
miRNAs and Other Epigenetic Changes as Biomarkers in Triple Negative Breast Cancer


Abstract
:1. Introduction
Clinical Trials in TNBC
2. Better Classification of TNBC to Find New Treatment Targets and Prognostic Indicators
3. Epigenetic Changes in TNBC—New Biomarkers?
4. MicroRNAs
4.1. MicroRNAs in Cancer
4.2. miRNAs Involved in Triple Negative Breast Cancer

{kind=link}
| miRNA | Result | Technology | References |
|---|---|---|---|
| miR-342, miR-299, miR-217, miR-190, miR-135b, miR-218 | Associated with ER status | Expression profiling of 453 miRNAs, 29 breast cancer cases (mixed receptor status) | [70] |
| miR-520g, miR-377, miR-527-518a, miR-520f-520c | Associated with PR status | ||
| miR-520d, miR-181c, miR-302c, miR-376b, miR-30e | Associated with HER2/neu status | ||
| miR-532-5p, miR-500, miR362-5p, miR-502-3p | Located at Xp11.23 and present in TNBC, compared to other subtypes | miRCURY LNA arrays (2090 miRNAs analysed) 103 lymph node negative cases (mixed breast cancer subtypes) | [71] |
| Signature of 41 miRNAs | Associated with TNBC subtype | ||
| Signature of 116 deregulated miRNAs | First study purely focused on miRNAs in TNBC | nanoString nCounter profiling (664 miRNAs analysed) 173 TNBC samples | [65] |
| miR-106b, miR-17/92 cluster, miR-200 family, miR-21, miR-155 | Most up-regulated | ||
| let-7b, let-7c, miR-126, miR-145, miR-205 | Most down-regulated | ||
| miR-424, miR-125a-5p, miR-627, miR-579, let-7g, miR-101 | Associated with metastasis | ||
| miR-130a | Second study purely on TNBC. Novel miRNAs, up-regulated in TNBC | Agilent miRNA microarrays (904 miRNAs analysed) 31 tumours, 13 lymph node metastasis, 23 normal adjacent tissues | [64,65] |
| miR-1280, miR-590-5p, miR-1308, miR-17* | Novel miRNAs, down-regulated in TNBC | ||
| 27 miRNA signature | Associated with lymph node metastasis, majority (25) are down-regulated | ||
| miR-145, miR-205 | ↓ in TNBC (preferentially expressed in normal myoepithelial cells) | Tissue microarrays 100 TNBC samples | [72] |
| miR-17-92 cluster, miR-106b-25 cluster | Associated with oncogenic processes EMT, PI3K/Akt/mTOR, MYC, PTEN | miRNA and gene expression arrays, prediction software, data integration (miRNA arrays, (based on Sanger miRBase release 12.0, containing probes for 866 miRNAs) (29 mixed breast cancer subtypes)) | [73] |
| miR-342, miR-299, miR-217, miR-190, miR-135b, miR-218 | Markers for ER status | miRNA microarray, network algorithms, qPCR (453 miRNAs analysed) (mixed breast cancer subtypes) | [70] |
| miR-520g, miR-377, miR-527-518a, miR-520f-520c | Markers for PR status | ||
| miR-520d, miR-181c, miR-302c, miR-376b, miR-30e | Markers for HER2 | ||
| miR-93 | Associated with ER and PR status | miRNA profiling, qPCR (3 miRNAs analysed) (TaqMan MicroRNA Assays) (37 mixed breast cancer subtypes) | [74] |
| miR-200c, miR-205 | Lower levels are associated with lymph node metastasis in TNBC | qPCR from tumour samples (16 miRNAs analysed) (32 TNBC samples) | [75] |
| miR-373, miR-10b | ↑ regulated in cases with lymph node metastasis | qPCR from tumour samples (2 miRNAs analysed) (TaqMan MicroRNA Assays) (60 mixed breast cancer subtypes) | [76] |
| miRNA | Result | Functional Evidence | References |
|---|---|---|---|
| miR-200a/b | Tumour suppressor-miR/targets ZEB1/ZEB2, Suz 12, EphA2/plays role during differentiation in mammary epithelial cells | Cell culture experiment (differentiation) and qPCR (non-TNBC cell line HC11 mouse mammary) | [66] |
| miR-200c | Tumour suppressor-miR/targets ZEB1/ZEB2, MSN, FN1, TrkB/inhibits EMT and migration | Dual luciferase reporter assays, wound healing assays, cell-death ELISAs, and viability assays (non-TNBC cell lines: Hec50, AN3CA, MCF7; TNBC cell lines: MDA-MB-231, BT549) | [67,68] |
| miR-205 | Tumour suppressor-miR/targets E2F1, LAMC1/supresses proliferation, cell cycle and tumour growth | Transfections, qPCR, colony formation assay, proliferation assay, cell cycle analysis, apoptosis assay, viability assay, senescence assay, western blot, chip assay (non-TNBC cell lines: HEK-293, MCF7, SAOS-2; TNBC cell lines: MDA-MB-231, BT549) | [77] |
| miR-203 | Tumour suppressor-miR/targets BIRC5, LASP1/inhibits proliferation and migration | qPCR, transfection, proliferation and migration assays, luciferase reporter assay (non-TNBC cell lines: MCF-10A (normal); TNBC cell lines: MDA-MB-231, MDA-MB-468) | [78] |
| miR-31 | Tumour suppressor-miR/targets WAVE3, RhoA, Radexin, PRKCE/suppresses metastatic potential, induction of apoptosis, increase of chemo-sensitivity | Transfection, qPCR, dual luciferase reporter assays, invasion assay, western blot, apoptosis assay, viability assay (non-TNBC cell lines: T-47D, MCF7, MCF-10A; TNBC cell lines: MDA-MB-231, MDA-MB-435, BT549) | [79,80] |
| miR-34a | Tumour suppressor-miR/targets AXL/inhibits migration | Target prediction, qPCR, dual luciferase reporter assays, DNA capture assay, western blot, proliferation and migration assays, cell cycle analysis (non-TNBC cell lines: MCF7, SK-BR-3, T47D; TNBC cell lines: MDA-MB-231, BT549, Hs578T) | [81] |
| miR-181a/b | Onco-miR/targets Bim, ATM/inhibits anoikisis, impairment of DNA double strand break repairs | Transfection, miRNA microarray, 3D cell culture, proliferation, migration and invasion assays, qPCR, dual luciferase reporter assays, tumour growth and metastasis assay, cell cycle analysis (non-TNBC cell lines: NMuMG, MCF7, HEK 293GP, SUM159PT, OVCAR,HT29, PANC1, SK-Br-3; TNBC cell lines: MDA-MB-231, MDA-MB-468) | [82,83] |
| miR-146 | Onco-miR/targets BRCA1/effects BRCA1-mediated proliferation and homologous recombination | Target prediction, transfections, qPCR, northern blot, western blot, dual luciferase reporter assays, proliferation assay (TNBC cell lines: MDA-MB-436, MDA-MB-157) | [84] |
| miR-182 | Onco-miR/targets PFN1/increases proliferation and invasion, decreases apoptosis | Transfections, proliferation assay (MTT and flow cytometry), apoptosis assay, invasion assay, dual luciferase reporter assays, western blot (TNBC cell line MDA-MB-231) | [85] |
| miR-200 family | Inhibits cancer cell migration, invasion, if low → poor response to chemotherapy and radiotherapy | qPCR, dual luciferase reporter assays, immunoblot and immunofluorescence assay, migration assay, ChIP assay, viability assay, clonogenic assay, western blot (non-TNBC cell lines: NMuMG, HeLa, MCF7; TNBC cell lines: MDA-MB-231) | [86,87,88,89] |
| Let-7 family | Tumour suppressor-miRs ↓ in TNBC/target onco-genes RAS, MYC, HMGA2 | c. elegans, mice, cell culture, transfections | [90,91] |
| miR-15a,b, miR-16, miR-128 | Target Smurf2 (tumour suppressive ubiquitin) which down-regulates retinoblastoma (tumour suppressor) in TNBC | Immunohistochemistry, qPCR, transfection (non-TNBC cell lines: MCF-10A (normal breast), MCF9, T47D, SK-Br-3, BT747; TNBC cell lines: MDA-MB-231, MDA-MB-468, BT549, MDA-MB-436, DU4475) | [92] |
| miR-200c | Targets X-linked inhibitor of apoptosis (XIAP), what then suppresses proliferation in TNBC | Transfections, qPCR, colony formation assay, proliferation assay, flow cytometry, western blot, luciferase reporter assays, mice tumour model (non-TNBC cell lines: MCF-10A (normal breast); TNBC cell lines: MDA-MB-231) | [69] |
| miR-221 | Onco-miR/promotes tumourigenesis in TNBC/if knocked-down cell cycle progression and induction of apoptosis is inhibited | Transfections, qPCR, immunoblotting, proliferation, migration, invasion, and apoptosis assays, cell cycle analysis, mice tumour analysis (non-TNBC cell lines: SKBR3, MDA-MB-361, T47D, ZR75-1, MCF-7; TNBC cell lines: MDA-MB-231, Hs-578T, BT-20, and MDA-MB-468) | [93] |
| miR-31 | Antimetastatic-miR/when ↓ regulated in TNBC more metastases/down-regulated due to promoter methylation | qPCR, bisulfite-modified DNA for methylation analysis, DNA sequencing, methylation specific PCR (non-TNBC cell lines: MCF-10A (normal breast), MCF7, SKBR3, T47D; TNBC cell lines: MDA-MB-231, BT549, MDA-MB-4355) | [94] |
| miR-200b | Targets protein kinase Cα and suppresses metastasis in TNBC | qPCR, transfection, luciferase reporter assays, migration assay, mouse xenograft model, immunohistochemistry, western blot, pulldown assay, MTT assay, colony formation assay (non-TNBC cell lines: MCF-7, T-47D, BT-474, SKBR-3; TNBC cell lines: MDA-MB-468, BT-20, Hs578T and BT-549, MDA-MB-453) | [95] |
| miR-22, miR-27a, miR-206, miR-221/222, miR-302c | Associated with ER signalling and endocrine resistance | Immunohistochemistry, qPCR, transfections, clonogenicity assay, microarray, western blot, viability assay, luciferase reporter assays (non-TNBC cell lines: MCF-7, BT-474, T47D, SK-BR-3; TNBC cell lines: MDA-MB-231) | [96,97,98,99] |
| miR-125b, miR-134, miR-193a-5p, miR-199b-5p, miR-331-3p, miR-342-5p, miR-744* | Associated with HER2 signalling and trastuzumab resistance | luciferase reporter assays, northern and western blot, proliferation, migration and invasion assays, microarray, qPCR, transfections (non-TNBC cell lines: MCF-10A (normal breast), SK-BR-3, KPL-4, JIMT-1, MCF-7, BT-474; TNBC cell lines: MDA-MB-231) | [100,101,102] |
| miR-23b/27b/24 cluster | Promotes metastasis by targeting prosaposin (=metastasis-suppressive gene) | Microarrays, qPCR, migration assay, tumour xenografts, luciferase reporter assays, western blot (non-TNBC cell lines: HeLa; TNBC cell lines: MDA-MB-231, 67NR, 168FARN, 4TO7, 66cl4, 4T1) | [103] |
| miRNA | Result | Predictive/Prognostic | References |
|---|---|---|---|
| miR-200 family | Inhibits cancer cell migration, invasion, if low → poor response to chemotherapy and radiotherapy (non-TNBC based studies & TNBC study [88]) | predictive | [86,87,88,89] |
| miR-21 | Onco-miR/associated with poor prognosis/↑ expressed in TNBC | prognostic | [104] |
| miR-155 | Onco-miR/associated with poor prognosis, angiogenesis, tumour growth, metastases/controlled epigenetically by BRCA1↑ expressed in TNBC | prognostic | [105] |
| miR-16, miR-155, miR-374 | Prognostic miR/if ↑ associated with better prognosis (overall survival) (TNBC based study) | prognostic | [65] |
| miR-125b | Prognostic miR/if ↓ associated with poor prognosis (overall survival) (TNBC based study) | ||
| miR-125b, miR-655, miR-421 | Risk associated miRs/associated with distant disease free survival (TNBC based study) | ||
| miR-16, miR-374a,b, miR-497 | Protective miRs/associated with distant disease free survival (TNBC based study) | ||
| miR-210 | ↑ regulated in TNBC compared to ER+ breast cancers/associated with early relapse/low levels are associated with better disease free survival in TNBC | prognostic | [104,106,107] |
| miR-34b | Associated with p53-pathway/negative correlation with disease free survival and overall survival (TNBC based study) | prognostic | [108,109] |
| miR-376b, miR-409-5p, miR-410miR-193a-3p | Associated with worse breast cancer specific survival (TNBC based study) | prognostic | [73] |
| miR-16-2* ↑, miR-381 ↓, miR-409-5p ↓, miR-766 ↑ | Associated with better distant metastasis free survival (TNBC based study) | ||
| miR-766, miR-33b*, miR-550, miR-1539, miR-548d-5p, miR-16-2*, miR-563, miR-155* | Positively correlation with prognosis (TNBC based study) | ||
| miR-193a-3p, miR-432, miR-376b, miR-381, miR-409-5p, miR-410 | Negatively correlated with prognosis (TNBC based study) | ||
| miR-342, miR-150 | miRNAs for good prognosis (TNBC based study) | prognostic | [110] |
| miR-27b, miR-210, miR-144 | miRNAs for poor prognosis (TNBC based study) | ||
| miR-21 | Onco-miR/↑ regulated in TNBC/associated with poor prognosis, shorter recurrence-free survival and increased proliferation | prognostic | [111,112] |
| miR-155 | Onco-miR ↑ regulated in TNBC/targets tumour suppressor VHL and promotes angiogenesis/associated with poor prognosis | prognostic | [113] |
| miR-200b-3p ↑, miR-190a ↑, miR-512-5p ↓ | In this combination associated with better response to chemotherapy (TNBC based study) | predictive | [114] |
| miR-155-5p, miR-21-3p, miR-181a-5p, miR-181b-5p, miR-183-5p | ↑ regulated in TNBC/associated with chemoresistance | predictive | [115] |
| miR-10b-5p, miR-451a, miR-125b-5p, miR-31-5p, miR-195-5p, miR-130a-3p | ↓ regulated in TNBC/associated with chemoresistance | predictive | [115] |
| miR-155, miR-30e, miR-27a, miR-493 | Biomarkers dividing TNBC into low and high level risk groups | prognostic | [116] |
| miR-10b | ↑ in TNBC/promotes tumour invasion and metastasis/shorter progression free and overall survival/by targeting HoxD10 (which depresses expression of prometastatic gene RhoC) | prognostic | [76,117,118,119] |
| miR-374b-5p, miR-218-5p, miR-126-3p | When ↑ in TNBC associated with good prognosis | prognostic | [120] |
| miR-27b-3p | When ↓ in TNBC associated with good prognosis | prognostic |
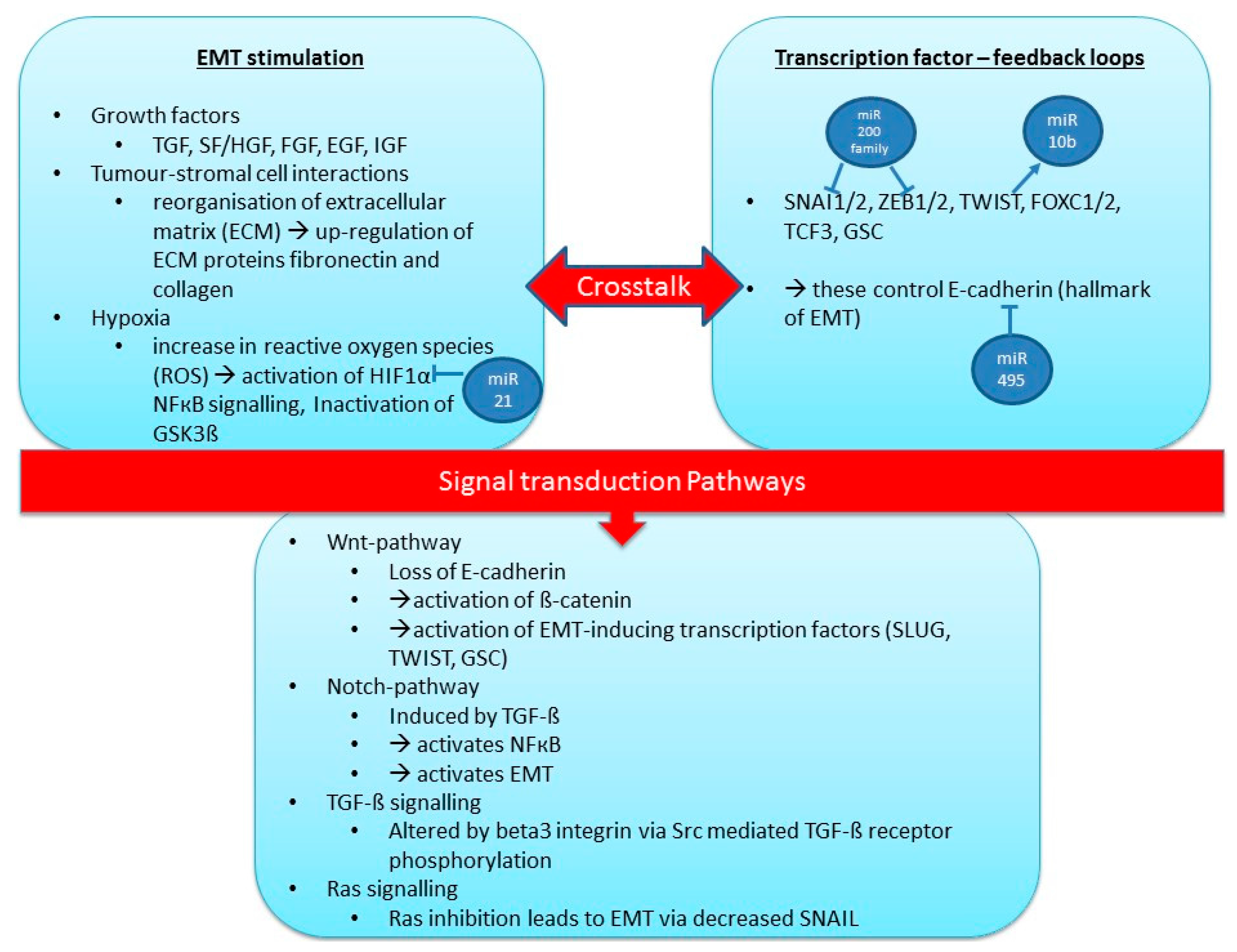
4.3. MicroRNAs and Metastasis/Epithelial to Mesenchymal Transition (EMT)

| EMT-Regulation | miRNA | Comments | References |
|---|---|---|---|
| Pro-EMT | miR-21 | Associated with invasive and metastatic breast cancer; regulates EMT and HIF-1a | [148,149,150] |
| miR-29 | Activates EMT by down-regulating peroxidasin homologue (cell adhesion molecule); down-regulation of DNA-methylation of tumour-suppressor genes; increasing chemosensitivity; targets EMT regulator N-myc | [151,152] | |
| miR-10b | Targets Tiam1-mediated Rac activation, which controls cell-cell adhesion and EMT through E-cadherin, leads to increased cell invasion and migration; Is activated by transcription factor TWIST (binds to promoter of miR-10b); expression increases during TGF-β induced EMT | [118,153,154] | |
| miR-9 | Is up-regulated in breast cancer, represses cadherin-1, which regulates cell adhesion and proliferation | [155,156] | |
| miR-206 | Suppresses proliferation, targets ER, SRC-1, SRC-2, GATA-3 (all estrogen signalling molecules) | [157,158,159] | |
| miR-221/222 | Increases proliferation in ER positive cell lines, targets ER, p27, p57 | [97,160,161] | |
| miR-495 | Targets E-cadherin, JAM-A, and REDD1 | [162,163] | |
| miR-181 | Targets PHLAD1 and ATM, associated with reduced survival in TNBC | [83,164] | |
| Pro-EMT (sometimes anti-EMT) | miR-17/92 cluster | Can act as tumour suppressor and oncogene, depending on microenvironment, mostly pro-metastatic, targets ER and SRC-3 | [165,166] |
| Anti-EMT | miR-130a | Targets ER, c-MET (onco-gene), down-regulates miR-221/222 | [167] |
| miR-145 | Acts as tumour suppressor, targets ER and MUC-1 (supports cell invasion) | [168,169,170] | |
| miR-7 | Targets SETDB1 → reduction of STAT1, Myc, Twist, and miR-9 | [171] | |
| miR-375 | Targets SHOX2 and IGFR, which leads to suppression of EMT | [172] |
4.4. Circulating miRNAs in Breast Cancer
| MicroRNA | Study Findings | References |
|---|---|---|
| miR-34a, miR-93, miR-373, miR-21, miR-155, miR-155, miR-181b, miR-24, miR-19a, miR-21, miR-106, miR-155, miR-29a, miR-21, miR-20a, miR-21 | Are up-regulated in breast cancer compared to healthy controls | [175,176,177,178,179,181,182,183] |
| miR-299-5p, miR-411, miR-126, miR-199a, miR-335, miR-181a, miR-1304 | Are down-regulated in breast cancer compared to healthy controls | [177,184,185] |
| miR-17, miR-155 (↑ in primary), miR-10b, miR-210, miR-214, miR-18b, miR-103, miR-107, miR-652, miR-101, miR-372, miR-373 | Discriminating primary tumour from metastatic tumour | [178,182,186,187,188,189] |
| miR-373, miR-17, miR-34a, miR-21, miR-126, miR-155, miR-199a, miR-335 | Associated with ER/PR/HER2 status | [177,178] |
| miR-210, miR-214, miR-10b, miR-373 | Associated with lymph node metastasis; miR-214 targets PTEN (tumour suppressor) | [76,182,187] |
| miR-200b, miR-18b, miR-103, miR-107, miR-652, miR-155 | Associated with survival | [180,188,190] |
| miR-210 ↓ (surgery), miR-214 ↓ (surgery), miR-155, miR-181b, miR-24 ↓ (surgery), miR-19a ↓ (therapy) | Levels of miRNA-expression change after surgery/therapy | [176,182,187] |
| miR-141, miR-200a,b,c, miR-203, miR-210, miR-375, miR-810 ↑, miR-768-3p ↓ | Altered in patients with circulating tumour cells (CTC) compared to patients without CTC | [190] |
| miR-210 | Higher in patients with residual disease than patients who achieved pathological complete response; Correlates with sensitivity to trastuzumab | [187] |
| miR-16, miR-21, miR-199a-5p | Lower in TNBC compared to non-TNBC; miR-199a-5p associated with tumour stage in TNBC | [191] |
| miR-373 | Exosomal levels higher in TNBC compared to luminal breast cancer | [186] |
| miR-127, miR-197, miR-222, miR-223 | Target CXCL12; are transposed via gap junctions from bone marrow to breast cancer cells and also through exosomes; → leading to cell quiescence, might contribute to dormancy of bone marrow metastasis | [192] |
| miR-223 | Macrophages secret microvesicles that contain this miRNA, promoting cell invasion | [193] |
| miR-222 | Chemoresistance is transmitted between breast cancer cells via exosomes with specific miRNAs | [194] |
| miR-105 | Is secreted in exosomes from metastatic breast cancer cells; targets ZO-1 (tight junction protein) → destroying tight junctions (barrier for metastasis). It’s over-expression induces metastasis | [195] |
| miR-155 | Targets TRF-1 (telomere sheltering function); high levels are associated with low TRF-1, metastasis-free survival, and relapse-free survival in ER+ cases. Reducing miR-155 improves telomere function and genomic stability | [180] |
4.5. miRNAs as Therapeutics
5. DNA Methylation
5.1. Epigenetic Gene Inactivation during Breast Cancer Development and Progression
5.2. DNA Methylation for Breast Cancer Subtype Classification
5.3. DNA Methylation in TNBC
6. Interactions between miRNAs and Epigenetic Mechanisms
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, P.; Savage, M.I.; Brown, P.H. Targeted therapy for breast cancer prevention. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Maxmen, A. The hard facts. Nature 2012, 485, S50–S51. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Traina, T.A. Triple-negative breast cancer: Adjuvant therapeutic options. Chemother. Res. Pract. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Wong-Brown, M.W.; Meldrum, C.J.; Carpenter, J.E.; Clarke, C.L.; Narod, S.A.; Jakubowska, A.; Rudnicka, H.; Lubinski, J.; Scott, R.J. Prevalence of BRCA1 and BRCA2 germline mutations in patients with triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 150, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Bihan, C.; Delaloge, S.; Andre, F. Triple-negative breast cancer: Are we making headway at least? Ther. Adv. Med. Oncol. 2012, 4, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.Y.; Lee, S.K.; Koo, M.Y.; Hur, S.M.; Choi, M.Y.; Cho, D.H.; Kim, S.; Choe, J.H.; Lee, J.E.; Kim, J.H.; et al. The prognoses of metaplastic breast cancer patients compared to those of triple-negative breast cancer patients. Breast Cancer Res. Treat. 2011, 126, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Kim, H.Y.; Nam, B.H.; Min, S.Y.; Lee, S.J.; Park, C.; Kwon, Y.; Kim, E.A.; Ko, K.L.; Shin, K.H.; et al. Worse prognosis of metaplastic breast cancer patients than other patients with triple-negative breast cancer. Breast Cancer Res. Treat. 2010, 120, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Honma, N.; Saji, S.; Kurabayashi, R.I.E.; Aida, J.; Arai, T.; Horii, R.I.E.; Akiyama, F.; Iwase, T.; Harada, N.; Younes, M.; et al. Oestrogen receptor-β1 but not oestrogen receptor-βcx is of prognostic value in apocrine carcinoma of the breast. APMIS 2008, 116, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Tutt, A.N. Triple negative tumours: A critical review. Histopathology 2008, 52, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the california cancer registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Hanna, W.M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Narod, S.A. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res. Treat. 2009, 115, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Parvin, F.; Peddi, M.J.E.; Cynthia, M. Molecular basis of triple negative breast cancer and implications for therapy. Int. J. Breast Cancer 2012, 2012. [Google Scholar] [CrossRef]
- Anders, C.K.; Winer, E.P.; Ford, J.M.; Dent, R.; Silver, D.P.; Sledge, G.W.; Carey, L.A. Poly(adp-ribose) polymerase inhibition: “Targeted” therapy for triple-negative breast cancer. Clin. Cancer Res. 2010, 16, 4702–4710. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Schwartzberg, L.; Danso, M.A.; Miller, K.D.; Rugo, H.S.; Neubauer, M.; Robert, N.; Hellerstedt, B.; Saleh, M.; Richards, P.; et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2014, 32, 3840–3847. [Google Scholar] [CrossRef] [PubMed]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtio, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Dieras, V.; Glaspy, J.; Brufsky, A.; Miller, K.; Miles, D.; Koralewski, P.; Phan, S.; Bhattacharya, S. Comparison of subgroup analyses of PFS from three phase III studies of bevacizumab in combination with chemotherapy in patients with HER2-negative metastatic breast cancer (MBC). Cancer Res. 2009, 69. [Google Scholar] [CrossRef]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gomez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Morgan, L.; Green, T.P.; Barrow, D.; Gee, J.; Nicholson, R.I. Elevated SRC activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res. Treat. 2006, 97, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S. Targeting src in breast cancer. Ann. Oncol. 2008, 19, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Fornier, M.N.; Morris, P.G.; Abbruzzi, A.; D’Andrea, G.; Gilewski, T.; Bromberg, J.; Dang, C.; Dickler, M.; Modi, S.; Seidman, A.D.; et al. A phase I study of dasatinib and weekly paclitaxel for metastatic breast cancer. Ann. Oncol. 2011, 22, 2575–2581. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.D.; Gucalp, A.; Traina, T.A. The role of the androgen receptor in triple-negative breast cancer. Womens Health 2013, 9, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Anestis, A.; Karamouzis, M.V.; Dalagiorgou, G.; Papavassiliou, A.G. Is androgen receptor targeting an emerging treatment strategy for triple negative breast cancer? Cancer Treat. Rev. 2015, 41, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor–positive, estrogen receptor–negative metastatic breast cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Barni, S. The value of platinum agents as neoadjuvant chemotherapy in triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2014, 144, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Eroles, P.; Bosch, A.; Alejandro Pérez-Fidalgo, J.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2011, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Lluch, A.; Albanell, J.; Barry, W.T.; Fan, C.; Chacon, J.I.; Parker, J.S.; Calvo, L.; Plazaola, A.; Arcusa, A.; et al. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br. J. Cancer 2014, 111, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Campone, M.; Valo, I.; Jezequel, P.; Moreau, M.; Boissard, A.; Campion, L.; Loussouarn, D.; Verriele, V.; Coqueret, O.; Guette, C. Prediction of recurrence and survival for triple-negative breast cancer by a protein signature in tissue samples. Mol. Cell. Proteom. 2015, 14, 2936–2946. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Sun, Q.; Liang, Z.; Cui, X.; Ren, X.; Chen, H.; Zhang, X.; Zhou, Y. A prognostic model of triple-negative breast cancer based on miR-27b-3p and node status. PLoS ONE 2014, 9, e100664. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.L.; Yang, W.T.; Rosen, D.G.; Landis, M.D.; Wong, H.; Lewis, M.T.; Creighton, C.J.; Sexton, K.R.; Hilsenbeck, S.G.; Sahin, A.A.; et al. Cancer stem cell markers are enriched in normal tissue adjacent to triple negative breast cancer and inversely correlated with DNA repair deficiency. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Leemans, C.R.; Brakenhoff, R.H. Using tissue adjacent to carcinoma as a normal control: An obvious but questionable practice. J. Pathol. 2004, 203, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.; Colacino, J.A.; Kim, J.H.; Rozek, L.S. Cancer epigenetics: A brief review. ILAR J. 2012, 53, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Beezhold, K.J.; Castranova, V.; Chen, F. Microprocessor of microRNAs: Regulation and potential for therapeutic intervention. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Andorfer, C.A.; Necela, B.M.; Thompson, E.A.; Perez, E.A. MicroRNA signatures: Clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- O’Day, E.; Lal, A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.Y.; Chang, D.C.; Lin, S.L. The microRNA (miRNA): Overview of the RNA genes that modulate gene function. Mol. Biotechnol. 2008, 38, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Diederichs, S. MicroRNA biogenesis and cancer. Methods Mol. Biol. 2011, 676, 3–22. [Google Scholar] [PubMed]
- McDaneld, T.G. MicroRNA: Mechanism of gene regulation and application to livestock. J. Anim. Sci. 2009, 87, E21–E28. [Google Scholar] [CrossRef] [PubMed]
- Rusk, N. When microRNAs activate translation. Nat. Methods 2008, 5, 122–123. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M. MicroRNAs: Oncogenes, tumor suppressors or master regulators of cancer heterogeneity? Biochim. Biophys. Acta 2010, 1805, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, M.; Tsuchiya, N.; Okamoto, K.; Nakagama, H. Systematic exploration of cancer-associated microRNA through functional screening assays. Cancer Sci. 2011, 102, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Baffa, R.; Fassan, M.; Volinia, S.; O’Hara, B.; Liu, C.G.; Palazzo, J.P.; Gardiman, M.; Rugge, M.; Gomella, L.G.; Croce, C.M.; et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009, 219, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Briskin, D.; Croce, C.M. MicroRNA in cancer: New hopes for antineoplastic chemotherapy. Ups. J. Med. Sci. 2012, 117, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, H.M.; Miller, N.; Lowery, A.J.; Sweeney, K.J.; Kerin, M.J. MicroRNAs as novel biomarkers for breast cancer. J. Oncol. 2009, 2009. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, S.S. Cancer biomarker profiling with microRNAs. Nat. Biotechnol. 2008, 26, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Braye, S.G.; Mathe, A.; Forbes, J.F.; Scott, R.J. Decreased expression of key tumour suppressor microRNAs is associated with lymph node metastases in triple negative breast cancer. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Cascione, L.; Gasparini, P.; Lovat, F.; Carasi, S.; Pulvirenti, A.; Ferro, A.; Alder, H.; He, G.; Vecchione, A.; Croce, C.M.; et al. Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLoS ONE 2013, 8, e55910. [Google Scholar] [CrossRef] [PubMed]
- Aydogdu, E.; Katchy, A.; Tsouko, E.; Lin, C.Y.; Haldosen, L.A.; Helguero, L.; Williams, C. MicroRNA-regulated gene networks during mammary cell differentiation are associated with breast cancer. Carcinogenesis 2012, 33, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Bert, A.G.; Goodall, G.J. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Han, X.; Yu, K.; Sun, S.; Zhen, L.; Li, Z.; Wang, S. MicroRNA-200c downregulates XIAP expression to suppress proliferation and promote apoptosis of triple-negative breast cancer cells. Mol. Med. Rep. 2014, 10, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Lowery, A.J.; Miller, N.; Devaney, A.; McNeill, R.E.; Davoren, P.A.; Lemetre, C.; Benes, V.; Schmidt, S.; Blake, J.; Ball, G.; et al. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res. 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.A.; Slewa, A.; Gudlaugsson, E.; Jonsdottir, K.; Skaland, I.; Soiland, H.; Baak, J.P. Biologic profiling of lymph node negative breast cancers by means of microRNA expression. Mod. Pathol. 2010, 23, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Christensen, M.; Silahtaroglu, A.; Bak, M.; Heath, C.V.; Schwartz, G.; Wells, W.; Kauppinen, S.; Cole, C.N. Altered microRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 2007, 67, 11612–11620. [Google Scholar] [CrossRef] [PubMed]
- De Rinaldis, E.; Gazinska, P.; Mera, A.; Modrusan, Z.; Fedorowicz, G.M.; Burford, B.; Gillett, C.; Marra, P.; Grigoriadis, A.; Dornan, D.; et al. Integrated genomic analysis of triple-negative breast cancers reveals novel microRNAs associated with clinical and molecular phenotypes and sheds light on the pathways they control. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Kolacinska, A.; Morawiec, J.; Pawlowska, Z.; Szemraj, J.; Szymanska, B.; Malachowska, B.; Morawiec, Z.; Morawiec-Sztandera, A.; Pakula, L.; Kubiak, R.; et al. Association of microRNA-93, 190, 200b and receptor status in core biopsies from stage III breast cancer patients. DNA Cell Biol. 2014, 33, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Berber, U.; Yilmaz, I.; Narli, G.; Haholu, A.; Kucukodaci, Z.; Demirel, D. miR-205 and miR-200c: Predictive micro RNAs for lymph node metastasis in triple negative breast cancer. J. Breast Cancer 2014, 17, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cai, F.; Zhang, B.; Barekati, Z.; Zhong, X.Y. The level of circulating miRNA-10b and miRNA-373 in detecting lymph node metastasis of breast cancer: Potential biomarkers. Tumour Biol. 2013, 34, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Piovan, C.; Palmieri, D.; di Leva, G.; Braccioli, L.; Casalini, P.; Nuovo, G.; Tortoreto, M.; Sasso, M.; Plantamura, I.; Triulzi, T.; et al. Oncosuppressive role of p53-induced miR-205 in triple negative breast cancer. Mol. Oncol. 2012, 6, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zheng, X.; Shen, C.; Shi, Y. MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2012, 31. [Google Scholar] [CrossRef] [PubMed]
- Koerner, C.; Keklikoglou, I.; Bender, C.; Woerner, A.; Muenstermann, E.; Wiemann, S. MicroRNA-31 sensitizes human breast cells to apoptosis by direct targeting of protein kinase C epsilon (PKCepsilon). J. Biol. Chem. 2013, 288, 8750–8761. [Google Scholar] [CrossRef] [PubMed]
- Sossey-Alaoui, K.; Downs-Kelly, E.; Das, M.; Izem, L.; Tubbs, R.; Plow, E.F. Wave3, an actin remodeling protein, is regulated by the metastasis suppressor microRNA, miR-31, during the invasion-metastasis cascade. Int. J. Cancer 2011, 129, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Huppi, K.; Pitt, J.J.; Dorsey, T.H.; Ambs, S.; Caplen, N.J. Identification of the receptor tyrosine kinase AXL in breast cancer as a target for the human miR-34a microRNA. Breast Cancer Res. Treat. 2011, 130, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Bisso, A.; Faleschini, M.; Zampa, F.; Capaci, V.; de Santa, J.; Santarpia, L.; Piazza, S.; Cappelletti, V.; Daidone, M.; Agami, R.; et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell Cycle 2013, 12, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Sossey-Alaoui, K.; Thompson, C.L.; Danielpour, D.; Schiemann, W.P. TGF-β upregulates mir-181a expression to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.I.; Buisson, M.; Bertrand, P.; Rimokh, R.; Rouleau, E.; Lopez, B.S.; Lidereau, R.; Mikaelian, I.; Mazoyer, S. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med. 2011, 3, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Li, X.; Zhang, Y.-J.; Li, J.; Zheng, Y.-Q.; Liu, M.; Song, X.; Li, X.-R. Expression and regulatory function of miRNA-182 in triple-negative breast cancer cells through its targeting of profilin 1. Tumor Biol. 2013, 34, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tian, W.; Cai, H.; He, H.; Deng, Y. Down-regulation of microRNA-200c is associated with drug resistance in human breast cancer. Med. Oncol. 2012, 29, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Liu, C.; Gao, F.; Mitchel, R.E.; Zhao, L.; Yang, Y.; Lei, J.; Cai, J. miR-200c enhances radiosensitivity of human breast cancer cells. J. Cell. Biochem. 2013, 114, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Boyerinas, B.; Park, S.M.; Hau, A.; Murmann, A.E.; Peter, M.E. The role of let-7 in cell differentiation and cancer. Endocr. Relat. Cancer 2010, 17, F19–F36. [Google Scholar] [CrossRef] [PubMed]
- Bussing, I.; Slack, F.J.; Grosshans, H. Let-7 microRNAs in development, stem cells and cancer. Trends Mol. Med. 2008, 14, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gu, X.; Sun, L.; Flowers, A.B.; Rademaker, A.W.; Zhou, Y.; Kiyokawa, H. Downregulation of Smurf2, a tumor-suppressive ubiquitin ligase, in triple-negative breast cancers: Involvement of the RB-microRNA axis. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Nassirpour, R.; Mehta, P.P.; Baxi, S.M.; Yin, M.J. miR-221 promotes tumorigenesis in human triple negative breast cancer cells. PLoS ONE 2013, 8, e62170. [Google Scholar] [CrossRef] [PubMed]
- Augoff, K.; McCue, B.; Plow, E.F.; Sossey-Alaoui, K. miR-31 and its host gene LncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol. Cancer 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Humphries, B.; Wang, Z.; Oom, A.L.; Fisher, T.; Tan, D.; Cui, Y.; Jiang, Y.; Yang, C. MicroRNA-200b targets protein kinase Cα and suppresses triple-negative breast cancer metastasis. Carcinogenesis 2014, 35, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Yang, Y.; Yang, X.; Zhao, L.; Lu, J.; Meng, Q.H. Downregulation of miR-221/222 enhances sensitivity of breast cancer cells to tamoxifen through upregulation of TIMP3. Cancer Gene Ther. 2014, 21, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; di Leva, G.; Li, M.; Fang, F.; Devlin, C.; Hartman-Frey, C.; Burow, M.E.; Ivan, M.; Croce, C.M.; Nephew, K.P. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 2011, 30, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Yu, D.; Wei, N.; Fu, H.; Cai, T.; Huang, Y.; Wu, C.; Zheng, X.; Du, Q.; Lin, D.; et al. An estrogen receptor α suppressor, microRNA-22, is downregulated in estrogen receptor alpha-positive human breast cancer cell lines and clinical samples. FEBS J. 2010, 277, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, N.; Toyama, T.; Takahashi, S.; Sugiura, H.; Endo, Y.; Iwasa, M.; Fujii, Y.; Yamashita, H. Distinct expressions of microRNAs that directly target estrogen receptor α in human breast cancer. Breast Cancer Res. Treat. 2011, 130, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhao, Y.; Guo, B. MiR-199b-5p targets HER2 in breast cancer cells. J. Cell. Biochem. 2013, 114, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Leivonen, S.K.; Sahlberg, K.K.; Makela, R.; Due, E.U.; Kallioniemi, O.; Borresen-Dale, A.L.; Perala, M. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol. Oncol. 2014, 8, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Goga, A.; Bhaumik, D.; Berger, C.E.; Sullivan, C.S.; Benz, C.C. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J. Biol. Chem. 2007, 282, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Qiu, Q.; Wei, Y.; Mercatali, L.; Ibrahim, T.; Amadori, D.; Kang, Y. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J. Biol. Chem. 2014, 289, 21888–21895. [Google Scholar] [CrossRef] [PubMed]
- Radojicic, J.; Zaravinos, A.; Vrekoussis, T.; Kafousi, M.; Spandidos, D.A.; Stathopoulos, E.N. MicroRNA expression analysis in triple-negative (ER, PR and HER2/neu) breast cancer. Cell Cycle 2011, 10, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Wang, R.H.; Akagi, K.; Kim, K.A.; Martin, B.K.; Cavallone, L.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer; Haines, D.C.; Basik, M.; Mai, P.; et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat. Med. 2011, 17, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Foekens, J.A.; Sieuwerts, A.M.; Smid, M.; Look, M.P.; de Weerd, V.; Boersma, A.W.; Klijn, J.G.; Wiemer, E.A.; Martens, J.W. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 13021–13026. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Kondo, N.; Endo, Y.; Sugiura, H.; Yoshimoto, N.; Iwasa, M.; Takahashi, S.; Fujii, Y.; Yamashita, H. High expression of microRNA-210 is an independent factor indicating a poor prognosis in Japanese triple-negative breast cancer patients. Jpn. J. Clin. Oncol. 2012, 42, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Sana, J.; Redova, M.; Navratil, J.; Palacova, M.; Fabian, P.; Slaby, O.; Vyzula, R. mir-34b is associated with clinical outcome in triple-negative breast cancer patients. Diagn. Pathol. 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Lowenstein, C.J. miR-34, SIRT1 and p53: The feedback loop. Cell Cycle 2009, 8, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.M.; Camps, C.; Winchester, L.; Snell, C.E.; Gee, H.E.; Sheldon, H.; Taylor, M.; Harris, A.L.; Ragoussis, J. MicroRNA-associated progression pathways and potential therapeutic targets identified by integrated mrna and microRNA expression profiling in breast cancer. Cancer Res. 2011, 71, 5635–5645. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Liang, X.; Wang, D.; Gao, H.; Wang, L.; Wang, L.; Liu, J.; Du, Z. High expression of miR-21 in triple-negative breast cancers was correlated with a poor prognosis and promoted tumor cell in vitro proliferation. Med. Oncol. 2014, 31. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, T.A.; Schwartz, G.N.; Calderone, H.M.; Graveel, C.R.; Winn, M.E.; Hostetter, G.; Wells, W.A.; Sempere, L.F. Stromal expression of miR-21 identifies high-risk group in triple-negative breast cancer. Am. J. Pathol. 2014, 184, 3217–3225. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; He, L.; Richards, E.J.; Challa, S.; Xu, C.X.; Permuth-Wey, J.; Lancaster, J.M.; Coppola, D.; Sellers, T.A.; Djeu, J.Y.; et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 2014, 33, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Kolacinska, A.; Morawiec, J.; Fendler, W.; Malachowska, B.; Morawiec, Z.; Szemraj, J.; Pawlowska, Z.; Chowdhury, D.; Choi, Y.E.; Kubiak, R.; et al. Association of microRNAs and pathologic response to preoperative chemotherapy in triple negative breast cancer: Preliminary report. Mol. Biol. Rep. 2014, 41, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Li, Y.; Ye, S.; Ma, J.; Lu, L.; Lv, W.; Chang, G.; Li, X.; Li, Q.; Wang, S.; et al. MicroRNA profiling implies new markers of chemoresistance of triple-negative breast cancer. PLoS ONE 2014, 9, e96228. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Cascione, L.; Fassan, M.; Lovat, F.; Guler, G.; Balci, S.; Irkkan, C.; Morrison, C.; Croce, C.M.; Shapiro, C.L.; et al. MicroRNA expression profiling identifies a four microRNA signature as a novel diagnostic and prognostic biomarker in triple negative breast cancers. Oncotarget 2014, 5, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef]
- Parrella, P.; Barbano, R.; Pasculli, B.; Fontana, A.; Copetti, M.; Valori, V.M.; Poeta, M.L.; Perrone, G.; Righi, D.; Castelvetere, M.; et al. Evaluation of microRNA-10b prognostic significance in a prospective cohort of breast cancer patients. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cai, Q.; Bao, P.P.; Su, Y.; Cai, H.; Wu, J.; Ye, F.; Guo, X.; Zheng, W.; Zheng, Y.; et al. Tumor tissue microRNA expression in association with triple-negative breast cancer outcomes. Breast Cancer Res. Treat. 2015, 152, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, Y.; Wang, Z.; Sarkar, F. Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: Are they cousins or twins? Cancers 2011, 3, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. β3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. BCR 2006, 8. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ueda, Y.; Akaboshi, S.; Hino, Y.; Sekita, Y.; Nakao, M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am. J. Pathol. 2009, 174, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Verschueren, K.; Yokoyama, K.; Nagayama, M.; Kamata, N. Involvement of ETS-1 transcription factor in inducing matrix metalloproteinase-2 expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Int. J. Oncol. 2006, 28, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Mamuya, F.A.; Duncan, M.K. aV integrins and TGF-β-induced EMT: A circle of regulation. J. Cell. Mol. Med. 2012, 16, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z. Role for α3 integrin in EMT and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, J.L.; Guala, A.S.; Leggett, S.E.; Sluimer, J.; Badura, E.C.; Janssen-Heininger, Y.M. Induction of a mesenchymal expression program in lung epithelial cells by Wnt/β-catenin requires the presence of c-Jun N-terminal kinase 1. Am. J. Respir. Cell Mol. Biol. 2012, 47, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011, 307, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Sarkar, S.H.; Bitar, B.; Ali, S.; Aboukameel, A.; Sethi, S.; Li, Y.; Bao, B.; Kong, D.; Banerjee, S.; et al. Garcinol regulates EMT and Wnt signaling pathways in vitro and in vivo leading to anticancer activity against breast cancer cells. Mol. Cancer Ther. 2012, 11, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Seike, M.; Soeno, C.; Mizutani, H.; Kitamura, K.; Minegishi, Y.; Noro, R.; Yoshimura, A.; Cai, L.; Gemma, A. MiR-23a regulates TGF-β-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells. Int. J. Oncol. 2012, 41, 869–875. [Google Scholar] [PubMed]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2012. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Liu, M.; Wang, Y.; Chen, X.; Xu, J.; Sun, Y.; Zhao, L.; Qu, H.; Fan, Y.; Wu, C. Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting pten. PLoS ONE 2012, 7, e39520. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. The miR-200 and miR-221/222 microRNA families: Opposing effects on epithelial identity. J. Mammary Gland Biol. Neoplasia 2012, 17, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Toiyama, Y.; Takahashi, M.; Balaguer, F.; Nagasaka, T.; Koike, J.; Hemmi, H.; Koi, M.; Boland, C.R.; Goel, A. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut 2013, 62, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Castilla, M.A.; Diaz-Martin, J.; Sarrio, D.; Romero-Perez, L.; Lopez-Garcia, M.A.; Vieites, B.; Biscuola, M.; Ramiro-Fuentes, S.; Isacke, C.M.; Palacios, J. MicroRNA-200 family modulation in distinct breast cancer phenotypes. PLoS ONE 2012, 7, e47709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volinia, S.; Galasso, M.; Sana, M.E.; Wise, T.F.; Palatini, J.; Huebner, K.; Croce, C.M. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, 3024–3029. [Google Scholar] [CrossRef] [PubMed]
- Dykxhoorn, D.M.; Wu, Y.; Xie, H.; Yu, F.; Lal, A.; Petrocca, F.; Martinvalet, D.; Song, E.; Lim, B.; Lieberman, J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS ONE 2009, 4, e7181. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Ell, B.J.; Buffa, F.M.; Ibrahim, T.; Blanco, M.A.; Celia-Terrassa, T.; Mercatali, L.; Khan, Z.; Goodarzi, H.; Hua, Y.; et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 2011, 17, 1101–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.N.; Cochrane, D.R.; Cittelly, D.M.; Richer, J.K. MiR-200c targets a NF-κB up-regulated Trkb/NTF3 autocrine signaling loop to enhance anoikis sensitivity in triple negative breast cancer. PLoS ONE 2012, 7, e49987. [Google Scholar] [CrossRef] [PubMed]
- Baulida, J.; Garcia de Herreros, A. Snail1-driven plasticity of epithelial and mesenchymal cells sustains cancer malignancy. Biochim. Biophys. Acta 2015, 1856, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.H.; Zhou, H.C.; Zhang, C.; Shang, L.R.; Zhang, L.; Xu, J.; Zheng, L.; Yuan, Y.; Guo, R.P.; Jia, W.H.; et al. A novel vascular pattern promotes metastasis of hepatocellular carcinoma in an epithelial-mesenchymal transition-independent manner. Hepatology 2015, 62, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, Y.; Nilsson, M.; Sundfeldt, K. TGF-β isoforms induce emt independent migration of ovarian cancer cells. Cancer Cell Int. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Wang, Y.; Liu, M.; Bi, X.; Bao, J.; Zeng, N.; Zhu, Z.; Mo, Z.; Wu, C.; Chen, X. MiR-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1α expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci. 2012, 103, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Wu, F.; Loeb, G.B.; Hsu, R.; Heidersbach, A.; Brincat, A.; Horiuchi, D.; Lebbink, R.J.; Mo, Y.Y.; Goga, A.; et al. Up-regulation of miR-21 by HER2/neu signaling promotes cell invasion. J. Biol.Chem. 2009, 284, 18515–18524. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.X.; Huang, X.F.; Shao, Q.; Huang, M.Y.; Deng, L.; Wu, Q.L.; Zeng, Y.X.; Shao, J.Y. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA 2008, 14, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, G.; Wu, J.H.; Jiang, C.P. Diverse roles of miR-29 in cancer (review). Oncol. Rep. 2014, 31, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.W., 3rd; Pruitt, H.C.; Metge, B.J.; Mitra, A.; Bailey, S.K.; Bae, S.; Singh, K.P.; Devine, D.J.; Dyess, D.L.; Richards, W.O.; et al. MicroRNA-29 negatively regulates EMT regulator N-myc interactor in breast cancer. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yan, S.; Weijie, Z.; Feng, W.; Liuxing, W.; Mengquan, L.; Qingxia, F. Critical role of miR-10b in transforming growth factor-β1-induced epithelial-mesenchymal transition in breast cancer. Cancer Gene Ther. 2014, 21, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Minard, M.E.; Ellis, L.M.; Gallick, G.E. Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin. Exp. Metastasis 2006, 23, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Gwak, J.M.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Yun, S.; Seo, A.N.; Lee, H.J.; Park, S.Y. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res. Treat. 2014, 147, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-α (ERα) and represses ERα messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 expression is down-regulated in estrogen receptor α-positive human breast cancer. Cancer Res. 2008, 68, 5004–5008. [Google Scholar] [CrossRef] [PubMed]
- Guttilla, I.K.; Phoenix, K.N.; Hong, X.; Tirnauer, J.S.; Claffey, K.P.; White, B.A. Prolonged mammosphere culture of MCF-7 cells induces an emt and repression of the estrogen receptor by microRNAs. Breast Cancer Res. Treat. 2012, 132, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptor α and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2008, 283, 31079–31086. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Nie, W.; Li, J.; Zhang, Y.; Yan, X.; Guan, X.; Chen, X.; Zen, K.; Zhang, C.Y.; Jiang, X.; et al. MicroRNA-495 induces breast cancer cell migration by targeting JAM-A. Protein Cell 2014, 5, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Hwang-Verslues, W.W.; Chang, P.H.; Wei, P.C.; Yang, C.Y.; Huang, C.K.; Kuo, W.H.; Shew, J.Y.; Chang, K.J.; Lee, E.Y.; Lee, W.H. MiR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and redd1. Oncogene 2011, 30, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Neel, J.C.; Lebrun, J.J. Activin and TGFβ regulate expression of the microRNA-181 family to promote cell migration and invasion in breast cancer cells. Cell Signal. 2013, 25, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Castellano, L.; Giamas, G.; Jacob, J.; Coombes, R.C.; Lucchesi, W.; Thiruchelvam, P.; Barton, G.; Jiao, L.R.; Wait, R.; Waxman, J.; et al. The estrogen receptor-α-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA 2009, 106, 15732–15737. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wang, C.; Wang, M.; Li, Z.; Casimiro, M.C.; Liu, M.; Wu, K.; Whittle, J.; Ju, X.; Hyslop, T.; et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J. Cell Biol. 2008, 182, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Acunzo, M.; Visone, R.; Romano, G.; Veronese, A.; Lovat, F.; Palmieri, D.; Bottoni, A.; Garofalo, M.; Gasparini, P.; Condorelli, G.; et al. miR-130a targets met and induces trail-sensitivity in NSCLC by downregulating miR-221 and 222. Oncogene 2012, 31, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Gotte, M.; Mohr, C.; Koo, C.Y.; Stock, C.; Vaske, A.K.; Viola, M.; Ibrahim, S.A.; Peddibhotla, S.; Teng, Y.H.; Low, J.Y.; et al. miR-145-dependent targeting of junctional adhesion molecule A and modulation of fascin expression are associated with reduced breast cancer cell motility and invasiveness. Oncogene 2010, 29, 6569–6580. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Mo, Y.Y. MicroRNA-145 suppresses cell invasion and metastasis by directly targeting mucin 1. Cancer Res. 2010, 70, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, R.; Nicoloso, M.S.; Lupini, L.; Lu, Y.; Fogarty, J.; Rossi, S.; Zagatti, B.; Fabbri, M.; Veronese, A.; Liu, X.; et al. miR-145 participates with TP53 in a death-promoting regulatory loop and targets estrogen receptor-α in human breast cancer cells. Cell Death Differ. 2010, 17, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, K.; Wang, J.; Wang, X.; Cheng, K.; Shi, F.; Jiang, L.; Zhang, Y.; Dou, J. MiR-7, inhibited indirectly by lincrna hotair, directly inhibits SETDB1 and reverses the emt of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 2014, 32, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Noh, H.; Teng, Y.; Shao, J.; Rehmani, H.; Ding, H.F.; Dong, Z.; Su, S.B.; Shi, H.; Kim, J.; et al. SHOX2 is a direct miR-375 target and a novel epithelial-to-mesenchymal transition inducer in breast cancer cells. Neoplasia 2014, 16, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Welsh, J.W.; Calin, G.A. Circulating microRNAs as noninvasive biomarkers in breast cancer. Recent Results Cancer Res. 2012, 195, 151–161. [Google Scholar] [PubMed]
- Roth, C.; Rack, B.; Muller, V.; Janni, W.; Pantel, K.; Schwarzenbach, H. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. BCR 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Sochor, M.; Basova, P.; Pesta, M.; Dusilkova, N.; Bartos, J.; Burda, P.; Pospisil, V.; Stopka, T. Oncogenic microRNAs: miR-155, miR-19a, miR-181b, and miR-24 enable monitoring of early breast cancer in serum. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zheng, Z.; Guo, J.; Ding, X. Correlation and quantitation of microRNA aberrant expression in tissues and sera from patients with breast tumor. Gynecol. Oncol. 2010, 119, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Eichelser, C.; Flesch-Janys, D.; Chang-Claude, J.; Pantel, K.; Schwarzenbach, H. Deregulated serum concentrations of circulating cell-free microRNAs miR-17, miR-34a, miR-155, and miR-373 in human breast cancer development and progression. Clin. Chem. 2013, 59, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Hou, J.; Jin, W.; Li, J.; Yue, Y.; Jin, H.; Wang, X. Increased circulating microRNA-155 as a potential biomarker for breast cancer screening: A meta-analysis. Molecules 2014, 19, 6282–6293. [Google Scholar] [CrossRef] [PubMed]
- Dinami, R.; Ercolani, C.; Petti, E.; Piazza, S.; Ciani, Y.; Sestito, R.; Sacconi, A.; Biagioni, F.; le Sage, C.; Agami, R.; et al. miR-155 drives telomere fragility in human breast cancer by targeting TRF1. Cancer Res. 2014, 74, 4145–4156. [Google Scholar] [CrossRef] [PubMed]
- Asaga, S.; Kuo, C.; Nguyen, T.; Terpenning, M.; Giuliano, A.E.; Hoon, D.S. Direct serum assay for microRNA-21 concentrations in early and advanced breast cancer. Clin. Chem. 2011, 57, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Milde-Langosch, K.; Steinbach, B.; Muller, V.; Pantel, K. Diagnostic potential of PTEN-targeting miR-214 in the blood of breast cancer patients. Breast Cancer Res. Treat. 2012, 134, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Lu, Z.; Li, H.; Lu, J.; Guo, L.; Ge, Q. Next-generation sequencing of microRNAs for breast cancer detection. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Van Schooneveld, E.; Wouters, M.C.; van der Auwera, I.; Peeters, D.J.; Wildiers, H.; van Dam, P.A.; Vergote, I.; Vermeulen, P.B.; Dirix, L.Y.; van Laere, S.J. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. BCR 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shen, J.; Medico, L.; Wang, D.; Ambrosone, C.B.; Liu, S. A pilot study of circulating miRNAs as potential biomarkers of early stage breast cancer. PLoS ONE 2010, 5, e13735. [Google Scholar] [CrossRef] [PubMed]
- Eichelser, C.; Stuckrath, I.; Muller, V.; Milde-Langosch, K.; Wikman, H.; Pantel, K.; Schwarzenbach, H. Increased serum levels of circulating exosomal microRNA-373 in receptor-negative breast cancer patients. Oncotarget 2014, 5, 9650–9663. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Santarpia, L.; Kim, J.; Esteva, F.J.; Moretti, E.; Buzdar, A.U.; di Leo, A.; Le, X.F.; Bast, R.C., Jr.; Park, S.T.; et al. Plasma microRNA 210 levels correlate with sensitivity to trastuzumab and tumor presence in breast cancer patients. Cancer 2012, 118, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- Kleivi-Sahlberg, K.; Bottai, G.; Naume, B.; Burwinkel, B.; Calin, G.A.; Borresen-Dale, A.L.; Santarpia, L. A serum microRNA signature predicts tumor relapse and survival in triple-negative breast cancer patients. Clin. Cancer Res. 2015, 21, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.L.; Hu, G.D.; Wang, X.F.; Zhang, X.H.; Zhang, Y.K.; Yu, Z.S. Serum overexpression of microRNA-10b in patients with bone metastatic primary breast cancer. J. Int. Med. Res. 2012, 40, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, D.; Zucknick, M.; Wallwiener, M.; Cuk, K.; Modugno, C.; Scharpff, M.; Schott, S.; Heil, J.; Turchinovich, A.; Yang, R.; et al. Circulating mirnas as surrogate markers for circulating tumor cells and prognostic markers in metastatic breast cancer. Clin. Cancer Res. 2012, 18, 5972–5982. [Google Scholar] [CrossRef] [PubMed]
- Shin, V.Y.; Siu, J.M.; Cheuk, I.; Ng, E.K.; Kwong, A. Circulating cell-free mirnas as biomarker for triple-negative breast cancer. Br. J. Cancer 2015, 112, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.X.; Liu, X.M.; Lv, M.M.; Chen, L.; Zhao, J.H.; Zhong, S.L.; Ji, M.H.; Hu, Q.; Luo, Z.; Wu, J.Z.; et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS ONE 2014, 9, e95240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Pecot, C.V.; Calin, G.A.; Coleman, R.L.; Lopez-Berestein, G.; Sood, A.K. RNA interference in the clinic: Challenges and future directions. Nat. Rev. Cancer 2011, 11, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Lee, S.K.; Wang, J.; Ince, N.; Ouyang, N.; Min, J.; Chen, J.; Shankar, P.; Lieberman, J. RNA interference targeting fas protects mice from fulminant hepatitis. Nat. Med. 2003, 9, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Nana-Sinkam, S.P.; Croce, C.M. MicroRNAs as therapeutic targets in cancer. Transl. Res. Lab. Clin. Med. 2011, 157, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. LncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Petrs-Silva, H.; Linden, R. Advances in recombinant adeno-associated viral vectors for gene delivery. Curr. Gene Ther. 2013, 13, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.W.; Chang, T.C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Trepel, M.; Korbelin, J.; Spies, E.; Heckmann, M.B.; Hunger, A.; Fehse, B.; Katus, H.A.; Kleinschmidt, J.A.; Muller, O.J.; Michelfelder, S. Treatment of multifocal breast cancer by systemic delivery of dual-targeted adeno-associated viral vectors. Gene Ther. 2015, 22, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Profumo, V.; Folini, M.; Zaffaroni, N. MicroRNAs as new therapeutic targets and tools in cancer. Expert Opin. Ther. Targets 2011, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Muthiah, M.; Park, I.K.; Cho, C.S. Nanoparticle-mediated delivery of therapeutic genes: Focus on miRNA therapeutics. Expert Opin. Drug Deliv. 2013, 10, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson, W.E., 3rd; Haque, F.; Xu, R.; Guo, P. Systemic delivery of anti-miRNA for suppression of triple negative breast cancer utilizing RNA nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.A.; Tang, T.; Wang, S.C.; Ptak, C.; Oh, G.H.; Wong, A.H.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C.; et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Altun, G.; Loring, J.F.; Laurent, L.C. DNA methylation in embryonic stem cells. J. Cell. Biochem. 2010, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Lopez-Serra, L.; Esteller, M. Proteins that bind methylated DNA and human cancer: Reading the wrong words. Br. J. Cancer 2008, 98, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.A.; Khamis, A.M.; Kulakovskiy, I.V.; Ba-Alawi, W.; Bhuyan, M.S.; Kawaji, H.; Lassmann, T.; Harbers, M.; Forrest, A.R.; Bajic, V.B. Effects of cytosine methylation on transcription factor binding sites. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.; Molloy, P.L. Cytosine methylation prevents binding to DNA of a hela cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Park, I.-H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific cpg island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Sanchez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Berman, H.; Zhang, J.; Crawford, Y.G.; Gauthier, M.L.; Fordyce, C.A.; McDermott, K.M.; Sigaroudinia, M.; Kozakiewicz, K.; Tlsty, T.D. Genetic and epigenetic changes in mammary epithelial cells identify a subpopulation of cells involved in early carcinogenesis. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.E.; Dallol, A.; Bieche, I.; Krex, D.; Morton, D.; Maher, E.R.; Latif, F. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br. J. Cancer 2004, 91, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.S.; Perry, M.R.; Laux, D.E.; Asare, A.L.; Caldwell, C.W.; Huang, T.H. CpG island arrays: An application toward deciphering epigenetic signatures of breast cancer. Clin. Cancer Res. 2000, 6, 1432–1438. [Google Scholar] [PubMed]
- Widschwendter, M.; Siegmund, K.D.; Muller, H.M.; Fiegl, H.; Marth, C.; Muller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef] [PubMed]
- Keshet, I.; Schlesinger, Y.; Farkash, S.; Rand, E.; Hecht, M.; Segal, E.; Pikarski, E.; Young, R.A.; Niveleau, A.; Cedar, H.; et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 2006, 38, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Ruike, Y.; Imanaka, Y.; Sato, F.; Shimizu, K.; Tsujimoto, G. Genome-wide analysis of aberrant methylation in human breast cancer cells using methyl-DNA immunoprecipitation combined with high-throughput sequencing. BMC Genom. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.M.; Dobrovic, A.; Fox, S.B. DNA methylation in ductal carcinoma in situ of the breast. Breast Cancer Res. BCR 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Muggerud, A.A.; Ronneberg, J.A.; Warnberg, F.; Botling, J.; Busato, F.; Jovanovic, J.; Solvang, H.; Bukholm, I.; Borresen-Dale, A.L.; Kristensen, V.N.; et al. Frequent aberrant DNA methylation of ABCB1, FOXC1, PPP2R2B and PTEN in ductal carcinoma in situ and early invasive breast cancer. Breast Cancer Res. BCR 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Van Hoesel, A.Q.; Sato, Y.; Elashoff, D.A.; Turner, R.R.; Giuliano, A.E.; Shamonki, J.M.; Kuppen, P.J.; van de Velde, C.J.; Hoon, D.S. Assessment of DNA methylation status in early stages of breast cancer development. Br. J. Cancer 2013, 108, 2033–2038. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, T.; Frigessi, A.; Johnson, K.C.; Edvardsen, H.; Touleimat, N.; Klajic, J.; Riis, M.L.; Haakensen, V.D.; Warnberg, F.; Naume, B.; et al. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014, 15. [Google Scholar] [CrossRef]
- Bediaga, N.G.; Acha-Sagredo, A.; Guerra, I.; Viguri, A.; Albaina, C.; Ruiz Diaz, I.; Rezola, R.; Alberdi, M.J.; Dopazo, J.; Montaner, D.; et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. BCR 2010, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, K.; Hegardt, C.; Staaf, J.; Vallon-Christersson, J.; Jonsson, G.; Olsson, H.; Borg, A.; Ringner, M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. BCR 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Ronneberg, J.A.; Fleischer, T.; Solvang, H.K.; Nordgard, S.H.; Edvardsen, H.; Potapenko, I.; Nebdal, D.; Daviaud, C.; Gut, I.; Bukholm, I.; et al. Methylation profiling with a panel of cancer related genes: Association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol. Oncol. 2011, 5, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Cocciardi, S.; Waddell, N.; Johnstone, C.N.; Marsh, A.; Henderson, S.; Simpson, P.; da Silva, L.; Khanna, K.; Lakhani, S.; et al. DNA methylome of familial breast cancer identifies distinct profiles defined by mutation status. Am. J. Hum. Genet. 2010, 86, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Edmiston, S.N.; May, R.; Kuan, P.F.; Chu, H.; Bryant, C.; Tse, C.K.; Swift-Scanlan, T.; Geradts, J.; Troester, M.A.; et al. DNA methylation profiling in the carolina breast cancer study defines cancer subclasses differing in clinicopathologic characteristics and survival. Breast Cancer Res. BCR 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, O.A.; Moran, S.; Gomez, A.; Sayols, S.; Arribas-Jorba, C.; Sandoval, J.; Hilmarsdottir, H.; Olafsdottir, E.; Tryggvadottir, L.; Jonasson, J.G.; et al. A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol. Oncol. 2015, 9, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Jing, F.; Yuping, W.; Yong, C.; Jie, L.; Jun, L.; Xuanbing, T.; Lihua, H. CpG island methylator phenotype of multigene in serum of sporadic breast carcinoma. Tumour Biol. 2010, 31, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Radpour, R.; Barekati, Z.; Kohler, C.; Lv, Q.; Burki, N.; Diesch, C.; Bitzer, J.; Zheng, H.; Schmid, S.; Zhong, X.Y. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS ONE 2011, 6, e16080. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeck, J.; Ropero, S.; Setien, F.; Gonzalez-Suarez, E.; Osorio, A.; Benitez, J.; Herman, J.G.; Esteller, M. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J. Clin. Oncol. 2010, 28, e563–e564. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, O.A.; Jonasson, J.G.; Olafsdottir, K.; Hilmarsdottir, H.; Olafsdottir, G.; Esteller, M.; Johannsson, O.T.; Eyfjord, J.E. CpG island hypermethylation of BRCA1 and loss of PRB as co-occurring events in basal/triple-negative breast cancer. Epigenetics 2011, 6, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Maeda, I.; Oikawa, R.; Wu, W.; Tsuchiya, K.; Miyoshi, Y.; Itoh, F.; Tsugawa, K.; Ohta, T. Aberrant DNA methylation status of DNA repair genes in breast cancer treated with neoadjuvant chemotherapy. Genes Cells 2013, 18, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Stecklein, S.R.; Kimler, B.F.; Sethi, G.; Petroff, B.K.; Phillips, T.A.; Tawfik, O.W.; Godwin, A.K.; Jensen, R.A. The prognostic value of promoter methylation in early stage triple negative breast cancer. J. Cancer Ther. Res. 2014, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Diao, L.; Chen, Y.; Liu, Y.; Wang, C.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; et al. Promoter methylation of BRCA1 in triple-negative breast cancer predicts sensitivity to adjuvant chemotherapy. Ann. Oncol. 2013, 24, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, T.; Poehlmann, A.; Ignatov, A.; Schinlauer, A.; Costa, S.D.; Roessner, A.; Kalinski, T.; Bischoff, J. BRCA1 promoter methylation is a marker of better response to anthracycline-based therapy in sporadic tnbc. Breast Cancer Res. Treat. 2013, 141, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Duursma, A.M.; Kedde, M.; Schrier, M.; le Sage, C.; Agami, R. miR-148 targets human DNMT3b protein coding region. RNA 2008, 14, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3a and 3b. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.Y.; Lin, S.L. Intronic microRNAs. Biochem. Biophys. Res. Commun. 2005, 326, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Wee, E.J.H.; Peters, K.; Nair, S.S.; Hulf, T.; Stein, S.; Wagner, S.; Bailey, P.; Lee, S.Y.; Qu, W.J.; Brewster, B.; et al. Mapping the regulatory sequences controlling 93 breast cancer-associated miRNA genes leads to the identification of two functional promoters of the hsa-miR-200b cluster, methylation of which is associated with metastasis or hormone receptor status in advanced breast cancer. Oncogene 2012, 31, 4182–4195. [Google Scholar] [PubMed]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Muller, M.; Romermann, D.; Langer, F.; Kreipe, H. Epigenetic inactivation of microRNA gene hsa-miR-9-1 in human breast cancer. J. Pathol. 2008, 214, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sanchez-Cespedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene bcl6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Calin, G.A. Epigenetics and miRNAs in human cancer. Adv. Genet. 2010, 70, 87–99. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathe, A.; Scott, R.J.; Avery-Kiejda, K.A. miRNAs and Other Epigenetic Changes as Biomarkers in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2015, 16, 28347-28376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226090
Mathe A, Scott RJ, Avery-Kiejda KA. miRNAs and Other Epigenetic Changes as Biomarkers in Triple Negative Breast Cancer. International Journal of Molecular Sciences. 2015; 16(12):28347-28376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226090
Chicago/Turabian StyleMathe, Andrea, Rodney J. Scott, and Kelly A. Avery-Kiejda. 2015. "miRNAs and Other Epigenetic Changes as Biomarkers in Triple Negative Breast Cancer" International Journal of Molecular Sciences 16, no. 12: 28347-28376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226090