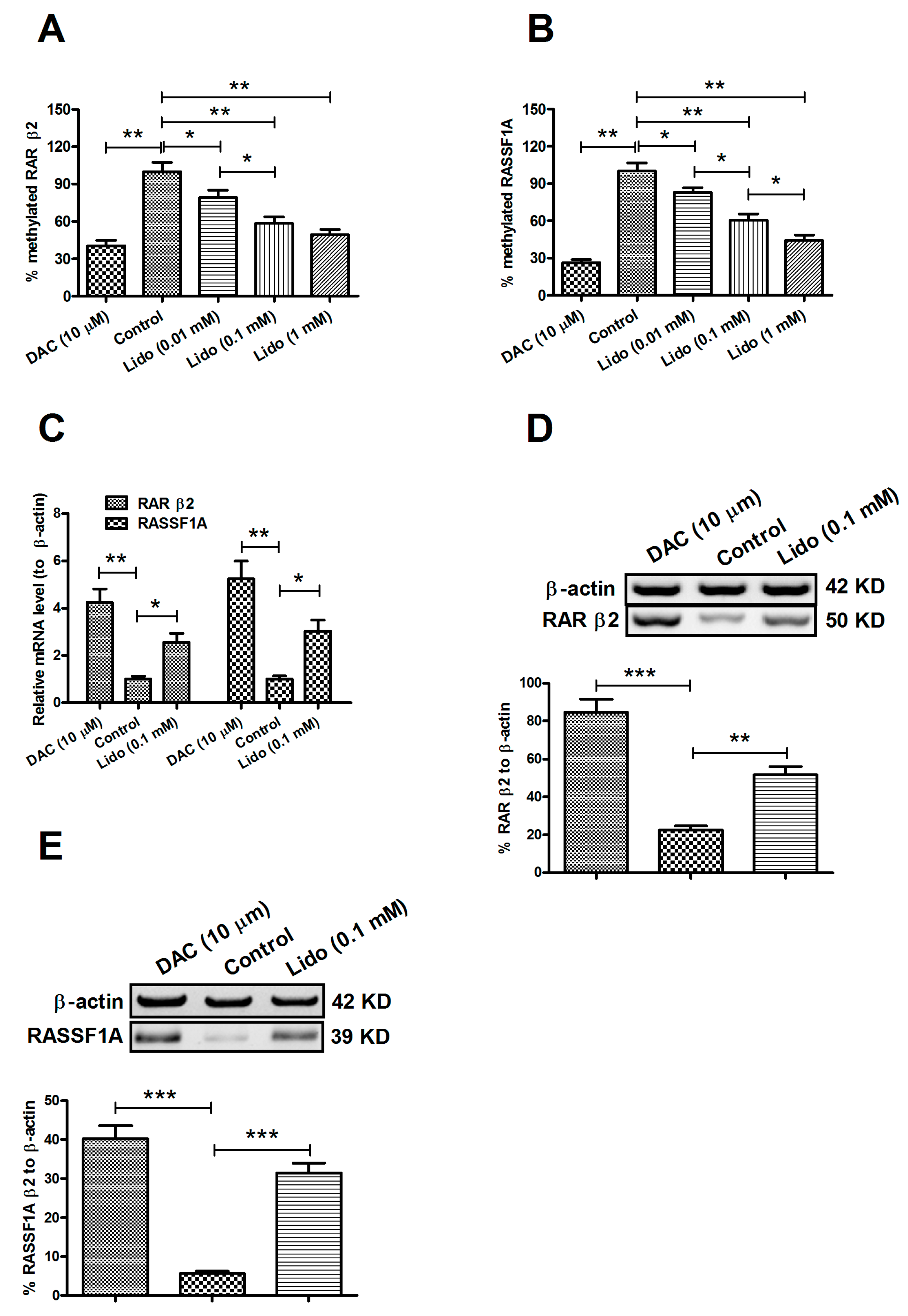
2.2. Lidocaine Ameliorates the Expression of RARβ2 and RASSF1A Genes in MCF-7 and MDA-MB-231 Cells
2.3. Regulation of Lidocaine on the Viability of MCF-7 and MDA-MB-231 Cells


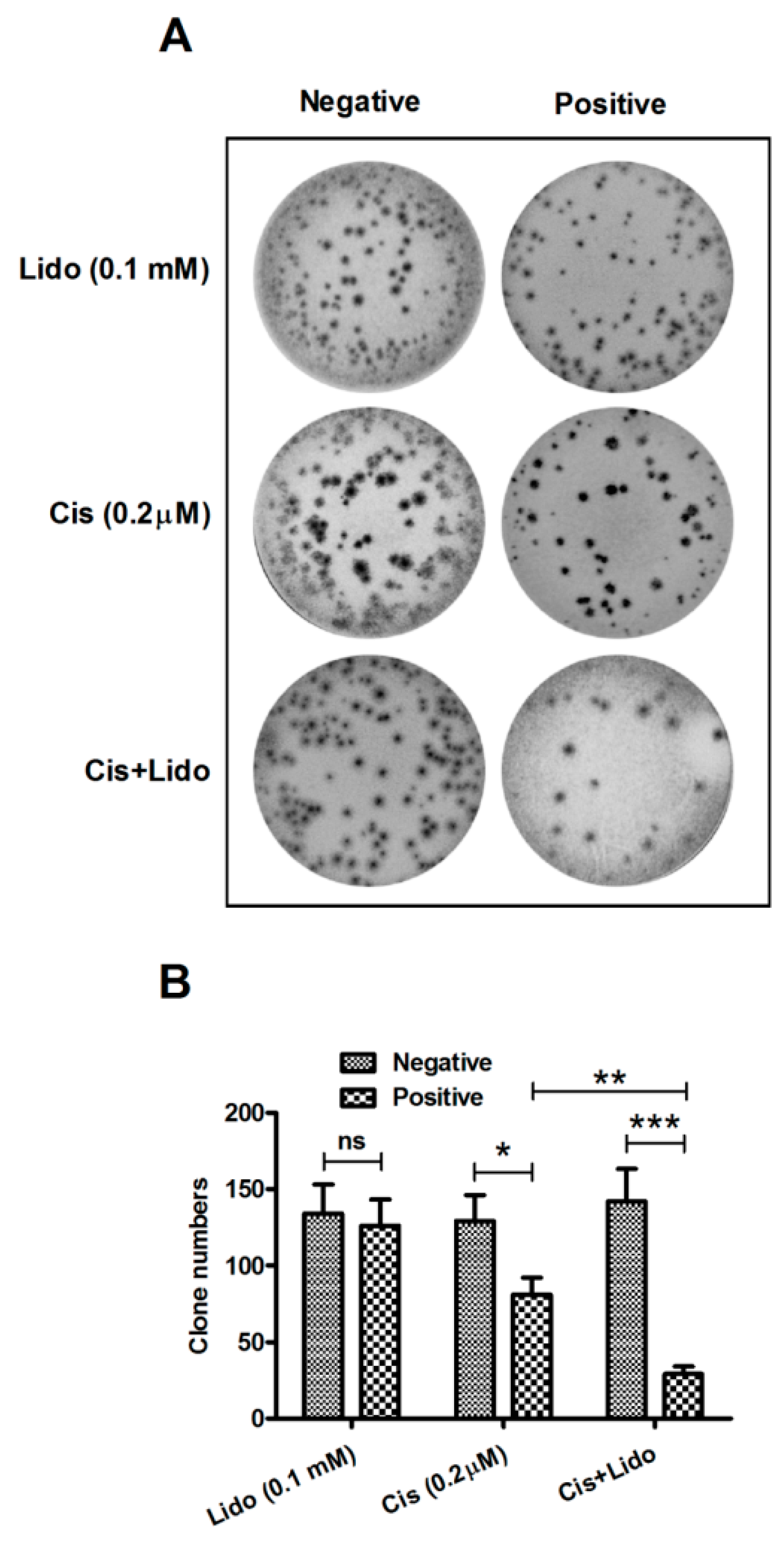
2.4. Lidocaine Enhances the Cytotoxicity of Cisplatin against MCF-7 Cells

2.5. Lidocaine Regulation of Apoptosis Promotion by Cisplatin in MCF-7 Cells
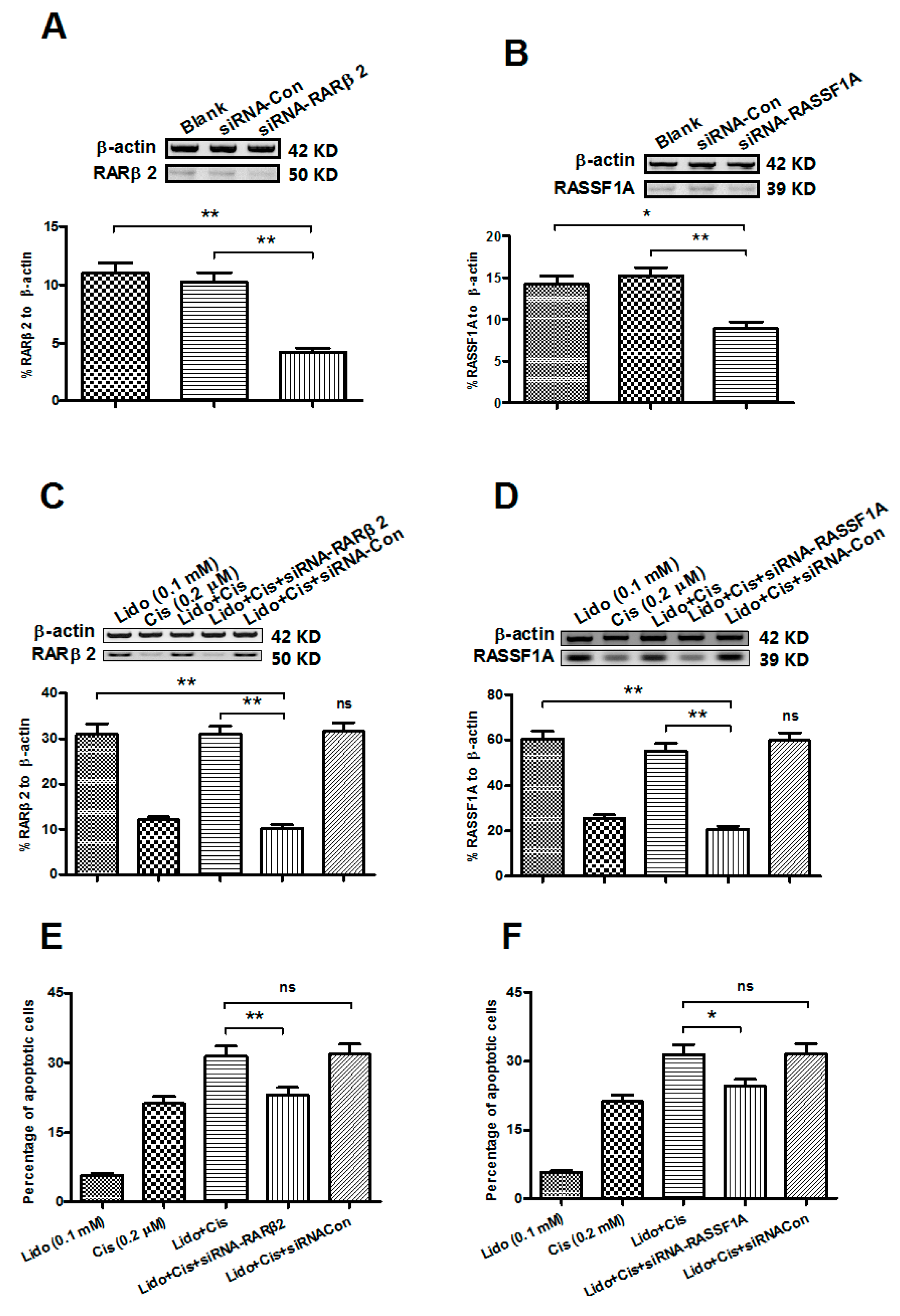
2.6. Abrogation of the RARβ2 and RASSF1A Genes Blocks the Apoptosis Promotion by Lidocaine and Cisplatin in MCF-7 Cells


3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment with Reagents
4.2. Methylation Analysis of Global Genomics and TSGs, RARβ2 and RASSF1A
4.3. RNA Extraction and RT-qPCR
4.4. Protein Isolation and Western Blot Analysis
4.5. MTT Assay and Cell Colony Formation Assay
4.6. Cell Apoptosis Assay and Fluorometric Analysis of Caspase 3 Activity
5. Conclusions
Supplementary Information
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lambert, L.A.; Lambert, D.H.; Strichartz, G.R. Irreversible conduction block in isolated nerve by high concentrations of local anesthetics. Anesthesiology 1994, 80, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Rigler, M.L.; Drasner, K.; Krejcie, T.C.; Yelich, S.J.; Scholnick, F.T.; DeFontes, J.; Bohner, D. Cauda equina syndrome after continuous spinal anesthesia. Anesth. Analg. 1991, 72, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Kasaba, T.; Onizuka, S.; Takasaki, M. Procaine and mepivacaine have less toxicity in vitro than other clinically used local anesthetics. Anesth. Analg. 2003, 97, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Onizuka, S.; Takasaki, M.; Syed, N.I. Long-term exposure to local but not inhalation anesthetics affects neurite regeneration and synapse formation between identified lymnaea neurons. Anesthesiology 2005, 102, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Werdehausen, R.; Braun, S.; Essmann, F.; Schulze-Osthoff, K.; Walczak, H.; Lipfert, P.; Stevens, M.F. Lidocaine induces apoptosis via the mitochondrial pathway independently of death receptor signaling. Anesthesiology 2007, 107, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.E.; Uhl, C.B.; Spittler, K.H.; Wang, H.; Gores, G.J. Mitochondrial injury and caspase activation by the local anesthetic lidocaine. Anesthesiology 2004, 101, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Onizuka, S.; Tamura, R.; Hosokawa, N.; Kawasaki, Y.; Tsuneyoshi, I. Local anesthetics depolarize mitochondrial membrane potential by intracellular alkalization in rat dorsal root ganglion neurons. Anesth. Analg. 2010, 111, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Onizuka, S.; Yonaha, T.; Tamura, R.; Kasiwada, M.; Shirasaka, T.; Tsuneyoshi, I. Lidocaine depolarizes the mitochondrial membrane potential by intracellular alkalization in rat dorsal root ganglion neurons. J. Anesth. 2011, 25, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Liu, C.L.; Chen, M.J.; Hsu, Y.W.; Chen, S.N.; Lin, C.H.; Chen, C.M.; Yang, F.M.; Hu, M.C. Local anesthetics induce apoptosis in human breast tumor cells. Anesth. Analg. 2014, 118, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Yagiela, J.A.; Benoit, P.W.; Fort, N.F. Mechanism of epinephrine enhancement of lidocaine-induced skeletal muscle necrosis. J. Dent. Res. 1982, 61, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, V.S.; Marte, E.; Brown, B.W.; van Bergen, F.H. Lidocaine, 2-chlorprocaine and hepatic necrosis. Anesth. Analg. 1966, 45, 55–58. [Google Scholar] [PubMed]
- Onizuka, S.; Tamura, R.; Yonaha, T.; Oda, N.; Kawasaki, Y.; Shirasaka, T.; Shiraishi, S.; Tsuneyoshi, I. Clinical dose of lidocaine destroys the cell membrane and induces both necrosis and apoptosis in an identified Lymnaea neuron. J. Anesth. 2012, 26, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Arrebola, F.; Zabiti, S.; Canizares, F.J.; Cubero, M.A.; Crespo, P.V.; Fernandez-Segura, E. Changes in intracellular sodium, chlorine, and potassium concentrations in staurosporine-induced apoptosis. J. Cell Physiol. 2005, 204, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Belaud-Rotureau, M.A.; Leducq, N.; Macouillard, P.D.G.F.; Diolez, P.; Lacoste, L.; Lacombe, F.; Bernard, P.; Belloc, F. Early transitory rise in intracellular pH leads to Bax conformation change during ceramide-induced apoptosis. Apoptosis 2000, 5, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Karpie, J.C.; Chu, C.R. Lidocaine exhibits dose- and time-dependent cytotoxic effects on bovine articular chondrocytes in vitro. Am. J. Sports Med. 2007, 35, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, A.W.; Kellen, J.; Tam, Y.K.; Wyse, D.G. Character of adverse effects of prophylactic lidocaine in the coronary care unit. Clin. Pharmacol. Ther. 1986, 40, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, J.; Bartoszek, A. DNA methylation in cancer development, diagnosis and therapy—Multiple opportunities for genotoxic agents to act as methylome disruptors or remediators. Mutagenesis 2011, 26, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, S.; Schotta, G.; Lachner, M.; Sengupta, R.; Kohlmaier, A.; Perez-Burgos, L.; Linderson, Y.; Martens, J.H.; OʼSullivan, R.J.; Fodor, B.D.; et al. The role of histone modifications in epigenetic transitions during normal and perturbed development. Ernst Scher. Res. Found. Workshop 2006, 57, 1–27. [Google Scholar]
- Ushijima, T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer 2005, 5, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Singal, R.; Ginder, G.D. DNA methylation. Blood 1999, 93, 4059–4070. [Google Scholar] [PubMed]
- Van Engeland, M.; Weijenberg, M.P.; Roemen, G.M.; Brink, M.; de Bruine, A.P.; Goldbohm, R.A.; van den Brandt, P.A.; Baylin, S.B.; de Goeij, A.F.; Herman, J.G. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: The Netherlands cohort study on diet and cancer. Cancer Res. 2003, 63, 3133–3137. [Google Scholar] [PubMed]
- Vaissiere, T.; Hung, R.J.; Zaridze, D.; Moukeria, A.; Cuenin, C.; Fasolo, V.; Ferro, G.; Paliwal, A.; Hainaut, P.; Brennan, P. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009, 69, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Villar-Garea, A.; Fraga, M.F.; Espada, J.; Esteller, M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003, 63, 4984–4989. [Google Scholar] [PubMed]
- Tada, M.; Imazeki, F.; Fukai, K.; Sakamoto, A.; Arai, M.; Mikata, R.; Tokuhisa, T.; Yokosuka, O. Procaine inhibits the proliferation and DNA methylation in human hepatoma cells. Hepatol. Int. 2007, 1, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Lirk, P.; Berger, R.; Hollmann, M.W.; Fiegl, H. Lidocaine time- and dose-dependently demethylates deoxyribonucleic acid in breast cancer cell lines in vitro. Br. J. Anaesth. 2012, 109, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.Y.; Kwon, K.; Lee, K.R.; Choi, Y.J.; Goo, T.W.; Yu, K.; Kim, S.W.; Kwon, O.Y. Lidocaine induces endoplasmic reticulum stress-associated apoptosis in vitro and in vivo. Int. J. Mol. Sci. 2011, 12, 7652–7661. [Google Scholar] [CrossRef] [PubMed]
- Sirchia, S.M.; Ren, M.; Pili, R.; Sironi, E.; Somenzi, G.; Ghidoni, R.; Toma, S.; Nicolò, G.; Sacchi, N. Endogenous reactivation of the RARbeta2 tumor suppressor gene epigenetically silenced in breast cancer. Cancer Res. 2002, 62, 2455–2461. [Google Scholar] [PubMed]
- Burbee, D.G.; Forgacs, E.; Zochbauer-Muller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 2011, 93, 691–699. [Google Scholar] [CrossRef]
- Agathanggelou, A.; Honorio, S.; Macartney, D.P.; Martinez, A.; Dallol, A.; Rader, J.; Fullwood, P.; Chauhan, A.; Walker, R.; Shaw, J.A.; et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumours. Oncogene 2001, 20, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Si, S.P.; Lee, X.; Tsou, H.C.; Buchsbaum, R.; Tibaduiza, E.; Peacocke, M. RAR beta 2-mediated growth inhibition in HeLa cells. Exp. Cell Res. 1996, 223, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Swisshelm, K.; Ryan, K.; Lee, X.; Tsou, H.C.; Peacocke, M.; Sager, R. Down-regulation of retinoic acid receptor beta in mammary carcinoma cell lines and its up-regulation in senescing normal mammary epithelial cells. Cell Growth Differ. 1994, 5, 133–141. [Google Scholar] [PubMed]
- Liu, Y.; Lee, M.O.; Wang, H.G.; Li, Y.; Hashimoto, Y.; Klaus, M.; Reed, J.C.; Zhang, X. Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol. Cell. Biol. 1996, 16, 1138–1149. [Google Scholar] [PubMed]
- Rabizadeh, S.; Xavier, R.J.; Ishiguro, K.; Bernabeortiz, J.; Lopez-Ilasaca, M.; Khokhlatchev, A.; Mollahan, P.; Pfeifer, G.P.; Avruch, J.; Seed, B. The scaffold protein CNK1 interacts with the tumor suppressor RASSF1A and augments RASSF1A-induced cell death. J. Biol. Chem. 2004, 279, 29247–29254. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Vega, S.; Khokhlatchev, A.; Nedwidek, M.; Zhang, X.F.; Dammann, R.; Pfeifer, G.P.; Avruch, J. The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene 2002, 21, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Song, S.J.; Kim, S.Y.; Oh, H.J.; Lim, D.S. The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 2008, 27, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Vo, A.; McKeehan, W.L. Specificity of the methylation-suppressed A isoform of candidate tumor suppressor RASSF1 for microtubule hyperstabilization is determined by cell death inducer C19ORF5. Cancer Res. 2005, 65, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Vassilomanolakis, M.; Koumakis, G.; Barbounis, V.; Demiri, M.; Pateras, H.; Efremidis, A.P. Vinorelbine and cisplatin in metastatic breast cancer patients previously treated with anthracyclines. Ann. Oncol. 2000, 11, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Nole, F.; Munzone, E.; Mandala, M.; Catania, C.; Orlando, L.; Zampino, M.G.; Minchella, I.; Colleoni, M.; Peruzzotti, G.; Marrocco, E.; et al. Vinorelbine, cisplatin and continuous infusion of 5-fluorouracil (ViFuP) in metastatic breast cancer patients: A phase II study. Ann. Oncol. 2001, 12, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Dixit, M.; Yang, J.L.; Poirier, M.C.; Price, J.O.; Andrews, P.A.; Arteaga, C.L. Abrogation of cisplatin-induced programmed cell death in human breast cancer cells by epidermal growth factor antisense RNA. J. Natl. Cancer Inst. 1997, 89, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, C.; Sweeney, E.A.; Shirahama, T.; Igarashi, Y.; Hakomori, S.; Tsujimoto, H.; Imanishi, T.; Ogaki, M.; Ohyama, T.; Yamazaki, J.; et al. Overexpression of bax sensitizes breast cancer MCF-7 cells to cisplatin and etoposide. Surg. Today 1997, 27, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Blanc, C.; Deveraux, Q.L.; Krajewski, S.; Janicke, R.U.; Porter, A.G.; Reed, J.C.; Jaggi, R.; Marti, A. Caspase-3 is essential for procaspase-9 processing and cisplatin-induced apoptosis of MCF-7 breast cancer cells. Cancer Res. 2000, 60, 4386–4390. [Google Scholar] [PubMed]
- Thomadaki, H.; Scorilas, A. Breast cancer cells response to the antineoplastic agents cisplatin, carboplatin, and doxorubicin at the mRNA expression levels of distinct apoptosis-related genes, including the new member, BCL2L12. Ann. N. Y. Acad. Sci. 2007, 1095, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yuan, F.; Li, P.; Chen, Z.; Chen, A.; Li, S.; Hu, C. Interferon regulatory factor 4 binding protein is a novel p53 target gene and suppresses cisplatin-induced apoptosis of breast cancer cells. Mol. Cancer 2012, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Shiu, L.Y.; Chang, L.C.; Liang, C.H.; Huang, Y.S.; Sheu, H.M.; Kuo, K.W. Solamargine induces apoptosis and sensitizes breast cancer cells to cisplatin. Food Chem. Toxicol. 2007, 45, 2155–2164. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.G.; Peek, R.J. Gastric cancer prevention by demethylation. Cancer Prev. Res. 2013, 6, 253–256. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Saini, S.; Shahryari, V.; Arora, S.; Zaman, M.S.; Chang, I.; Yamamura, S.; Tanaka, Y.; Chiyomaru, T. miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin. Cancer Res. 2013, 19, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Kim, J.H.; Lee, Y.S.; Lee, Y.C. Change in gene expression profiles of secreted frizzled-related proteins (SFRPs) by sodium butyrate in gastric cancers: Induction of promoter demethylation and histone modification causing inhibition of Wnt signaling. Int. J. Oncol. 2012, 40, 1533–1542. [Google Scholar] [PubMed]
- Liu, J.; Xie, Y.S.; Wang, F.L.; Zhang, L.J.; Zhang, Y.; Luo, H.S. Cytotoxicity of 5-aza-2'-deoxycytidine against gastric cancer involves DNA damage in an ATM-P53 dependent signaling pathway and demethylation of P16(INK4A). Biomed. Pharmacother. 2013, 67, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Treas, J.; Tyagi, T.; Gao, W. DNA demethylation by 5-aza-2-deoxycytidine treatment abrogates 17 beta-estradiol-induced cell growth and restores expression of DNA repair genes in human breast cancer cells. Cancer Lett. 2012, 316, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Seewaldt, V.L.; Johnson, B.S.; Parker, M.B.; Collins, S.J.; Swisshelm, K. Expression of retinoic acid receptor beta mediates retinoic acid-induced growth arrest and apoptosis in breast cancer cells. Cell Growth Differ. 1995, 6, 1077–1088. [Google Scholar] [PubMed]
- Levesley, J.; Lusher, M.E.; Lindsey, J.C.; Clifford, S.C.; Grundy, R.; Coyle, B. RASSF1A and the BH3-only mimetic ABT-737 promote apoptosis in pediatric medulloblastoma cell lines. Neuro Oncol. 2011, 13, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Yang, J.; Chen, X.; Li, J.; Li, X.; Wang, L.; Tan, Y.; Xiong, W.; Zhou, M.; McCarthy, J.B.; et al. RASSF1A suppresses melanoma development by modulating apoptosis and cell-cycle progression. J. Cell Physiol. 2011, 226, 2360–2369. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; OʼNeill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Yang, G.; Pfeifer, G.P. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res. 2011, 61, 3105–3109. [Google Scholar]
- Niknafs, B. Induction of apoptosis and non-apoptosis in human breast cancer cell line (MCF-7) by cisplatin and caffeine. Iran. Biomed. J. 2011, 15, 130–133. [Google Scholar] [PubMed]
- Mir, R.; Tortosa, A.; Martinez-Soler, F.; Vidal, A.; Condom, E.; Perez-Perarnau, A.; Ruiz-Larroya, T.; Gil, J.; Giménez-Bonafé, P. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. Int. J. Cancer 2013, 132, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Barkinge, J.; Hawkins, S.; Gudi, R.; Salgia, R.; Kanteti, P.V. Expression of Siva-1 protein or its putative amphipathic helical region enhances cisplatin-induced apoptosis in breast cancer cells: Effect of elevated levels of BCL-2. Cancer Res. 2005, 65, 5301–5309. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Hosokawa, K.; Dantes, A.; Tajima, K.; Kotsuji, F.; Amsterdam, A. Theophylline and cisplatin synergize in down regulation of BCL-2 induction of apoptosis in human granulosa cells transformed by a mutated p53 (p53 val135) and Ha-ras oncogene. Int. J. Oncol. 2000, 17, 227–235. [Google Scholar] [PubMed]
- Cho, N.Y.; Kim, B.H.; Choi, M.; Yoo, E.J.; Moon, K.C.; Cho, Y.M.; Kim, D.; Kang, G.H. Hypermethylation of CpG island loci and hypomethylation of LINE-1 and Alu repeats in prostate adenocarcinoma and their relationship to clinicopathological features. J. Pathol. 2007, 211, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed]
- Farias, E.F; Arapshian, A.; Bleiweiss, I.J.; Waxman, S.; Zelent, A.; Mira-Y-Lopez, R. Retinoic acid receptor alpha2 is a growth suppressor epigenetically silenced in MCF-7 human breast cancer cells. Cell Growth Differ. 2002, 13, 335–341. [Google Scholar] [PubMed]
- Yan, P.S.; Shi, H.; Rahmatpanah, F.; Hsiau, T.H.; Hsiau, A.H.; Leu, Y.W.; Liu, J.C.; Huang, T.H. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res. 2003, 63, 6178–6186. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Yang, J.; Han, X. Lidocaine Sensitizes the Cytotoxicity of Cisplatin in Breast Cancer Cells via Up-Regulation of RARβ2 and RASSF1A Demethylation. Int. J. Mol. Sci. 2014, 15, 23519-23536. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151223519
Li K, Yang J, Han X. Lidocaine Sensitizes the Cytotoxicity of Cisplatin in Breast Cancer Cells via Up-Regulation of RARβ2 and RASSF1A Demethylation. International Journal of Molecular Sciences. 2014; 15(12):23519-23536. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151223519
Chicago/Turabian StyleLi, Kehan, Jianxue Yang, and Xuechang Han. 2014. "Lidocaine Sensitizes the Cytotoxicity of Cisplatin in Breast Cancer Cells via Up-Regulation of RARβ2 and RASSF1A Demethylation" International Journal of Molecular Sciences 15, no. 12: 23519-23536. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms151223519