
Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases


Abstract
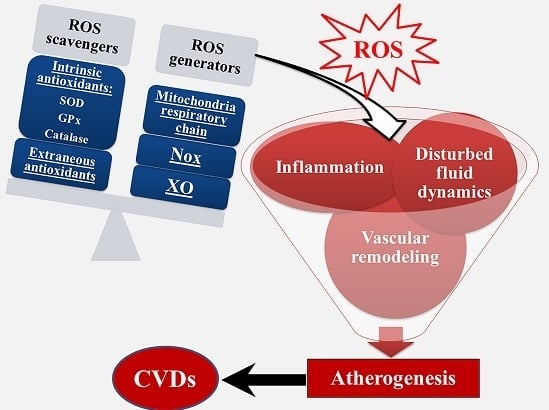
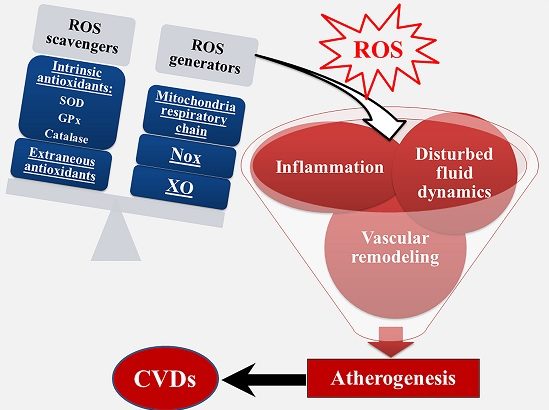
:
{kind=link}
1. Introduction
2. Potential Sources of ROS (Reactive Oxygen Species)
3. ROS in Ischemia-Reperfusion Damage
4. ROS and Atherosclerosis
5. Pharmacological Interventions Targeting ROS Sources in CVD (Cardiovascular Disease)
6. Technological Advance and Challenges in Genetic Antioxidant Therapies in CVD
7. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fearon, I.M.; Faux, S.P. Oxidative stress and cardiovascular disease: Novel tools give (free) radical insight. J. Mol. Cell. Cardiol. 2009, 47, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the american heart association. Circulation 2015, 131, e29–e322. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Pasternak, R.; Greenland, P.; Smith, S., Jr.; Fuster, V. Assessment of cardiovascular risk by use of multiple-risk-factor assessment equations: A statement for healthcare professionals from the american heart association and the american college of cardiology. Circulation 1999, 100, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Youtz, D.J.; Wold, L.E. Particulate matter exposure exacerbates high glucose-induced cardiomyocyte dysfunction through ROS generation. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Thanassoulis, G.; Vasan, R.S. Genetic cardiovascular risk prediction: Will we get there? Circulation 2010, 122, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.W. The jeremiah metzger lecture. Pathogenesis of atherosclerosis: Redox as a unifying mechanism. Trans. Am. Clin. Climatol. Assoc. 2003, 114, 273–304. [Google Scholar] [PubMed]
- Zuo, L.; Rose, B.A.; Roberts, W.J.; He, F.; Banes-Berceli, A.K. Molecular characterization of reactive oxygen species in systemic and pulmonary hypertension. Am. J. Hypertens. 2014, 27, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Ushio-Fukai, M. Redox control of vascular smooth muscle proliferation. J. Lab. Clin. Med. 1998, 132, 9–15. [Google Scholar] [CrossRef]
- Blomhoff, R. Dietary antioxidants and cardiovascular disease. Curr. Opin. Lipidol. 2005, 16, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.; Hayek, T.; Raz, A.; Coleman, R.; Dornfeld, L.; Vaya, J.; Aviram, M. Pomegranate juice supplementation to atherosclerotic mice reduces macrophage lipid peroxidation, cellular cholesterol accumulation and development of atherosclerosis. J. Nutr. 2001, 131, 2082–2089. [Google Scholar] [PubMed]
- Hayek, T.; Fuhrman, B.; Vaya, J.; Rosenblat, M.; Belinky, P.; Coleman, R.; Elis, A.; Aviram, M. Reduced progression of atherosclerosis in apolipoprotein E-deficient mice following consumption of red wine, or its polyphenols quercetin or catechin, is associated with reduced susceptibility of LDl to oxidation and aggregation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.; Egri, Z.; Tanna, G.; Colman, R.; Newton, G.E. Vitamin C prevents hyperoxia-mediated vasoconstriction and impairment of endothelium-dependent vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2414–H2421. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxygen radicals: A commonsense look at their nature and medical importance. Med. Biol. 1984, 62, 71–77. [Google Scholar] [PubMed]
- Zuo, L.; Best, T.M.; Roberts, W.J.; Diaz, P.T.; Wagner, P.D. Characterization of reactive oxygen species in diaphragm. Acta Physiol. 2015, 213, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—The good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Liu, C.Y.; Merola, A.J.; Berliner, L.J.; Clanton, T.L. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am. J. Physiol. Cell Physiol. 2000, 279, C1058–C1066. [Google Scholar] [PubMed]
- Wattanapitayakul, S.K.; Bauer, J.A. Oxidative pathways in cardiovascular disease: Roles, mechanisms, and therapeutic implications. Pharmacol. Ther. 2001, 89, 187–206. [Google Scholar] [CrossRef]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Adam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ros generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Taverne, Y.J.; Bogers, A.J.; Duncker, D.J.; Merkus, D. Reactive oxygen species and the cardiovascular system. Oxid. Med. Cell Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Nogueira, L.; Hogan, M.C. Reactive oxygen species formation during tetanic contractions in single isolated xenopus myofibers. J. Appl. Physiol. 2011, 111, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Shiah, A.; Roberts, W.J.; Chien, M.T.; Wagner, P.D.; Hogan, M.C. Low Po2 conditions induce reactive oxygen species formation during contractions in single skeletal muscle fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R1009–R1016. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Papa, S.; Bolanos, J.; Bruckdorfer, R.; Carlsen, H.; Elliott, R.M.; Flier, J.; Griffiths, H.R.; Heales, S.; Holst, B.; et al. Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol. Asp. Med. 2002, 23, 209–285. [Google Scholar] [CrossRef]
- Dale, D.C.; Boxer, L.; Liles, W.C. The phagocytes: Neutrophils and monocytes. Blood 2008, 112, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pasniciuc, S.; Wright, V.P.; Merola, A.J.; Clanton, T.L. Sources for superoxide release: Lessons from blockade of electron transport, nadph oxidase, and anion channels in diaphragm. Antioxid. Redox Signal. 2003, 5, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. Nadph oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.; Grieve, D.; Johar, S.; Zhang, M.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Judkins, C.P.; Diep, H.; Broughton, B.R.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H24–H32. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. Nox4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, E.; Baldassari, F.; Bononi, A.; Wieckowski, M.R.; Pinton, P. Oxidative stress in cardiovascular diseases and obesity: Role of p66shc and protein kinase C. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R.; Jeroudi, M.O.; Patel, B.S.; DuBose, C.M.; Lai, E.K.; Roberts, R.; McCay, P.B. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc. Natl. Acad. Sci. USA 1989, 86, 4695–4699. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zuo, L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Majidi, M.; Kosinski, A.S.; Al-Khatib, S.M.; Lemmert, M.E.; Smolders, L.; van Weert, A.; Reiber, J.H.; Tzivoni, D.; Bar, F.W.; Wellens, H.J.; et al. Reperfusion ventricular arrhythmia 'bursts' predict larger infarct size despite timi 3 flow restoration with primary angioplasty for anterior st-elevation myocardial infarction. Eur. Heart J. 2009, 30, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Di Stolfo, G.; Sicuro, S.; Dragoni, S.; Lisi, M.; Forconi, S.; Parker, J.D. Nitroglycerin protects the endothelium from ischaemia and reperfusion: Human mechanistic insight. Br. J. Clin. Pharmacol. 2007, 64, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, A.; Melis, F.; Tocco, F.; Santoboni, U.M.; Lai, C.; Angioy, G.; Lorrai, L.; Pittau, G.; Concu, A.; Pagliaro, P. Exercise-induced and nitroglycerin-induced myocardial preconditioning improves hemodynamics in patients with angina. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H235–H242. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Roberts, W.; Tolomello, R.; Goins, A. Ischemic and hypoxic preconditioning protect cardiac muscles via intracellular ros signaling. Front. Biol. 2013, 8, 305–311. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Lebuffe, G.; Schumacker, P.T.; Shao, Z.H.; Anderson, T.; Iwase, H.; Vanden Hoek, T.L. ROS and NO trigger early preconditioning: Relationship to mitochondrial katp channel. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H299–H308. [Google Scholar] [CrossRef] [PubMed]
- Benetos, A.; Laurent, S.; Hoeks, A.P.; Boutouyrie, P.H.; Safar, M.E. Arterial alterations with aging and high blood pressure. A noninvasive study of carotid and femoral arteries. Arterioscler. Thromb. 1993, 13, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Mattace-Raso, F.U.S.; van der Cammen, T.J.M.; Hofman, A.; van Popele, N.M.; Bos, M.L.; Schalekamp, M.A.D.H.; Asmar, R.; Reneman, R.S.; Hoeks, A.P.G.; Breteler, M.M.B.; et al. Arterial stiffness and risk of coronary heart disease and stroke—The rotterdam study. Circulation 2006, 113, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Froehlich, J.; Galis, Z.S.; Lakatta, E.G. Increased expression of matrix metalloproteinase-2 in the thickened intima of aged rats. Hypertension 1999, 33, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Iiyama, K.; Hajra, L.; Iiyama, M.; Li, H.; DiChiara, M.; Medoff, B.D.; Cybulsky, M.I. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 1999, 85, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.S.; Al Mheid, I.; Morris, A.A.; Ahmed, Y.; Kavtaradze, N.; Ali, S.; Dabhadkar, K.; Brigham, K.; Hooper, W.C.; Alexander, R.W.; et al. Oxidative stress is associated with impaired arterial elasticity. Atherosclerosis 2011, 218, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lakatta, E.G. Role of inflammation in the pathogenesis of arterial stiffness. Yonsei Med. J. 2012, 53, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [PubMed]
- Fukai, T.; Siegfried, M.R.; Ushio-Fukai, M.; Cheng, Y.; Kojda, G.; Harrison, D.G. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Investig. 2000, 105, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 942. [Google Scholar] [CrossRef]
- Hajra, L.; Evans, A.I.; Chen, M.; Hyduk, S.J.; Collins, T.; Cybulsky, M.I. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA 2000, 97, 9052–9057. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Saha, A.; Boo, Y.C.; Sorescu, G.P.; McNally, J.S.; Holland, S.M.; Dikalov, S.; Giddens, D.P.; Griendling, K.K.; Harrison, D.G.; et al. Oscillatory shear stress stimulates endothelial production of O2− from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J. Biol. Chem. 2003, 278, 47291–47298. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free-radicals, reactive oxygen species and human-disease—A critical-evaluation with special reference to atherosclerosis. Br. J. Exp. Pathol. 1989, 70, 737–757. [Google Scholar] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Shah, A.M. Intracellular localization and preassembly of the nadph oxidase complex in cultured endothelial cells. J. Biol. Chem. 2002, 277, 19952–19960. [Google Scholar] [CrossRef] [PubMed]
- Kojda, G.; Harrison, D. Interactions between no and reactive oxygen species: Pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc. Res. 1999, 43, 562–571. [Google Scholar] [CrossRef]
- Navab, M.; Berliner, J.A.; Watson, A.D.; Hama, S.Y.; Territo, M.C.; Lusis, A.J.; Shih, D.M.; Van Lenten, B.J.; Frank, J.S.; Demer, L.L.; et al. The yin and yang of oxidation in the development of the fatty streak. A review based on the 1994 george lyman duff memorial lecture. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [PubMed]
- Collaborators, C. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1358. [Google Scholar] [PubMed]
- Takemoto, M.; Node, K.; Nakagami, H.; Liao, Y.L.; Grimm, M.; Takemoto, Y.; Kitakaze, M.; Liao, J.K. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J. Clin. Investig. 2001, 108, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Shishehbor, M.H.; Brennan, M.L.; Aviles, R.J.; Fu, X.M.; Penn, M.S.; Sprecher, D.L.; Hazen, S.L. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003, 108, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Fujita, M.; Kitakaze, M.; Hori, M.; Liao, J.K. Short-term statin therapy improves cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy. Circulation 2003, 108, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Mcquillan, L.P.; Leung, G.K.; Marsden, P.A.; Kostyk, S.K.; Kourembanas, S. Hypoxia inhibits expression of enos via transcriptional and posttranscriptional mechanisms. Am. J. Physiol. Heart C 1994, 267, H1921–H1927. [Google Scholar]
- Endres, M.; Laufs, U.; Liao, J.K.; Moskowitz, M.A. Targeting enos for stroke protection. Trends Neurosci. 2004, 27, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, E.; Dernbach, E.; Hermann, C.; Mondorf, U.F.; Zeiher, A.M.; Dimmeler, S. Oxidized LDl inhibits vascular endothelial growth factor-induced endothelial cell migration by an inhibitory effect on the Akt/endothelial nitric oxide synthase pathway. Circulation 2001, 103, 2102–2107. [Google Scholar] [CrossRef] [PubMed]
- Jantzen, F.; Konemann, S.; Wolff, B.; Barth, S.; Staudt, A.; Kroemer, H.K.; Dahm, J.B.; Felix, S.B.; Landsberger, M. Isoprenoid depletion by statins antagonizes cytokine-induced down-regulation of endothelial nitric oxide expression and increases no synthase activity in human umbilical vein endothelial cells. J. Physiol. Pharmacol. 2007, 58, 503–514. [Google Scholar] [PubMed]
- Rapola, J.M.; Virtamo, J.; Ripatti, S.; Huttunen, J.K.; Albanes, D.; Taylor, P.R.; Heinonen, O.P. Randomised trial of α-tocopherol and β-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. Lancet 1997, 349, 1715–1720. [Google Scholar] [CrossRef]
- Singh, R.B.; Wander, G.S.; Rastogi, A.; Shukla, P.K.; Mittal, A.; Sharma, J.P.; Mehrotra, S.K.; Kapoor, R.; Chopra, R.K. Randomized, double-blind placebo-controlled trial of coenzyme q10 in patients with acute myocardial infarction. Cardiovasc. Drug Ther. 1998, 12, 347–353. [Google Scholar] [CrossRef]
- Das, S.; Otani, H.; Maulik, N.; Das, D.K. Lycopene, tomatoes, and coronary heart disease. Free Radic. Res. 2005, 39, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Su, J.F.; Guo, C.J.; Wei, J.Y.; Yang, J.J.; Jiang, Y.G.; Li, Y.F. Protection against hepatic ischemia-reperfusion injury in rats by oral pretreatment with quercetin. Biomed. Environ. Sci. 2003, 16, 1–8. [Google Scholar] [PubMed]
- Schmidt-Ott, K.M.; Kagiyama, S.; Phillips, M.I. The multiple actions of angiotensin II in atherosclerosis. Regul. Pept. 2000, 93, 65–77. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Pryor, T.; Givvimani, S.; Lederer, E.; Tyagi, S.C.; Sen, U. Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. J. Hypertens. 2013, 31, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of superoxide in angiotensin ii-induced but not catecholamine-induced hypertension. Circulation 1997, 95, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Konig, I.; Meyer, W.; Kojda, G. Inhibition of vascular oxidative stress in hypercholesterolemia by eccentric isosorbide mononitrate. J. Am. Coll. Cardiol. 2004, 44, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Napoli, C. Novel features of nitric oxide, endothelial nitric oxide synthase, and atherosclerosis. Curr. Diabetes Rep. 2005, 5, 17–23. [Google Scholar] [CrossRef]
- Cosentino, F.; Savoia, C.; de Paolis, P.; Francia, P.; Russo, A.; Maffei, A.; Venturelli, V.; Schiavoni, M.; Lembo, G.; Volpe, M. Angiotensin II type 2 receptors contribute to vascular responses in spontaneously hypertensive rats treated with angiotensin II type 1 receptor antagonists. Am. J. Hypertens. 2005, 18, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Minatoguchi, S.; Watanabe, K.; Yamada, Y.; Mizukusa, T.; Kawasaki, H.; Takahashi, H.; Uno, T.; Tsukamoto, T.; Hiei, K.; et al. Candesartan decreases carotid intima-media thickness by enhancing nitric oxide and decreasing oxidative stress in patients with hypertension. Hypertens. Res. 2008, 31, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Yagi, S.; Morita, T.; Katayama, S. Combined treatment with an AT1 receptor blocker and angiotensin converting enzyme inhibitor has an additive effect on inhibiting neointima formation via improvement of nitric oxide production and suppression of oxidative stress. Hypertens. Res. 2004, 27, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.H.; Armas-Hernandez, M.J.; Velasco, M.; Israili, Z.H.; Armas-Padilla, M.C. Calcium antagonists and atherosclerosis protection in hypertension. Am. J. Ther. 2003, 10, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.; Perrone-Filardi, P.; Petretta, M.; Marciano, C.; Vassallo, E.; Gargiulo, P.; Paolillo, S.; Petretta, A.; Chiariello, M. Calcium channel blockers and cardiovascular outcomes: A meta-analysis of 175,634 patients. J. Hypertens. 2009, 27, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Survase, S.; Ivey, M.E.; Nigro, J.; Osman, N.; Little, P.J. Actions of calcium channel blockers on vascular proteoglycan synthesis: Relationship to atherosclerosis. Vasc. Health Risk Manag. 2005, 1, 199–208. [Google Scholar] [PubMed]
- Ganji, S.H.; Qin, S.; Zhang, L.; Kamanna, V.S.; Kashyap, M.L. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis 2009, 202, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of action of niacin. Am. J. Cardiol. 2008, 101, 20B–26B. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W. Hypertension: Is it time to replace drugs with nutrition and nutraceuticals? Pharm. Ther. 2014, 39, 291–295. [Google Scholar]
- Houston, M.C. The role of cellular micronutrient analysis, nutraceuticals, vitamins, antioxidants and minerals in the prevention and treatment of hypertension and cardiovascular disease. Ther. Adv. Cardiovasc. Dis. 2010, 4, 165–183. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; FitzGerald, G.A. Oxidative stress and cardiovascular injury: Part II: Animal and human studies. Circulation 2003, 108, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.S.; Magno, C.P.; Lane, K.T.; Chan, T.; Hoyt, D.B.; Greenfield, S. Nutrition impacts the prevalence of peripheral arterial disease in the united states. J. Vasc. Surg. 2008, 48, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.G.; Parsons, A.; Schofield, P.M.; Kelly, F.; Cheeseman, K.; Mitchinson, M.J. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge heart antioxidant study (CHAOS). Lancet 1996, 347, 781–786. [Google Scholar] [CrossRef]
- Ashor, A.W.; Siervo, M.; Lara, J.; Oggioni, C.; Mathers, J.C. Antioxidant vitamin supplementation reduces arterial stiffness in adults: A systematic review and meta-analysis of randomized controlled trials. J. Nutr. 2014, 144, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., III; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Farmakis, D.; Kremastinos, D.T. To prevent, protect and save the ischemic heart: Antioxidants revisited. Expert Opin. Ther. Targets 2009, 13, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Jaxa-Chamiec, T.; Bednarz, B.; Herbaczynska-Cedro, K.; Maciejewski, P.; Ceremuzynski, L. Effects of vitamins C and E on the outcome after acute myocardial infarction in diabetics: A retrospective, hypothesis-generating analysis from the mivit study. Cardiology 2009, 112, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Inkala, M.; Heikura, T.; Jauhiainen, S.; Jyrkkanen, H.K.; Kansanen, E.; Maatta, K.; Romppanen, E.; Turunen, P.; Rutanen, J.; et al. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Brasen, J.H.; Leppanen, O.; Inkala, M.; Heikura, T.; Levin, M.; Ahrens, F.; Rutanen, J.; Pietsch, H.; Bergqvist, D.; Levonen, A.L.; et al. Extracellular superoxide dismutase accelerates endothelial recovery and inhibits in-stent restenosis in stented atherosclerotic watanabe heritable hyperlipidemic rabbit aorta. J. Am. Coll. Cardiol. 2007, 50, 2249–2253. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, G.; Yoshida, T.; Noguchi, M. Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 2005, 338, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Soares, M.P.; Yamashita, K.; Bach, F.H. Heme oxygenase-1: Unleashing the protective properties of heme. Trends Immunol. 2003, 24, 449–455. [Google Scholar] [CrossRef]
- Jain, A.K.; Mehra, N.K.; Swarnakar, N.K. Role of antioxidants for the treatment of cardiovascular diseases: Challenges and opportunities. Curr. Pharm. Des. 2015, 21, 4441–4455. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, F.; Zuo, L. Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 27770-27780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126059
He F, Zuo L. Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases. International Journal of Molecular Sciences. 2015; 16(11):27770-27780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126059
Chicago/Turabian StyleHe, Feng, and Li Zuo. 2015. "Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases" International Journal of Molecular Sciences 16, no. 11: 27770-27780. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161126059