Novel Therapeutic GPCRs for Psychiatric Disorders

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. GPCRs in CNS
3. Medium-Sized Spiny Neurons (MSNs) in the Striatum Control Psychiatric Symptoms
4. Antipsychotics Exert Therapeutic Action through Dopamine D1 and D2 Receptors
5. Striatal-Enriched GPCRs Are Potential Drug Targets for Psychiatric Disorders
5.1. Adenosine A2a Receptor

5.2. GPR88
5.3. GPR6
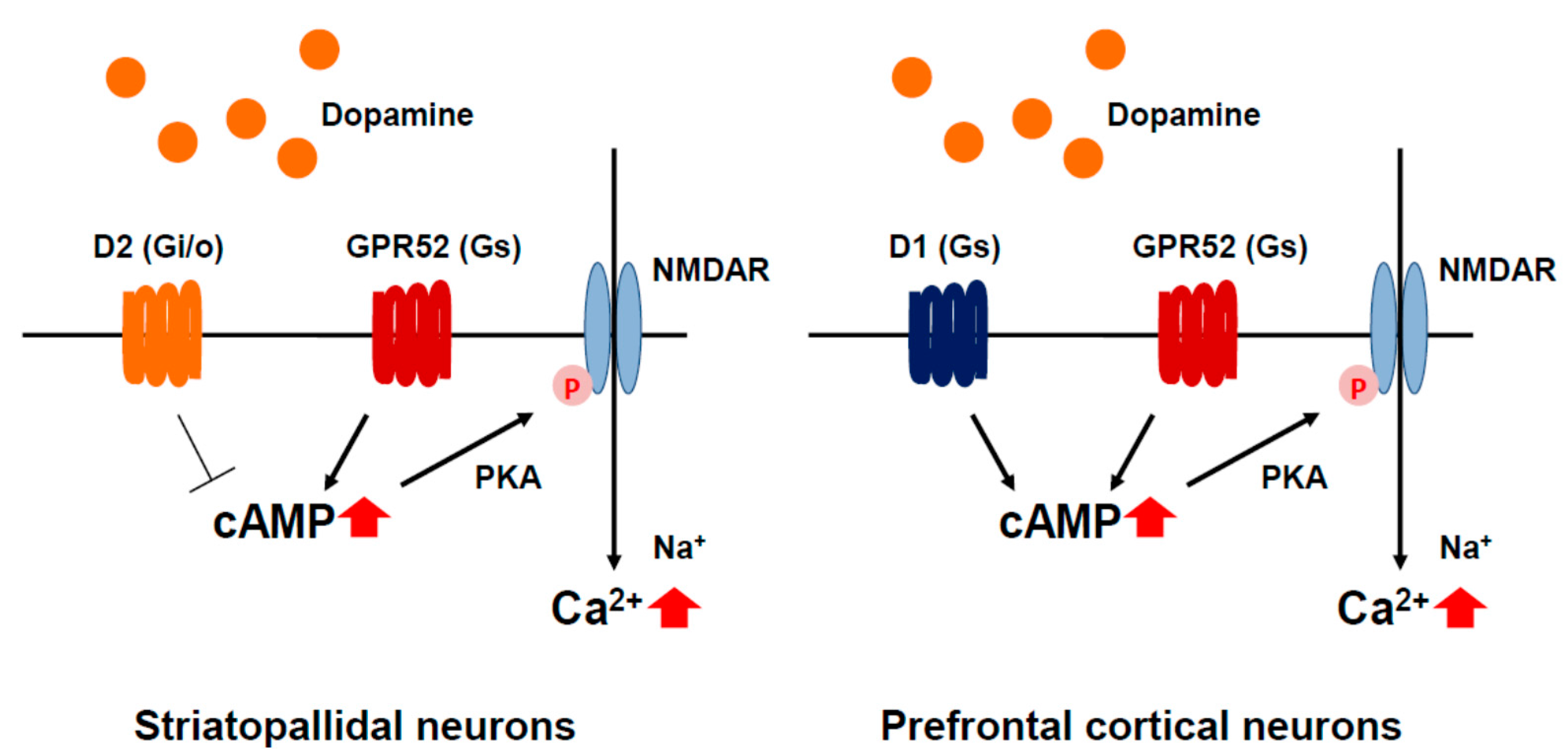
5.4. GPR52

6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Greengard, P. The neurobiology of slow synaptic transmission. Science 2001, 294, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Jessell, T.M.; Kandel, E.R. Synaptic transmission: A bidirectional and self-modifiable form of cell-cell communication. Cell 1993, 72, 1–30. [Google Scholar] [CrossRef]
- Sakuma, K.; Komatsu, H.; Maruyama, M.; Imaichi, S.; Habata, Y.; Mori, M. Temporal and spatial transcriptional fingerprints by antipsychotic or propsychotic drugs in mouse brain. PLoS ONE 2015, 10, e0118510. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Maeso, J.; Sealfon, S.C. Agonist-trafficking and hallucinogens. Curr. Med. Chem. 2009, 16, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Schioth, H.B. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharmacol. 2005, 67, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical profiling of G protein-coupled receptor expression. Cell 2008, 135, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Maruyama, M.; Yao, S.; Shinohara, T.; Sakuma, K.; Imaichi, S.; Chikatsu, T.; Kuniyeda, K.; Siu, F.K.; Peng, L.S.; et al. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS ONE 2014, 9, e90134. [Google Scholar] [CrossRef] [PubMed]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef] [PubMed]
- Ena, S.; de Kerchove d’Exaerde, A.; Schiffmann, S.N. Unraveling the differential functions and regulation of striatal neuron sub-populations in motor control, reward, and motivational processes. Front. Behav. Neurosci. 2011, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Ding, J.; Day, M.; Wang, Z.; Shen, W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007, 30, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Cachope, R.; Cheer, J.F. Local control of striatal dopamine release. Front. Behav. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Wilson, C.J.; Augood, S.J.; Emson, P.C. Striatal interneurones: Chemical, physiological and morphological characterization. Trends Neurosci. 1995, 18, 527–535. [Google Scholar] [CrossRef]
- Bolam, J.P.; Hanley, J.J.; Booth, P.A.; Bevan, M.D. Synaptic organisation of the basal ganglia. J. Anat. 2000, 196, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Tepper, J.M.; Bolam, J.P. Functional diversity and specificity of neostriatal interneurons. Curr. Opin. Neurobiol. 2004, 14, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Canales, J.J.; Capper-Loup, C. Levodopa-induced dyskinesias and dopamine-dependent stereotypies: A new hypothesis. Trends Neurosci. 2000, 23, S71–S77. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Young, W.S., III. Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: An in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988, 460, 161–167. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J., Jr.; Sibley, D.R. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, S.N.; Jacobs, O.; Vanderhaeghen, J.J. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: An in situ hybridization histochemistry study. J. Neurochem. 1991, 57, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, S.N.; Fisone, G.; Moresco, R.; Cunha, R.A.; Ferre, S. Adenosine A2A receptors and basal ganglia physiology. Prog. Neurobiol. 2007, 83, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Young, A.B.; Penney, J.B. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989, 12, 366–375. [Google Scholar] [CrossRef]
- DeLong, M.R.; Wichmann, T. Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 2007, 64, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C.; Carpenter, W.T., Jr. Diagnosis and classification of schizophrenia. Schizophr. Bull. 1993, 19, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y.; Matsubara, S.; Lee, J.C. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharmacol. Exp. Ther. 1989, 251, 238–246. [Google Scholar] [PubMed]
- Hirose, T.; Kikuchi, T. Aripiprazole, a novel antipsychotic agent: Dopamine D2 receptor partial agonist. J. Med. Investig. I 2005, 52, 284–290. [Google Scholar] [CrossRef]
- Wood, M.; Reavill, C. Aripiprazole acts as a selective dopamine D2 receptor partial agonist. Exp. Opin. Investig. Drugs 2007, 16, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; van Gompel, P.; Lesage, A.S.; de Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Ananth, J.; Burgoyne, K.S.; Gadasalli, R.; Aquino, S. How do the atypical antipsychotics work? J. Psychiatry Neurosci. 2001, 26, 385–394. [Google Scholar] [PubMed]
- Okubo, Y.; Suhara, T.; Suzuki, K.; Kobayashi, K.; Inoue, O.; Terasaki, O.; Someya, Y.; Sassa, T.; Sudo, Y.; Matsushima, E.; et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature 1997, 385, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, A.M.; Ribeiro, J.A. Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog. Neurobiol. 1996, 48, 167–189. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Agnati, L.; Fuxe, K.; Chen, J.F.; Morelli, M. Targeting adenosine A2A receptors in Parkinson’s disease. Trends Neurosci. 2006, 29, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Merello, M. Two new adenosine receptor antagonists for the treatment of Parkinson’s disease: Istradefylline versus tozadenant. Exp. Opin. Pharmacother. 2014, 15, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Shindou, T. Modulation of GABAergic transmission in the striatopallidal system by adenosine A2A receptors: A potential mechanism for the antiparkinsonian effects of A2A antagonists. Neurology 2003, 61, S44–S48. [Google Scholar] [CrossRef] [PubMed]
- Rimondini, R.; Ferre, S.; Ogren, S.O.; Fuxe, K. Adenosine A2A agonists: A potential new type of atypical antipsychotic. Neuropsychopharmacology 1997, 17, 82–91. [Google Scholar] [CrossRef]
- Justinova, Z.; Ferre, S.; Segal, P.N.; Antoniou, K.; Solinas, M.; Pappas, L.A.; Highkin, J.L.; Hockemeyer, J.; Munzar, P.; Goldberg, S.R. Involvement of adenosine A1 and A2A receptors in the adenosinergic modulation of the discriminative-stimulus effects of cocaine and methamphetamine in rats. J. Pharmacol. Exp. Ther. 2003, 307, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Lucas, P.B.; Pickar, D.; Kelsoe, J.; Rapaport, M.; Pato, C.; Hommer, D. Effects of the acute administration of caffeine in patients with schizophrenia. Biol. Psychiatry 1990, 28, 35–40. [Google Scholar] [CrossRef]
- Mikkelsen, E.J. Caffeine and schizophrenia. J. Clin. Psychiatry 1978, 39, 732–736. [Google Scholar] [PubMed]
- Deckert, J.; Nothen, M.M.; Bryant, S.P.; Schuffenhauer, S.; Schofield, P.R.; Spurr, N.K.; Propping, P. Mapping of the human adenosine A2a receptor gene: Relationship to potential schizophrenia loci on chromosome 22q and exclusion from the CATCH 22 region. Human Genet. 1997, 99, 326–328. [Google Scholar] [CrossRef]
- Hong, C.J.; Liu, H.C.; Liu, T.Y.; Liao, D.L.; Tsai, S.J. Association studies of the adenosine A2a receptor (1976T>C) genetic polymorphism in Parkinson’s disease and schizophrenia. J. Neural Transm. 2005, 112, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, A.M.; Ribeiro, J.A. Tuning and fine-tuning of synapses with adenosine. Curr. Neuropharmacol. 2009, 7, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Ferre, S.; Fredholm, B.B.; Morelli, M.; Popoli, P.; Fuxe, K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487. [Google Scholar] [CrossRef]
- Popoli, P.; Reggio, R.; Pezzola, A. Adenosine A1 and A2 receptor agonists significantly prevent the electroencephalographic effects induced by MK-801 in rats. Eur. J. Pharmacol. 1997, 333, 2143–2146. [Google Scholar] [CrossRef]
- Sills, T.L.; Azampanah, A.; Fletcher, P.J. The adenosine A1 receptor agonist N6-cyclopentyladenosine blocks the disruptive effect of phencyclidine on prepulse inhibition of the acoustic startle response in the rat. Eur. J. Pharmacol. 1999, 369, 325–329. [Google Scholar] [CrossRef]
- De Mendonca, A.; Sebastiao, A.M.; Ribeiro, J.A. Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport 1995, 6, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Gerevich, Z.; Wirkner, K.; Illes, P. Adenosine A2A receptors inhibit the N-methyl-d-aspartate component of excitatory synaptic currents in rat striatal neurons. Eur. J. Pharmacol. 2002, 451, 161–164. [Google Scholar] [CrossRef]
- Shen, H.Y.; Coelho, J.E.; Ohtsuka, N.; Canas, P.M.; Day, Y.J.; Huang, Q.Y.; Rebola, N.; Yu, L.; Boison, D.; Cunha, R.A.; et al. A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J. Neurosci. 2008, 28, 2970–2975. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Sanz, E.; Wang, W.; Storey, G.P.; Guler, A.D.; Wanat, M.J.; Roller, B.A.; La Torre, A.; Amieux, P.S.; McKnight, G.S.; et al. Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat. Neurosci. 2012, 15, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.F.; Grauer, S.M.; Paulsen, J.; Graf, R.; Taylor, N.; Sung, M.A.; Zhang, L.; Hughes, Z.; Pulito, V.L.; Liu, F.; et al. The orphan GPCR, GPR88, modulates function of the striatal dopamine system: A possible therapeutic target for psychiatric disorders? Mol. Cell. Neurosci. 2009, 42, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Ingallinesi, M.; Le Bouil, L.; Faucon Biguet, N.; Do Thi, A.; Mannoury la Cour, C.; Millan, M.J.; Ravassard, P.; Mallet, J.; Meloni, R. Local inactivation of Gpr88 in the nucleus accumbens attenuates behavioral deficits elicited by the neonatal administration of phencyclidine in rats. Mol. Psychiatry 2014. [Google Scholar] [CrossRef] [PubMed]
- Del Zompo, M.; Deleuze, J.F.; Chillotti, C.; Cousin, E.; Niehaus, D.; Ebstein, R.P.; Ardau, R.; Mace, S.; Warnich, L.; Mujahed, M.; et al. Association study in three different populations between the GPR88 gene and major psychoses. Mol. Genet. Genomic Med 2014, 2, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.; Maier, R.; Barr, A.M.; Morale, M.C.; Lu, X.; Sanna, P.P.; Bilbe, G.; Hoyer, D.; Bartfai, T. Region-specific transcriptional changes following the three antidepressant treatments electro convulsive therapy, sleep deprivation and fluoxetine. Mol. Psychiatry 2007, 12, 167–189. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Jockers, R. Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep. 2006, 7, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Decker, A.M.; Huang, X.P.; Gilmour, B.P.; Blough, B.E.; Roth, B.L.; Hu, Y.; Gill, J.B.; Zhang, X.P. Synthesis, pharmacological characterization, and structure-activity relationship studies of small molecular agonists for the orphan GPR88 receptor. ACS Chem. Neurosci. 2014, 5, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.K.; Cui, Y.; Ostlund, S.B.; Balleine, B.W.; Yang, X.W. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nat. Neurosci. 2007, 10, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbrock, K.; Gassenhuber, H.; Kostenis, E. Sphingosine 1-phosphate is a ligand of the human GPR3, GPR6 and GPR12 family of constitutively active G protein-coupled receptors. Cell Signal. 2002, 14, 941–953. [Google Scholar] [CrossRef]
- Yin, H.; Chu, A.; Li, W.; Wang, B.; Shelton, F.; Otero, F.; Nguyen, D.G.; Caldwell, J.S.; Chen, Y.A. Lipid G protein-coupled receptor ligand identification using β-arrestin PathHunter assay. J. Biol. Chem. 2009, 284, 12328–12338. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.G.; Frank, M.J. How much of reinforcement learning is working memory, not reinforcement learning? A behavioral, computational, and neurogenetic analysis. Eur. J. Neurosci. 2012, 35, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Ishii, K.; Kasai, K.; Yoon, S.O.; Saeki, Y. Neural expression of G protein-coupled receptors GPR3, GPR6, and GPR12 up-regulates cyclic AMP levels and promotes neurite outgrowth. J. Biol. Chem. 2007, 282, 10506–10515. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Hengerer, B.; Ferger, B. G-protein coupled receptor 6 deficiency alters striatal dopamine and cAMP concentrations and reduces dyskinesia in a mouse model of Parkinson’s disease. Exp. Neurol. 2014, 257, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Nishi, A.; Fisone, G.; Girault, J.A.; Nairn, A.C.; Greengard, P. DARPP-32: An integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 269–296. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Svenningsson, P.; Kuroiwa, M.; Gong, S.; Nishi, A.; Heintz, N.; Greengard, P. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat. Neurosci. 2008, 11, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Dudman, J.T.; Eaton, M.E.; Rajadhyaksha, A.; Macias, W.; Taher, M.; Barczak, A.; Kameyama, K.; Huganir, R.; Konradi, C. Dopamine D1 receptors mediate CREB phosphorylation via phosphorylation of the NMDA receptor at Ser897-NR1. J. Neurochem. 2003, 87, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Greengard, P.; Yan, Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc. Natl. Acad. Sci. USA 2004, 101, 2596–2600. [Google Scholar] [CrossRef] [PubMed]
- Setoh, M.; Ishii, N.; Kono, M.; Miyanohana, Y.; Shiraishi, E.; Harasawa, T.; Ota, H.; Odani, T.; Kanzaki, N.; Aoyama, K.; et al. Discovery of the first potent and orally available agonist of the orphan G-protein-coupled receptor 52. J. Med. Chem. 2014, 57, 5226–5237. [Google Scholar] [CrossRef] [PubMed]
- Aggleton, J.P.; Brown, M.W. Episodic memory, amnesia, and the hippocampal-anterior thalamic axis. Behav. Brain Sci. 1999, 22, 425–444, discussion 444–489. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hikosaka, O. Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 2007, 447, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Ouagazzal, A.M.; Jenck, F.; Moreau, J.L. Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: A model for detecting antipsychotic activity? Psychopharmacology 2001, 156, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W.; Shipe, W.D.; Wolkenberg, S.E.; Theberge, C.R.; Williams, D.L., Jr.; Sur, C.; Kinney, G.G. Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr. Top. Med. Chem. 2006, 6, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Hartling, L.; Abou-Setta, A.M.; Dursun, S.; Mousavi, S.S.; Pasichnyk, D.; Newton, A.S. Antipsychotics in adults with schizophrenia: Comparative effectiveness of first-generation versus second-generation medications: A systematic review and meta-analysis. Ann. Int. Med. 2012, 157, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Abou-Setta, A.M.; Mousavi, S.S.; Spooner, C.; Schouten, J.R.; Pasichnyk, D.; Armijo-Olivo, S.; eaith, A.; Seida, J.C.; Dursun, S.; Newton, A.S.; et al. First-Generation Versus Second-Generation Antipsychotics in Adults: Comparative Effectiveness; University of Alberta Evidence-based Practice Center: Rockville, MD, USA, 2012. [Google Scholar]
- Dunlop, J.; Brandon, N.J. Schizophrenia drug discovery and development in an evolving era: Are new drug targets fulfilling expectations? J. Psychopharmacol. 2015, 29, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cui, X.; Al-Ramahi, I.; Sun, X.; Li, B.; Hou, J.; Difiglia, M.; Palacino, J.; Wu, Z.Y.; Ma, L.; et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komatsu, H. Novel Therapeutic GPCRs for Psychiatric Disorders. Int. J. Mol. Sci. 2015, 16, 14109-14121. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160614109
Komatsu H. Novel Therapeutic GPCRs for Psychiatric Disorders. International Journal of Molecular Sciences. 2015; 16(6):14109-14121. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160614109
Chicago/Turabian StyleKomatsu, Hidetoshi. 2015. "Novel Therapeutic GPCRs for Psychiatric Disorders" International Journal of Molecular Sciences 16, no. 6: 14109-14121. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms160614109