
Transcriptome-Wide Identification of miRNA Targets under Nitrogen Deficiency in Populus tomentosa Using Degradome Sequencing


Abstract
:1. Introduction
2. Results
2.1. Construction of the Degradome Library and Sequencing Analysis in P. tomentosa
2.2. Systematic Identification of miRNA Targets in P. tomentosa
2.3. Annotation and Validation of Targets for Known Conserved miRNAs in P. tomentosa
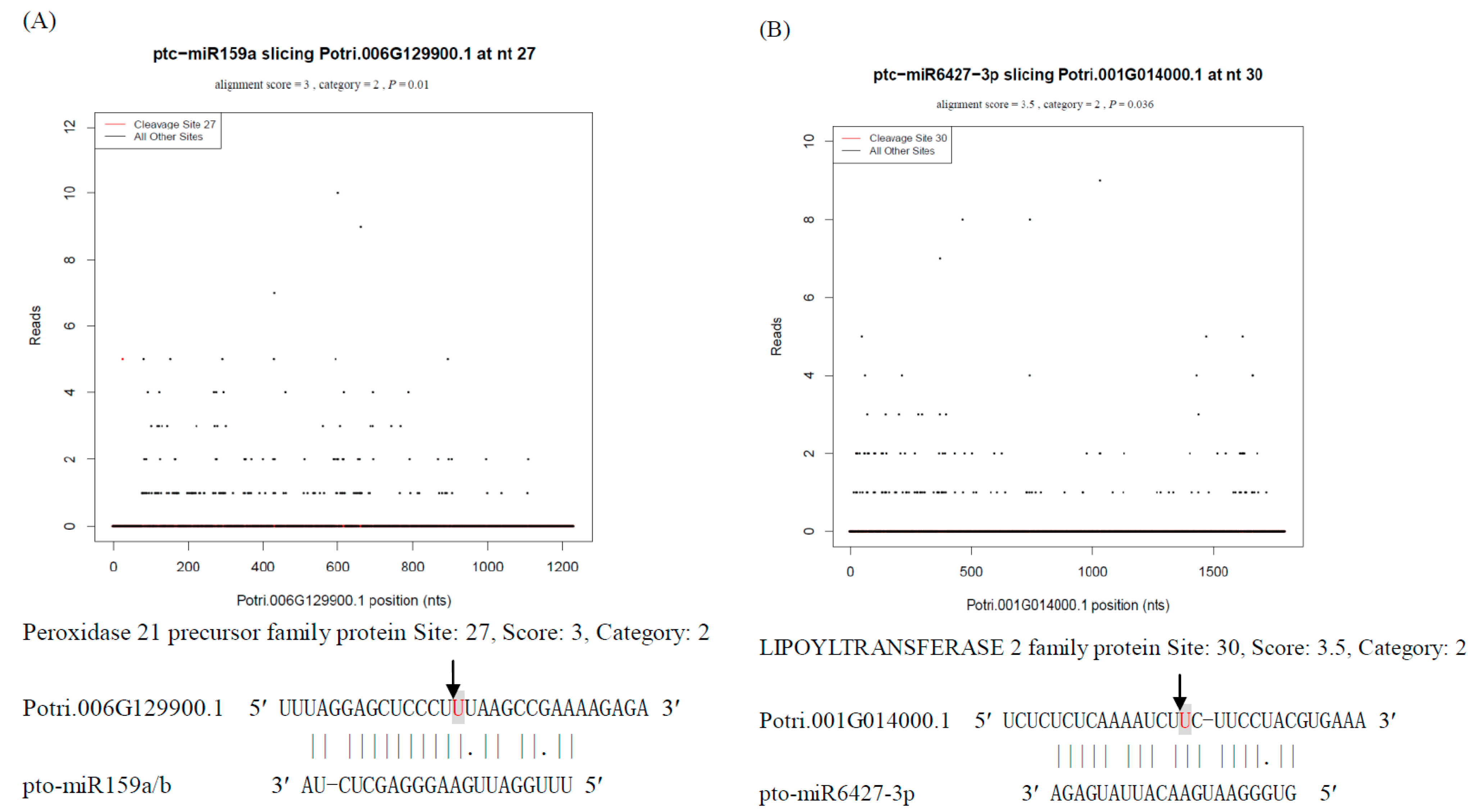
2.4. Annotation and Validation of Targets of Known Non-Conserved miRNAs in P. tomentosa
2.5. Annotation and Validation of the Targets of Novel miRNAs in P. tomentosa
2.6. Detection and Validation of Targets of N-Responsive mRNAs in P. tomentosa
2.7. Specificity and Diversity of Cleavage Sites


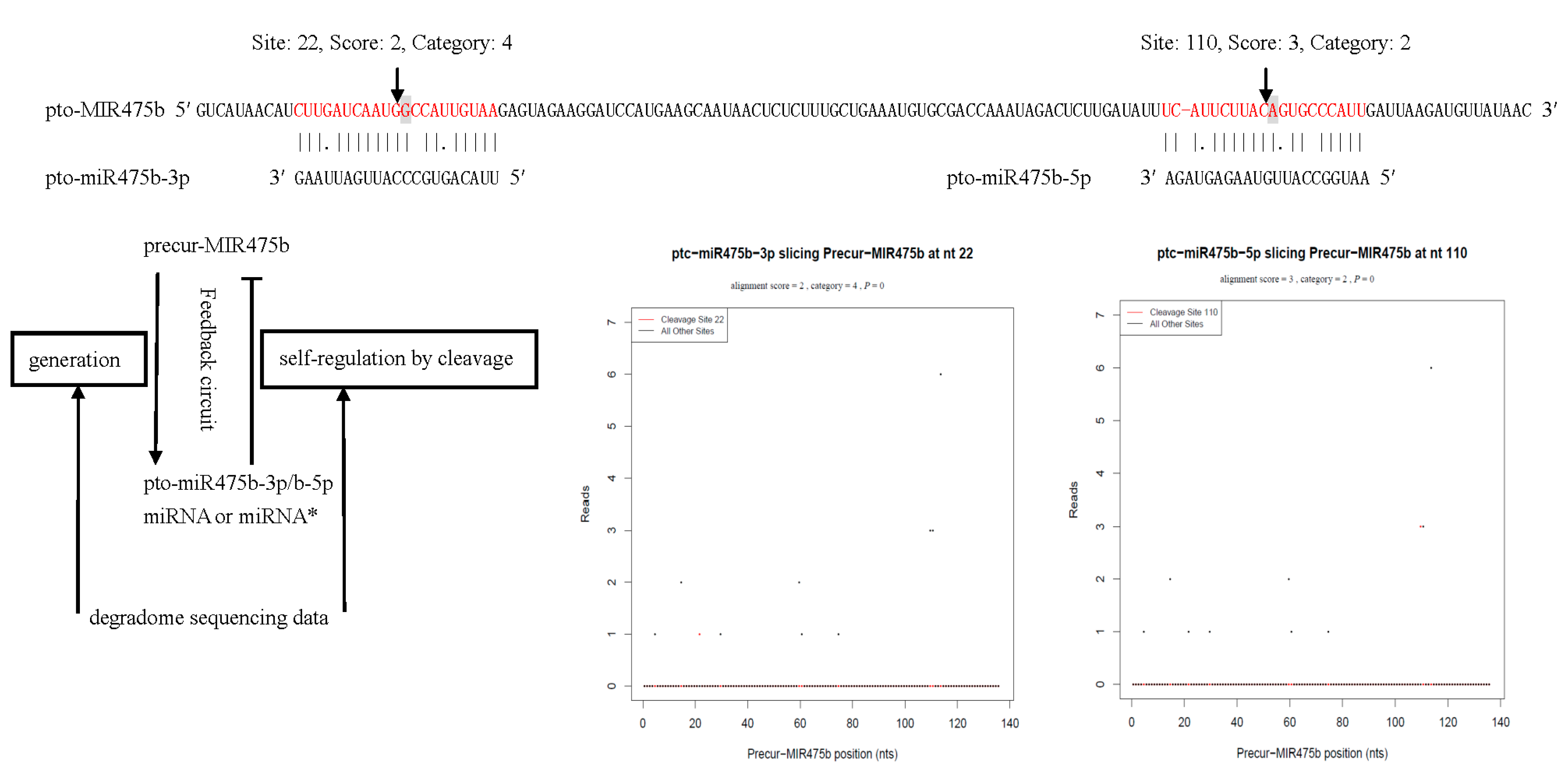
2.8. Identification of miRNA Precursor in P. tomentosa Using Degradome Sequencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-miRNA | MiRNA Sequence | Count | Letters | Cleavage Sites |
|---|---|---|---|---|
| pto-MIR156a | TGACAGAAGAGAGTGAGCAC | 10 | 20 | 11–30 |
| pto-MIR156b | TGACAGAAGAGAGTGAGCAC | 10 | 20 | 11–30 |
| pto-MIR156c | TGACAGAAGAGAGTGAGCAC | 10 | 20 | 11–30 |
| pto-MIR156d | TGACAGAAGAGAGTGAGCAC | 10 | 20 | 11–30 |
| pto-MIR156e | TGACAGAAGAGAGTGAGCAC | 10 | 20 | 11–30 |
| pto-MIR156f | TGACAGAAGAGAGTGAGCAC | 10 | 20 | 11–30 |
| pto-MIR156g | TTGACAGAAGATAGAGAGCAC | 84 | 21 | 11–31 |
| pto-MIR156h | TTGACAGAAGATAGAGAGCAC | 84 | 21 | 11–31 |
| pto-MIR156i | TTGACAGAAGATAGAGAGCAC | 84 | 21 | 11–31 |
| pto-MIR156j | TTGACAGAAGATAGAGAGCAC | 84 | 21 | 11–31 |
| pto-MIR156k | TGACAGAAGAGAGGGAGCA | 2 | 20 | 11–29 |
| pto-MIR159a | TTTGGATTGAAGGGAGCTCTA | 408 | 21 | 154–174 |
| pto-MIR159d | CTTGGATTGAAGGGAGCTCCT | 35 | 21 | 165–185 |
| pto-MIR162a | TCGATAAACCTCTGCATCCAG | 34 | 21 | 78–98 |
| pto-MIR167e | TGAAGCTGCCAGCATGATCTG | 2 | 21 | 11–31 |
| pto-MIR167h | TGAAGCTGCCAACATGATCTG | 2 | 21 | 11–31 |
| pto-MIR168a | TCGCTTGGTGCAGGTCGGGAA | 2 | 21 | 11–31 |
| pto-MIR168b | TCGCTTGGTGCAGGTCGGGAA | 2 | 21 | 11–31 |
| pto-MIR169o | AAGCCAAGGATGACTTGCCTG | 10 | 21 | 11–31 |
| pto-MIR171e | TGATTGAGCCGTGCCAATATC | 1 | 21 | 65–85 |
| pto-MIR171f | TGATTGAGCCGTGCCAATATC | 1 | 21 | 101–121 |
| pto-MIR171g | TGATTGAGCCGTGCCAATATC | 1 | 21 | 71–91 |
| pto-MIR171h | TGATTGAGCCGTGCCAATATC | 1 | 21 | 71–91 |
| pto-MIR171i | TGATTGAGCCGTGCCAATATC | 1 | 21 | 106–126 |
| pto-MIR172d | GGAATCTTGATGATGCTGCAT | 2 | 21 | 153–173 |
| pto-MIR172e | GGAATCTTGATGATGCTGCAT | 2 | 21 | 104–124 |
| pto-MIR319a | TTGGACTGAAGGGAGCTCCC | 11 | 20 | 160–179 |
| pto-MIR319b | TTGGACTGAAGGGAGCTCCC | 11 | 20 | 157–176 |
| pto-MIR319c | TTGGACTGAAGGGAGCTCCC | 11 | 20 | 168–187 |
| pto-MIR319d | TTGGACTGAAGGGAGCTCCC | 11 | 20 | 165–184 |
| pto-MIR394a | TTGGCATTCTGTCCACCTCC | 2 | 20 | 38–57 |
| pto-MIR394a | CTGTTGGTCTCTCTTTGTAA | 1 | 20 | 117–136 |
| pto-MIR394b | TTGGCATTCTGTCCACCTCC | 2 | 20 | 38–57 |
| pto-MIR394b | CTGTTGGTCTCTCTTTGTAA | 1 | 20 | 117–136 |
| pto-MIR396a | TTCCACAGCTTTCTTGAACTG | 33 | 21 | 11–31 |
| pto-MIR396b | TTCCACAGCTTTCTTGAACTG | 33 | 21 | 11–31 |
| pto-MIR396c | TTCCACAGCTTTCTTGAACTT | 9 | 21 | 11–31 |
| pto-MIR396d | TTCCACAGCTTTCTTGAACTT | 9 | 21 | 20–40 |
| pto-MIR396e | TTCCACAGCTTTCTTGAACTT | 9 | 21 | 11–31 |
| pto-MIR397a | TCATTGAGTGCAGCGTTGATG | 6 | 21 | 11–31 |
| pto-MIR408 | ATGCACTGCCTCTTCCCTGGC | 149 | 21 | 76–96 |
| pto-MIR475d | TTACAGAGTCCATTGATTAAG | 2 | 21 | 77–97 |
| pto-MIR482a | TCTTGCCTACTCCTCCCATT | 3 | 20 | 68–87 |
| pto-MIR1444a | TCCACATTCGGTCAATGTTC | 2 | 20 | 59–78 |
| pto-MIR6421 | TCCCTTACAATCTACTCTTTC | 1 | 21 | 18–38 |
| pto-MIR6457b | TTAGTTTGGCAGCCTCTTCTC | 8 | 21 | 151–171 |
| pto-MIR6460 | TGATATGTGGCATTCAATCGA | 1 | 21 | 93–113 |
2.9. Identification of Biologically Functional miRNA* Strands in P. tomentosa by Degradome Analysis
| MiRNA | MiRNA or miRNA* Sequence | Target Gene | C-Sites | Category | Raw Tags | Score |
|---|---|---|---|---|---|---|
| pto-miR169b-5p1 | CAGCCAAGGATGACTTGCCGA | Potri.007G127100.1 | 31 | 1 | 2 | 4.5 |
| pto-miR169b-3p | GGCAGGTTGTTCTTGGCTAC | Potri.007G085200.1 | 60 | 2 | 8 | 3.5 |
| pto-miR169b-3p | GGCAGGTTGTTCTTGGCTAC | Potri.010G155300.1 | 45 | 2 | 2 | 4.5 |
| pto-miR169b-3p | GGCAGGTTGTTCTTGGCTAC | Potri.014G142600.1 | 38 | 2 | 2 | 4 |
| pto-miR396e-5p1 | TTCCACAGCTTTCTTGAACTT | Precur-sR6a | 127 | 2 | 7 | 3.5 |
| pto-miR396e-3p2 | CTCAAGAAAGCTGTGGGAGA | Precur-MIR396a | 19 | 2 | 5 | 1.5 |
| pto-miR396e-3p2 | CTCAAGAAAGCTGTGGGAGA | Precur-MIR396b | 19 | 2 | 5 | 1.5 |
| pto-miR396e-3p2 | CTCAAGAAAGCTGTGGGAGA | Precur-MIR396e | 19 | 4 | 1 | 2 |
| pto-miR396e-3p2 | CTCAAGAAAGCTGTGGGAGA | Precur-sR3 | 22 | 2 | 5 | 1.5 |
| pto-miR396e-3p2 | CTCAAGAAAGCTGTGGGAGA | Precur-sR6a | 19 | 4 | 1 | 2 |
| pto-miR398c-5p | GGAGCGACCTGAAATCACATG | Precur-MIR398b | 65 | 1 | 2 | 3 |
| pto-miR398c-5p | GGAGCGACCTGAAATCACATG | Precur-MIR398c | 77 | 2 | 2 | 3 |
| pto-miR398c-3p | TGTGTTCTCAGGTCGCCCCTG | Precur-MIR398c | 22 | 0 | 49 | 3 |
| pto-miR475a-3p/b-3p1 | TTACAGTGCCCATTGATTAAG | Precur-MIR475a | 15 | 4 | 1 | 2 |
| pto-miR475a-3p/b-3p1 | TTACAGTGCCCATTGATTAAG | Precur-MIR475b | 22 | 4 | 1 | 2 |
| pto-miR475a-3p/b-3p1 | TTACAGTGCCCATTGATTAAG | Precur-MIR475d | 19 | 4 | 1 | 3 |
| pto-miR475a-5p/b-5p | AATGGCCATTGTAAGAGTAGA | Precur-MIR475b | 110 | 2 | 3 | 3 |


3. Discussion
3.1. N-Responsive miRNA-Mediated Targets


3.2. Analysis of miRNA Precursor Processing in P. tomentosa Using Degradome Sequencing
3.3. Identification of Biologically Functional miRNA* Strands in P. tomentosa Based on Degradome Analysis
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and Degradome Sequencing
4.3. Initial Processing and Analysis of Reads for the Degradome Library
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Data Archiving Statement
References
- Crawford, N.M.; Forde, B.G. Molecular and developmental biology of inorganic nitrogen nutrition. Arabidopsis Book 2002, 1, e0011. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Coschigano, K.T.; Oliveira, I.C.; Melo-Oliveira, R.; Coruzzi, G.M. The molecular-geneticsof nitrogen assimilation into amino acids in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 569–593. [Google Scholar] [CrossRef] [PubMed]
- Phua, Y.L.; Ho, J. MicroRNAs: In the pathogenesis of cystic kidney disease. Curr. Opin. Pediatr. 2015, 27, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Loss-Morais, G.; Ferreira, D.C.; Margis, R.; Alves-Ferreira, M.; Correa, R.L. Identification of novel and conserved microRNAs in Coffea canephora and Coffea arabica. Genet. Mol. Biol. 2014, 37, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Chiou, T.J. The role of microRNAs in sensing nutrient stress. Plant Cell Environ. 2007, 30, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, L.C.; Lin, S.I.; Shih, A.C.; Chen, J.W.; Lin, W.Y.; Tseng, C.Y.; Li, W.H.; Chiou, T.J. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009, 151, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rong, H.; Pilbeam, D. Signalling mechanisms underlying the morphological responses of the root system to nitrogen in Arabidopsis thaliana. J. Exp. Bot. 2007, 58, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Secco, D.; Jabnoune, M.; Walker, H.; Shou, H.; Wu, P.; Poirier, Y.; Whelan, J. Spatio-temporal transcript profiling of rice roots and shoots in response to phosphate starvation and recovery. Plant Cell 2013, 25, 4285–4304. [Google Scholar] [CrossRef] [PubMed]
- Nischal, L.; Mohsin, M.; Khan, I.; Kardam, H.; Wadhwa, A.; Abrol, Y.P.; Iqbal, M.; Ahmad, A. Identification and comparative analysis of microRNAs associated with low-N tolerance in rice genotypes. PLoS ONE 2012, 7, e50261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, H.; Shen, Y.; Gao, J.; Xiang, K.; Liu, L.; Ding, H.; Yuan, G.; Lan, H.; Zhou, S.; et al. Cloning and characterization of miRNAs from maize seedling roots under low phosphorus stress. Mol. Biol. Rep. 2012, 39, 8137–8146. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, S.; Nonis, A.; Begheldo, M.; Manoli, A.; Palme, K.; Caporale, G.; Ruperti, B.; Quaggiotti, S. Expression and tissue-specific localization of nitrate-responsive miRNAs in roots of maize seedlings. Plant Cell Environ. 2012, 35, 1137–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ding, H.; Zhu, J.K.; Zhang, F.; Li, W.X. Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol. 2011, 190, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Gifford, M.L.; Dean, A.; Gutierrez, R.A.; Coruzzi, G.M.; Birnbaum, K.D. Cell-specific nitrogen responses mediate developmental plasticity. Proc. Natl. Acad. Sci. USA 2008, 105, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Good, A.G.; Shrawat, A.K.; Muench, D.G. Can less yield more? Is reducing nutrient input into the environment compatible with maintaining crop production? Trends Plant. Sci. 2004, 9, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Rennenberg, H.; Wildhagen, H.; Ehlting, B. Nitrogen nutrition of poplar trees. Plant Biol. 2010, 12, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Ma, K.; Ci, D.; Zhang, Z.; Zhang, D. Biochemical, physiological and gene expression analysis reveals sex-specific differences in Populus tomentosa floral development. Physiol. Plant. 2014, 150, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Gong, C.; Pan, W.; Zhang, D. Development and application of microsatellites in candidate genes related to wood properties in the Chinese white poplar (Populus tomentosa Carr.). DNA Res. 2013, 20, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Sun, F.; Hou, J.; Chen, L.; Zhang, Y.; Kang, X.; Wang, Y. Differential profiling analysis of miRNAs reveals a regulatory role in low N stress response of Populus. Funct. Integr. Genomics 2015, 15, 93–105. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Pillay, M.; Jeong, D.H.; Hetawal, A.; Luo, S.; Janardhanan, P.; Kannan, V.; Rymarquis, L.A.; Nobuta, K.; German, R.; et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008, 26, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; He, Y.; Li, J.; Wang, X.; Chen, J. Genome-Wide characterization of Rice black streaked dwarfvirus-responsive microRNAs in rice leaves and roots by small RNA and degradome sequencing. Plant Cell Physiol. 2014. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Chen, D.; Wu, X.; Huang, D.; Chen, L.; Li, L.; Deng, X.; Xu, Q. Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange. BMC Genomics 2014, 15, 695. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Z.; Deng, M.; Liu, R.; Niu, S.; Fan, G. Identification and functional analysis of microRNAs and their targets in Platanus acerifolia under Lead (Pb) stress. Int. J. Mol. Sci. 2015, 16, 7098–7111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Chen, X.; Song, C.; Zou, Z.; Wang, Y.; Wang, M.; Fang, W.; Li, X. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014, 14, 271. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Zhang, L.; Li, W.W.; Hu, X.L.; Wang, M.B.; Fan, Y.L.; Zhang, C.Y.; Wang, L. Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Luan, M.; Xu, M.; Lu, Y.; Zhang, L.; Fan, Y.; Wang, L. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 2015, 555, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.R.; Chung, K.M.; Park, J.H.; Oh, S.A.; Ahn, T.; Hong, S.H.; Jang, S.K.; Nam, H.G. ORE9, an F-box protein that regulates leaf senescence in Arabidopsis. Plant Cell 2001, 13, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Sano, H. Transactivation of wound-responsive genes containing the core sequence of the auxin-responsive element by a wound-induced protein kinase-activated transcription factor in tobacco plants. Plant Mol. Biol. 2007, 65, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, J.X.; Terce-Laforgue, T.; Armengaud, P.; Clement, G.; Renou, J.P.; Pelletier, S.; Catterou, M.; Azzopardi, M.; Gibon, Y.; Lea, P.J.; et al. Characterization of a NADH-dependent glutamate dehydrogenase mutant of Arabidopsis demonstrates the key role of this enzyme in root carbon and nitrogen metabolism. Plant Cell 2012, 24, 4044–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, K.; Kanno, T.; Akita, M.; Ikejiri-Kanno, Y.; Wakasa, K.; Tozawa, Y. Identification of three shikimate kinase genes in rice characterization of their differential expression during panicle development and of the enzymatic activities of the encoded proteins. Planta 2005, 222, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.; Hauptmann, M.; Niessing, D.; Lloyd, C.W.; Schaffner, A.R. Helical growth of the Arabidopsis mutant tortifolia2 does not depend on cell division patterns but involves handed twisting of isolated cells. Plant Cell 2009, 21, 2090–2106. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Kaboshi, M.; Muto, H.; Takeda, A.; Murata, T.; Hasebe, M.; Watanabe, Y. Localization of tobacco germin-like protein 1 in leaf intercellular space. Plant Physiol. Biochem. 2014, 85, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Knecht, K.; Seyffarth, M.; Desel, C.; Thurau, T.; Sherameti, I.; Lou, B.G.; Oelmuller, R.; Cai, D.G. Expression of BvGLP-1 encoding a germin-like protein from sugar beet in Arabidopsis thaliana leads to resistance against phytopathogenic fungi. Mol. Plant-Microbe Interact. 2010, 23, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Aravind, L. Identification of novel families and classification of the C2 domain superfamily elucidate the origin and evolution of membrane targeting activities in eukaryotes. Gene 2010, 469, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Li, Y.; Hua, J. The ArabidopsisBAP1 and BAP2 genes are general inhibitors of programmed cell death. Plant Physiol. 2007, 145, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Lorkovic, Z.J. Role of plant RNA-binding proteins in development, stress response and genome organization. Trends Plant Sci. 2009, 14, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.H.; Kwak, K.J.; Kim, M.K.; Park, S.J.; Yang, K.Y.; Kang, H. Expression of Arabidopsis glycine-rich RNA-binding protein AtGRP2 or AtGRP7 improves grain yield of rice (Oryza sativa) under drought stress conditions. Plant Sci. 2014, 214, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Zhao, Y.X.; Xiao, H.L.; Zheng, Y.L.; Yue, B. Genome-wide identification, evolution, and expression analysis of RNA-binding glycine-rich protein family in maize. J. Integr. Plant Biol. 2014, 56, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, Y.; Zhang, Y.; Xu, J.; Zhang, Z.; Wang, Y. Genome-wide profiling of novel and conserved Populus microRNAs involved in pathogen stress response by deep sequencing. Planta 2012, 235, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.; Ren, Y.; Xu, J.; Zhang, Z.; Wang, Y. Genome-wide identification of cold-responsive and new microRNAs in Populus tomentosa by high-throughput sequencing. Biochem. Biophys. Res. Commun. 2012, 417, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Gou, L.; Chen, D.; Wu, P.; Chen, M. High-throughput degradome sequencing can be used to gain insights into microRNA precursor metabolism. J. Exp. Bot. 2010, 61, 3833–3837. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Zheng, Y.; Addo-Quaye, C.; Zhang, L.; Saini, A.; Jagadeeswaran, G.; Axtell, M.J.; Zhang, W.; Sunkar, R. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010, 62, 742–759. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Peral, M.M.; Li, J.; Li, Y.; Allen, R.S.; Schnippenkoetter, W.; Ohms, S.; White, R.G.; Millar, A.A. The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 2010, 154, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, N.; Mi, X.; Wu, L.; Ma, R.; Zhu, X.; Yao, L.; Jin, X.; Si, H.; Wang, D. Identification of miR159s and their target genes and expression analysis under drought stress in potato. Comput. Biol. Chem. 2014, 53PB, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Jeong, H.K.; Lee, E.; Natarajan, S. Characterization of a lipoate-protein ligase A gene of rice (Oryza sativa L.). Gene 2007, 393, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Yasuno, R.; Jordan, S.W.; Cronan, J.E., Jr.; Wada, H. Lipoic acid metabolism in Arabidopsis thaliana cloning and characterization of a cDNA encoding lipoyltransferase. Plant Cell Physiol. 2001, 42, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Grotz, N.; Guerinot, M.L. Molecular aspects of Cu, Fe and Zn homeostasis in plants. Biochim. Biophys. Acta 2006, 1763, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Haydon, M.J.; Cobbett, C.S. Transporters of ligands for essential metal ions in plants. New Phytol. 2007, 174, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Li, Y.F.; Sunkar, R. Redox signaling mediates the expression of a sulfate-deprivation-inducible microRNA395 in Arabidopsis. Plant J. 2014, 77, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Hayashi, N.; Yamaya, T.; Takahashi, H. Root-to-shoot transport of sulfate in Arabidopsis. Evidence for the role of SULTR3; 5 as a component of low-affinity sulfate transport system in the root vasculature. Plant Physiol. 2004, 136, 4198–4204. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, X.M.; Yang, Z.M.; Li, S.Q.; Wang, J.; Wang, S.H. Expression of Brassica napus L. γ-glutamyleysteine synthetase and low-and high-affinity sulfate transporters in response to excess cadmium. J. Integr. Plant Biol. 2005, 47, 243–250. [Google Scholar] [CrossRef]
- Tagami, Y.; Inaba, N.; Kutsuna, N.; Kurihara, Y.; Watanabe, Y. Specific enrichment of miRNAs in Arabidopsis thaliana infected with Tobacco mosaic virus. DNA Res. 2007, 14, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Ma, X.; Xu, X.; Meng, Y. Identification of the highly accumulated microRNA*s in Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa). Gene 2013, 515, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Lv, Q.; Zhang, J.; Li, J.; Du, Y.; Zhao, Q. Differential expression of the microRNAs in superior and inferior spikelets in rice (Oryza sativa). J. Exp. Bot. 2011, 62, 4943–4954. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Shao, C.; Gou, L.; Jin, Y.; Chen, M. Construction of microRNA- and microRNA*-mediated regulatory networks in plants. RNA Biol. 2011, 8, 1124–1148. [Google Scholar] [CrossRef] [PubMed]
- Pant, B.D.; Musialak-Lange, M.; Nuc, P.; May, P.; Buhtz, A.; Kehr, J.; Walther, D.; Scheible, W.R. Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Plant Physiol. 2009, 150, 1541–1555. [Google Scholar] [CrossRef] [PubMed]
- Buhtz, A.; Springer, F.; Chappell, L.; Baulcombe, D.C.; Kehr, J. Identification and characterization of small RNAs from the phloem of Brassica napus. Plant J. 2008, 53, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, M.; Shi, B.J.; Gustafson, P.; Langridge, P. Characterization of phosphorus-regulated miR399 and miR827 and their isomirs in barley under phosphorus-sufficient and phosphorus-deficient conditions. BMC Plant Biol. 2013, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Luo, S.; Schroth, G.; Meyers, B.C.; Green, P.J. Construction of parallel analysis of RNA ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009, 4, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2 an improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Bao, H.; Wu, Q.; Wang, Y. Transcriptome-Wide Identification of miRNA Targets under Nitrogen Deficiency in Populus tomentosa Using Degradome Sequencing. Int. J. Mol. Sci. 2015, 16, 13937-13958. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613937
Chen M, Bao H, Wu Q, Wang Y. Transcriptome-Wide Identification of miRNA Targets under Nitrogen Deficiency in Populus tomentosa Using Degradome Sequencing. International Journal of Molecular Sciences. 2015; 16(6):13937-13958. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613937
Chicago/Turabian StyleChen, Min, Hai Bao, Qiuming Wu, and Yanwei Wang. 2015. "Transcriptome-Wide Identification of miRNA Targets under Nitrogen Deficiency in Populus tomentosa Using Degradome Sequencing" International Journal of Molecular Sciences 16, no. 6: 13937-13958. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160613937