
MicroRNA-16 Modulates HuR Regulation of Cyclin E1 in Breast Cancer Cells

Abstract
:
1. Introduction
2. Results and Discussion
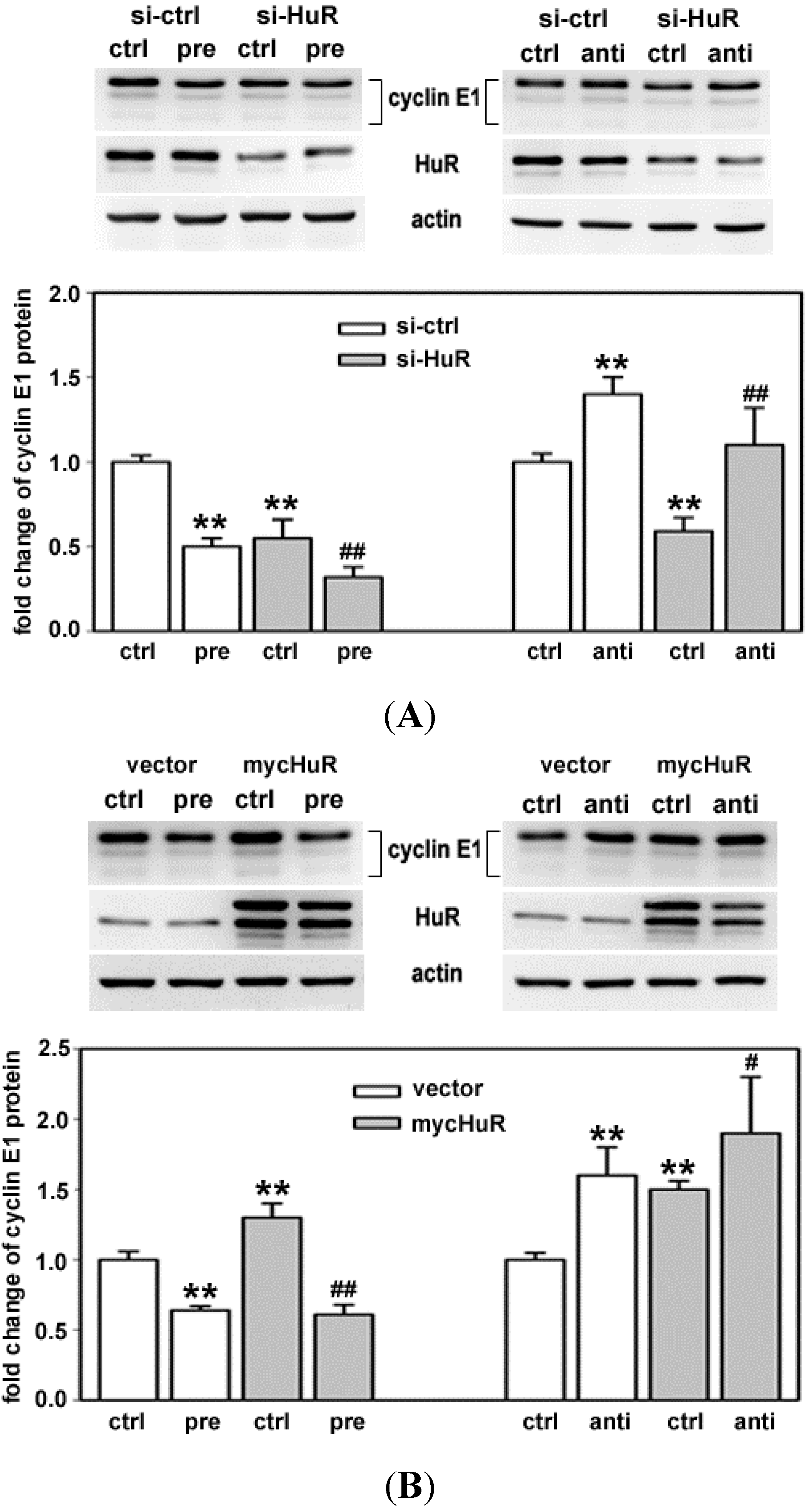
2.1. Both HuR and miR-16 Regulate Cyclin E1 in Breast Cancer Cells




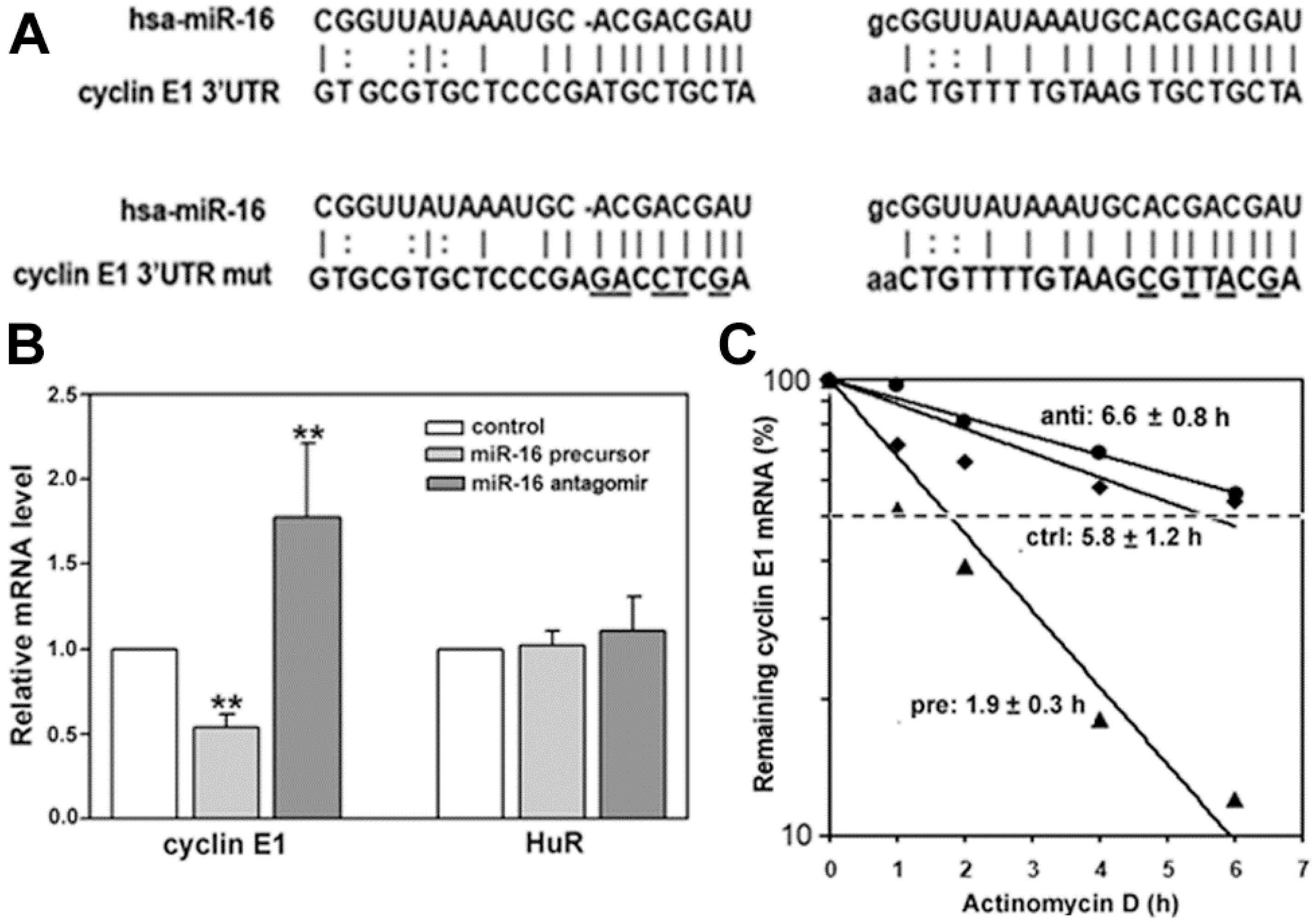
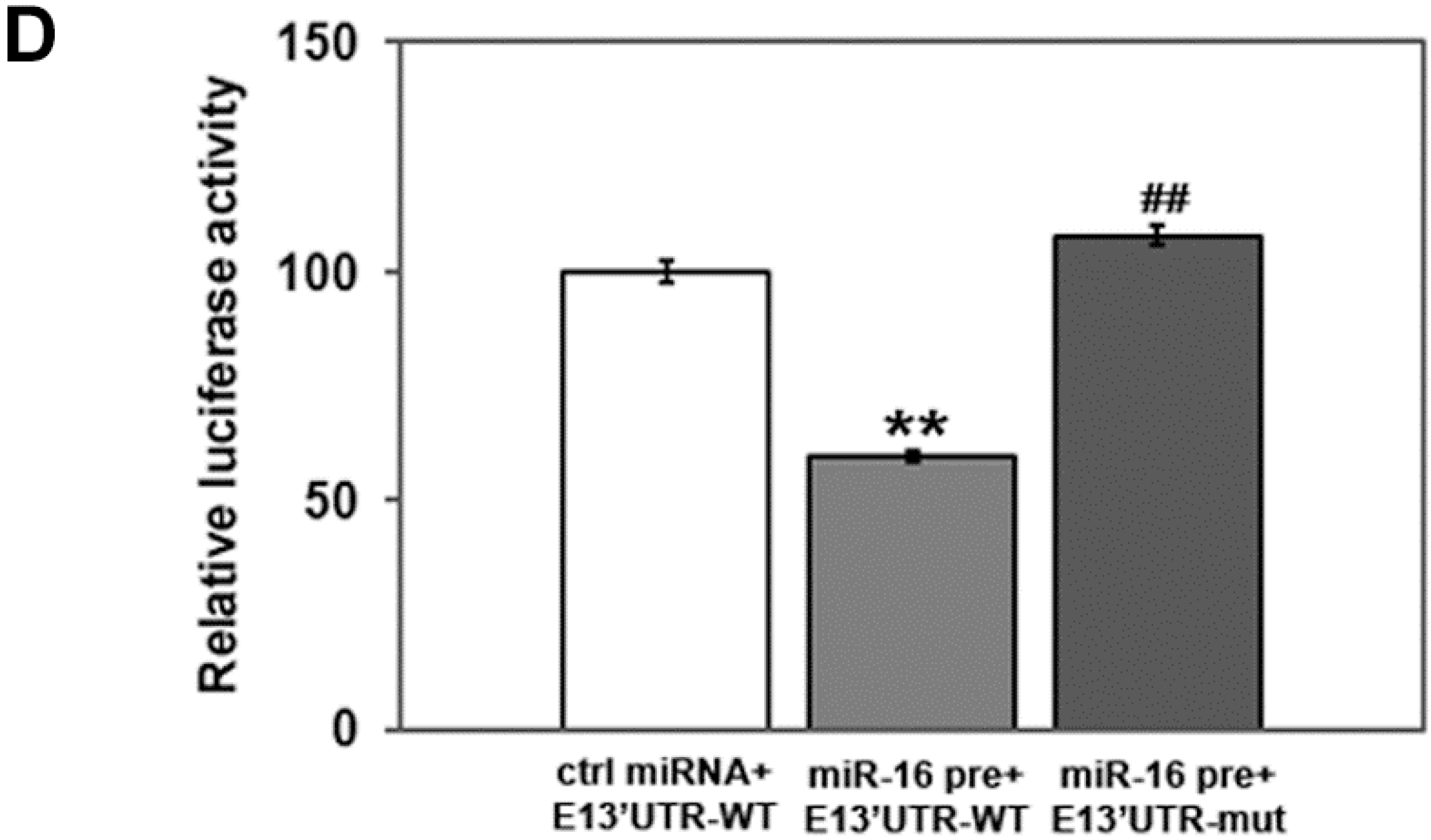
2.2. miR-16 Represses Cyclin E1 Dependent on Cognate Binding Sites within the 3'UTR of Its mRNA
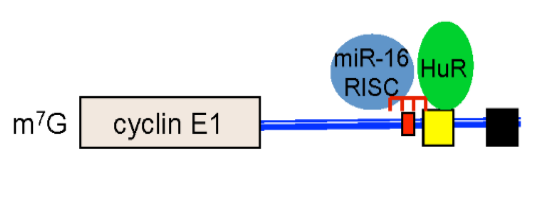
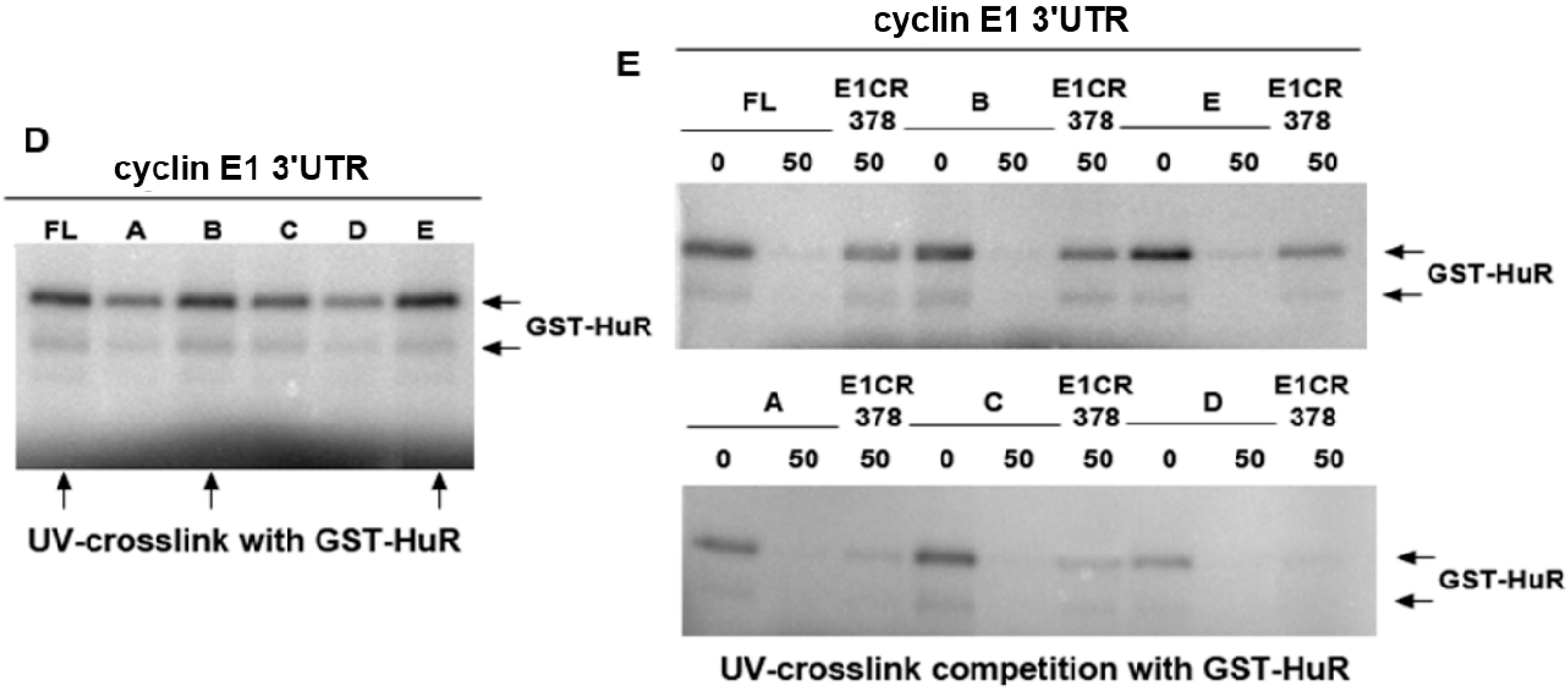
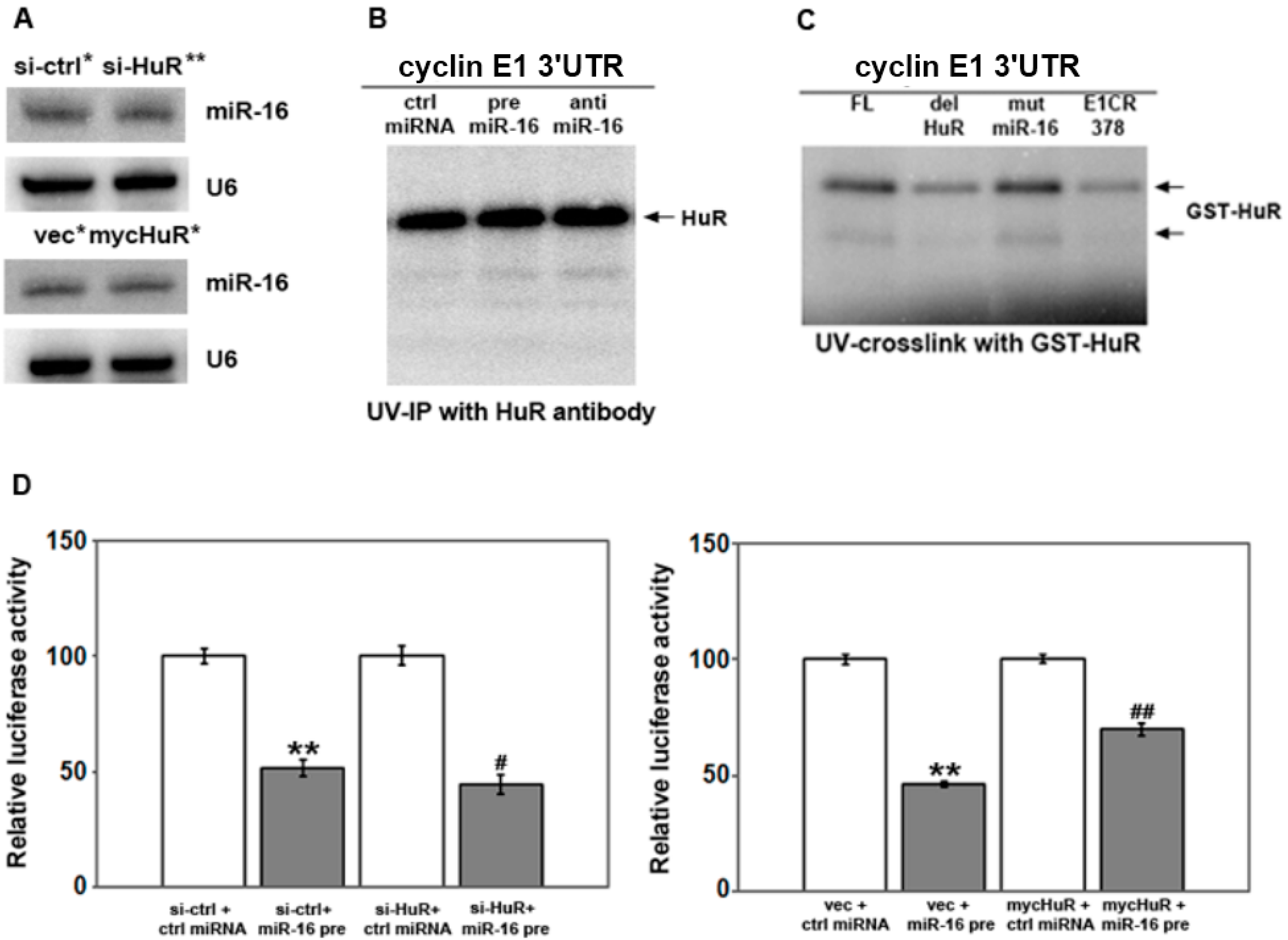
2.3. HuR Does not Affect miR-16 Expression nor Do HuR and miR-16 Affect the Other’s Binding



2.4. HuR Partially Blocks miR-16 Repression of a Reporter mRNA Containing the Cyclin E1 3'UTR
2.5. HuR Does not Block miR-16 Repression of Endogenous Cyclin E1 mRNA

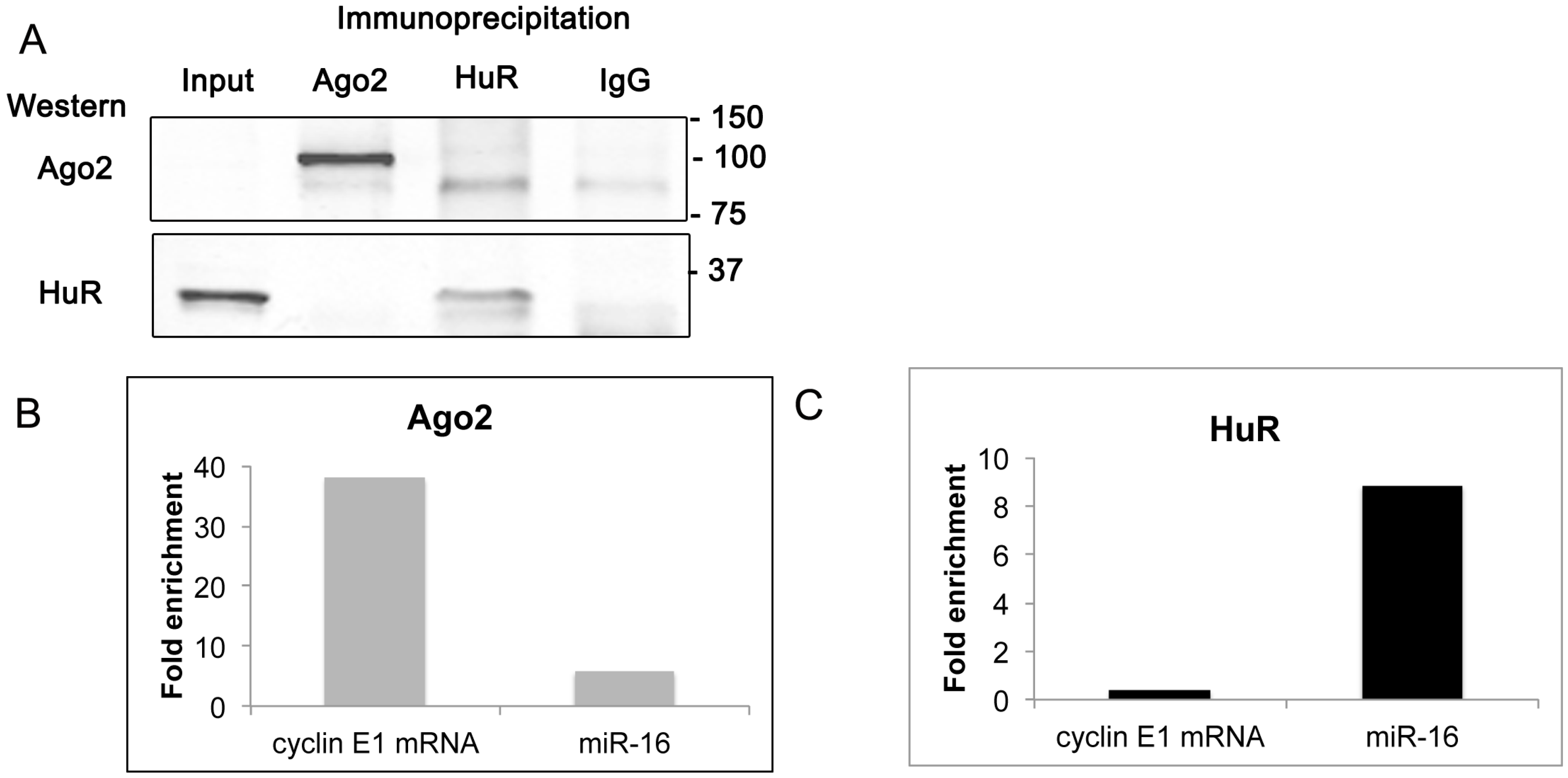
2.6. Cyclin E1 mRNA Associates with Ago2

2.7. Discussion
3. Experimental Section
3.1. Cell Culture and Transfections
3.2. Constructs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Primers (from 5' to 3') |
|---|---|
| A (1–157) | Forward: GCAGTCTAGACCACCCCATCCTTCTCCACCA |
| Reverse: CGACGATATCTGCCCTGTTTGATGCCATCCACA | |
| B (135–291) | Forward: GCAGTCTAGATGTGGATGGCATCAAACAGGGCA |
| Reverse: GCAGGATATCTGTTGTGGGAGTCCCTTAGGTCAA | |
| C (229–385) | Forward: GCAGTCTAGAACCAGTGCGTGCTCCCGATG |
| Reverse: CACCGATATCCCCGCAACCACCTGCTCCAC | |
| D (323–445) | Forward: GCAGTCTAGAGGCGTGGCTCTCCTCGCAG |
| Reverse: GAGACGGATATCCTGATAATGTGGAGAGGGCAGCCC | |
| E (388–534) | Forward: GCAGTCTAGAAGCGTTGTGCAGAGCCCATAGC |
| Reverse: GGACGATATCGTCTCAAAAACAGTATTATC | |
| miR-16 site 1 | Forward: ACACCAGTGCGTGCTCCCGAGACCTCGATGGAAGGTGCTACTTGACC |
| Reverse: GGTCAAGTAGCACCTTCCATCGAGGTCTCGGGAGCACGCACTGGTGT | |
| miR-16 site 2 | Forward: GTGTACAATGCCTTTGATGAACTGTTTTGTAAGCGTTACGATATCTATCCATTTTTTAATAAAGATAATACTG |
| Reverse: CAGTATTATCTTTATTAAAAAATGGATAGATATCGTAACGCTTACAAAACAGTTCATCAAAGGCATTGTACAC | |
| HuR site 1 | Forward: GATGGCATCAAACAGGGCAAAGTGGGTCAAGTAC |
| Reverse: GTACTTGACCCACTTTGCCCTGTTTGATGCCATC | |
| HuR site 2 | Forward: GTGCTGCTATATCTATCCAAATAAAGATAATACTG |
| Reverse: CAGTATTATCTTTATTTGGATAGATATAGCAGCAC | |
| U6 | Forward: CGCAAGGATGACACGCAAATTC |
| Reverse: QuantiMir Universal Reverse primer | |
| miR-16 | Forward: TAGCAGCACGTAAATATTGGCG |
| Reverse: QuantiMir Universal Reverse primer | |
| Cyclin E1 mRNA | Forward: CGGCTCGCTCCAGGAA |
| Reverse: TCATCTGGATCCTGCAAAAAAA | |
| GAPDH mRNA | Forward: GGCCTCCAAGGAGTAAGACC |
| Reverse: AGGGGTCTACATGGAAACTG |
3.3. Northern Blotting and qRT-PCR
3.4. Western Blotting
3.5. Reporter Gene Assay
3.6. UV Cross-Link Competition Assays and Immunoprecipitation
3.7. Ribonucleoprotein Immunoprecipitation
3.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clurman, B.E.; Sheaff, R.J.; Thress, K.; Groudine, M.; Roberts, J.M. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996, 10, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Zack, T.I.; Morris, L.G.; Lin, K.; Hukkelhoven, E.; Raheja, R.; Tan, I.L.; Turcan, S.; Veeriah, S.; Meng, S.; et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 2014, 46, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Moberg, K.H.; Bell, D.W.; Wahrer, D.C.; Haber, D.A.; Hariharan, I.K. Archipelago regulates cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 2001, 413, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Strohmaier, H.; Spruck, C.H.; Kaiser, P.; Won, K.A.; Sangfelt, O.; Reed, S.I. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Lee, Y.M.; Welcker, M.; Swanger, J.; Zagozdzon, A.; Winer, J.D.; Roberts, J.M.; Kaldis, P.; Clurman, B.E.; Sicinski, P. Kinase-independent function of cyclin E. Mol. Cell 2007, 25, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Yu, Q.; Sicinska, E.; Das, M.; Schneider, J.E.; Bhattacharya, S.; Rideout, W.M.; Bronson, R.T.; Gardner, H.; Sicinski, P. Cyclin E ablation in the mouse. Cell 2003, 114, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Parisi, T.; Beck, A.R.; Rougier, N.; McNeil, T.; Lucian, L.; Werb, Z.; Amati, B. Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J. 2003, 22, 4794–4803. [Google Scholar] [CrossRef] [PubMed]
- Geisen, C.; Moroy, T. The oncogenic activity of cyclin E is not confined to cdk2 activation alone but relies on several other, distinct functions of the protein. J. Biol. Chem. 2002, 277, 39909–39918. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, K.J.; Swarbrick, A.; Sutherland, R.L.; Musgrove, E.A. Lack of relationship between CDK activity and G1 cyclin expressionin breast cancer cells. Oncogene 1998, 16, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Herzinger, T.; Hansen, K.; Moroni, M.C.; Resnitzky, D.; Helin, K.; Reed, S.I.; Bartek, J. Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev. 1997, 11, 1479–1492. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Resnitzky, D.; Gossen, M.; Bujard, H.; Reed, S.I. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol. Cell. Biol. 1994, 14, 1669–1679. [Google Scholar] [PubMed]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Bortner, D.M.; Rosenberg, M.P. Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol. Cell. Biol. 1997, 17, 453–459. [Google Scholar] [PubMed]
- Liang, Y.; Gao, H.; Lin, S.-Y.; Goss, J.A.; Brunicardi, F.C.; Li, K. siRNA-based targeting of cyclin E overexpression inhibits breast cancer cell growth and suppresses tumor development in breast cancer mouse model. PLoS ONE 2010, 5, e12860. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.K.; Keyomarsi, K. Cyclin E as a prognostic and predictive marker in breast cancer. Semin. Cancer Biol. 2005, 15, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Beltran, A.; MacLennan, G.T.; Montironi, R. Cyclin E as molecular marker in the management of breast cancer: A review. Anal. Quant. Cytol. Histol. 2006, 28, 111–114. [Google Scholar] [PubMed]
- Porter, D.C.; Keyomarsi, K. Novel splice variants of cyclin E with altered substrate specificity. Nucleic Acids Res. 2000, 28, e101. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Ouriaghli, F.E.; Durbecq, V.; Soree, A.; Colozza, M.A.; Azambuja, E.; Paesmans, M.; Larsimont, D.; Buyse, M.; Harris, A.; et al. Impact of cyclins E, neutrophil elastase and proteinase 3 expression levels on clinical outcome in primary breast cancer patients. Int. J. Cancer 2006, 119, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Sieuwerts, A.M.; Look, M.P.; Meijer-van Gelder, M.E.; Timmermans, M.; Trapman, A.M.; Garcia, R.R.; Arnold, M.; Goedheer, A.J.; de Weerd, V.; Portengen, H.; et al. Which cyclin E prevails as prognostic marker for breast cancer? Results from a retrospective study involving 635 lymph node—Negative breast cancer patients. Clin. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Kreike, B.; Hart, G.; Bartelink, H.; van de Vijver, M.J. Analysis of breast cancer related gene expression using natural splines and the Cox proportional hazard model to identify prognostic associations. Breast Cancer Res. Treat. 2010, 122, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Keyomarsi, K.; Pardee, A.B. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc. Natl. Acad. Sci. USA 1993, 90, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Weichert, W.; Winzer, K.J.; Muller, B.M.; Noske, A.; Niesporek, S.; Kristiansen, G.; Guski, H.; Dietel, M.; Hauptmann, S. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin. Cancer Res. 2004, 10, 5580–5586. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Fagerholm, R.; Aaltonen, K.; Kilpivaara, O.; Aittomaki, K.; Blomqvist, C.; Heikkila, P.; Haglund, C.; Nevanlinna, H.; Ristimaki, A. Prognostic role of HuR in hereditary breast cancer. Clin. Cancer Res. 2007, 13, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Wang, B.; Wu, Y. Predictive and prognostic significance of cytoplasmic expression of ELAV-like protein HuR in invasive breast cancer treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2013, 141, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, B.; Bi, J.; Zhang, C.; Guo, Y.; Chu, H.; Liang, X.; Zhong, C.; Wang, J. Cytoplasmic HuR expression correlates with P-gp, HER-2 positivity, and poor outcome in breast cancer. Tumour Biol. 2013, 34, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Hartley, R.S. HuR contributes to cyclin E1 deregulation in MCF-7 breast cancer cells. Cancer Res. 2006, 66, 7948–7956. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fu, H.; Sun, F.; Zhang, H.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008, 36, 5391–5404. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fu, X.D.; Zhou, Y.; Zhang, Y. Down-regulation of the cyclin E1 oncogene expression by microRNA-16-1 induces cell cycle arrest in human cancer cells. BMB Rep. 2009, 42, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Venturutti, L.; Huang, Y.W.; Schillaci, R.; Huang, T.H.; Elizalde, P.V. Downregulation of the tumor-suppressor miR-16 via progestin-mediated oncogenic signaling contributes to breast cancer development. Breast Cancer Res. 2012, 14, R77. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhausser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M., Jr.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, X.; Lei, Y.; Liu, X.; Liu, Z.; Tong, T.; Wang, W. Loss of repression of HuR translation by miR-16 may be responsible for the elevation of HuR in human breast carcinoma. J. Cell Biochem. 2010, 111, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Srikantan, S.; Tominaga, K.; Gorospe, M. Functional interplay between RNA-binding protein HuR and microRNAs. Curr. Protein Pept. Sci. 2012, 13, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012, 10, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wu, Y.; Hartley, R.S. MicroRNA-125a represses cell growth by targeting HuR in breast cancer. RNA Biol. 2009, 6, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Epis, M.R.; Barker, A.; Giles, K.M.; Beveridge, D.J.; Leedman, P.J. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J. Biol. Chem. 2011, 286, 41442–41454. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Srikantan, S.; Lee, E.K.; Subaran, S.S.; Martindale, J.L.; Abdelmohsen, K.; Gorospe, M. Competitive regulation of nucleolin expression by HuR and miR-494. Mol. Cell. Biol. 2011, 31, 4219–4231. [Google Scholar] [CrossRef] [PubMed]
- Glorian, V.; Maillot, G.; Poles, S.; Iacovoni, J.S.; Favre, G.; Vagner, S. HuR-dependent loading of miRNA RISC to the mRNA encoding the Ras-related small GTPase RhoB controls its translation during UV-induced apoptosis. Cell Death Differ. 2011, 18, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Yi, J.; Guo, G.; Liu, X.; Shang, Y.; Tong, T.; Cui, Q.; Zhan, M.; Gorospe, M.; Wang, W. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Mol. Cell. Biol. 2010, 30, 3875–3886. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, C.; Ahlin, C.; Holmberg, L.; Amini, R.M.; Fjallskog, M.L.; Blomqvist, C. Cyclin E1 is a strong prognostic marker for death from lymph node negative breast cancer. A population-based case-control study. Acta Oncol. 2014, 54, 1–7. [Google Scholar] [PubMed]
- Yuan, Z.; Sanders, A.J.; Ye, L.; Jiang, W.G. HuR, a key post-transcriptional regulator, and its implication in progression of breast cancer. Histol. Histopathol. 2010, 25, 1331–1340. [Google Scholar] [PubMed]
- Li, X.; Kazan, H.; Lipshitz, H.D.; Morris, Q.D. Finding the target sites of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2014, 5, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Quon, G.; Lipshitz, H.D.; Morris, Q. Predicting in vivo binding sites of RNA-binding proteins using mRNA secondary structure. RNA 2010, 16, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Dahm, G.M.; Gubin, M.M.; Magee, J.D.; Techasintana, P.; Calaluce, R.; Atasoy, U. Method for the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts using RIP-Chip. J. Vis. Exp. 2012. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.; Connick, M.C.; Vanderhoof, J.; Ishak, M.-A.; Hartley, R.S. MicroRNA-16 Modulates HuR Regulation of Cyclin E1 in Breast Cancer Cells. Int. J. Mol. Sci. 2015, 16, 7112-7132. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16047112
Guo X, Connick MC, Vanderhoof J, Ishak M-A, Hartley RS. MicroRNA-16 Modulates HuR Regulation of Cyclin E1 in Breast Cancer Cells. International Journal of Molecular Sciences. 2015; 16(4):7112-7132. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16047112
Chicago/Turabian StyleGuo, Xun, Melanie C. Connick, Jennifer Vanderhoof, Mohammad-Ali Ishak, and Rebecca S. Hartley. 2015. "MicroRNA-16 Modulates HuR Regulation of Cyclin E1 in Breast Cancer Cells" International Journal of Molecular Sciences 16, no. 4: 7112-7132. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms16047112