
The Intrinsic Dynamics and Unfolding Process of an Antibody Fab Fragment Revealed by Elastic Network Model

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion
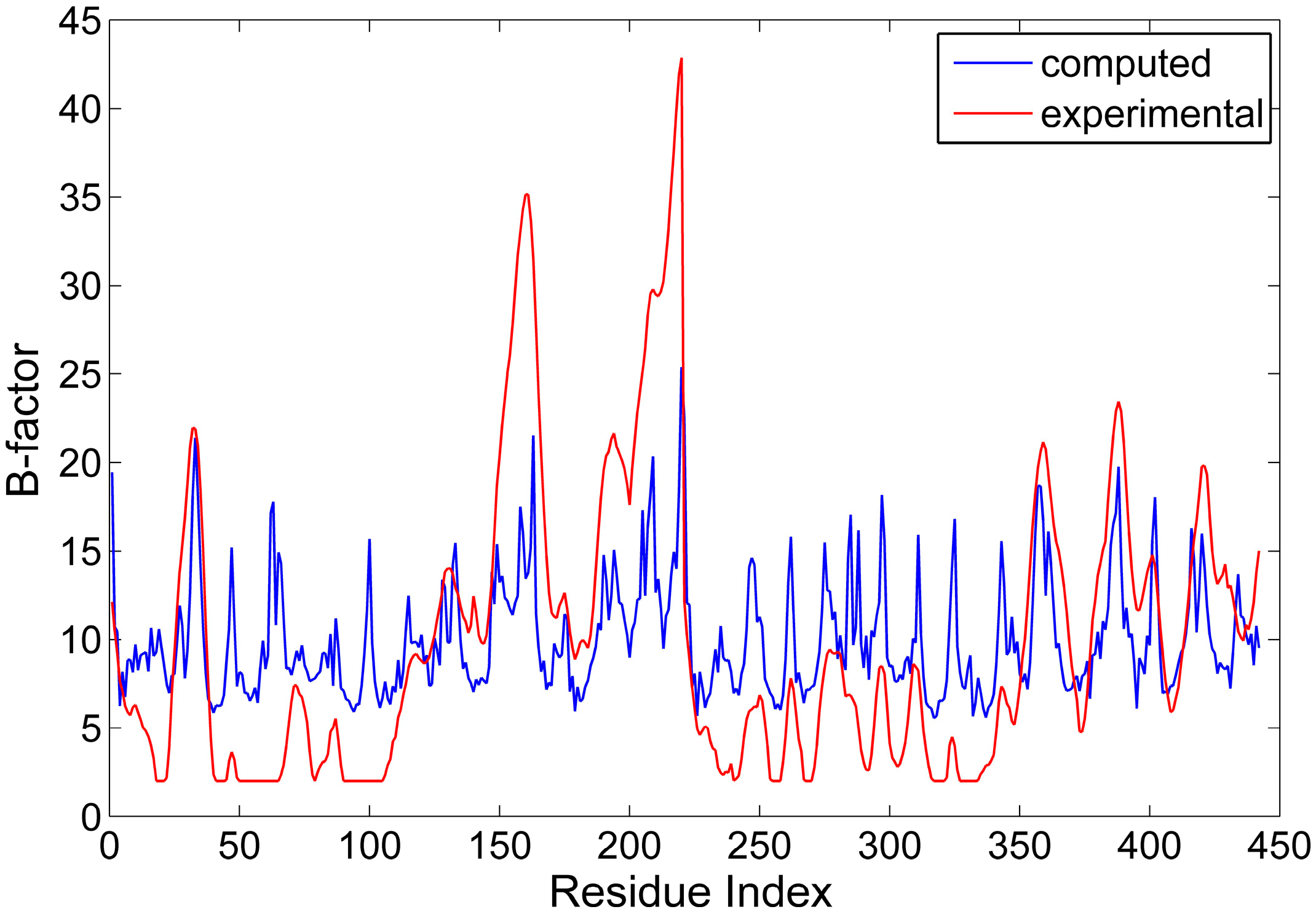
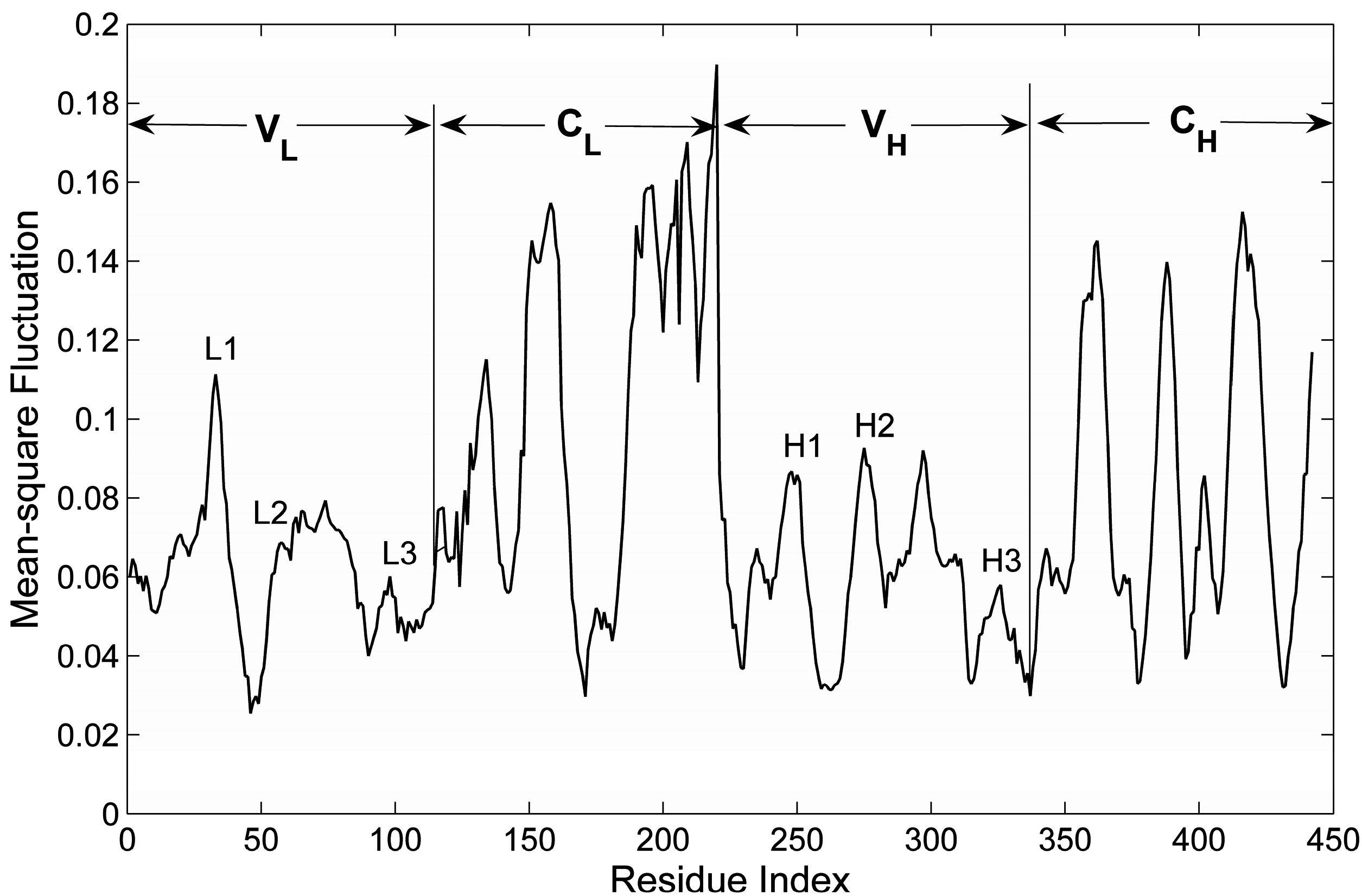
2.1. The Comparison between the Computed and Experimental B-Factors

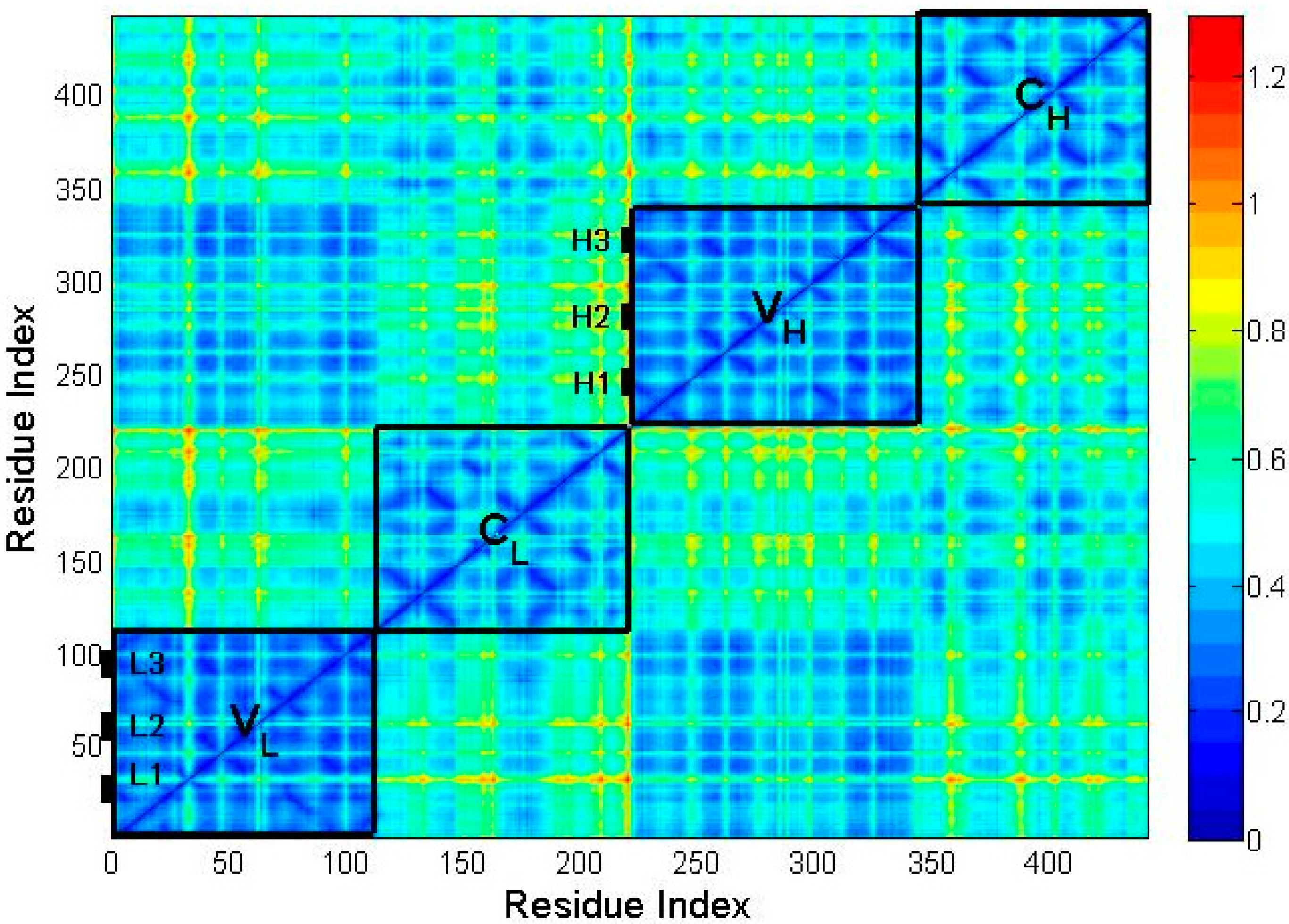
2.2. The Intrinsic Collective Dynamics of the Fab Fragment of McPC603

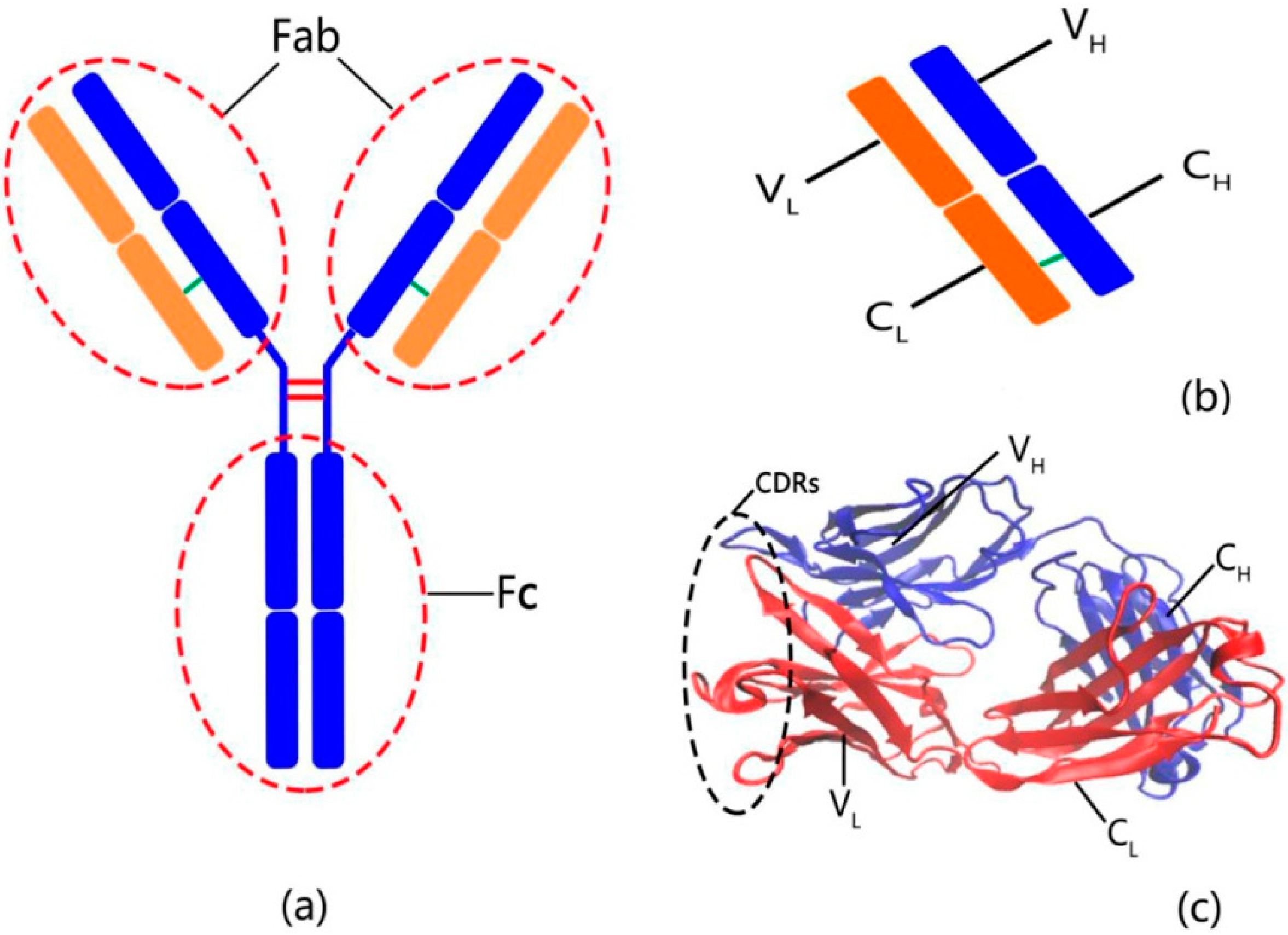
2.3. The Flexibility of Different Parts in the Structure of the Fab Fragment of McPC603

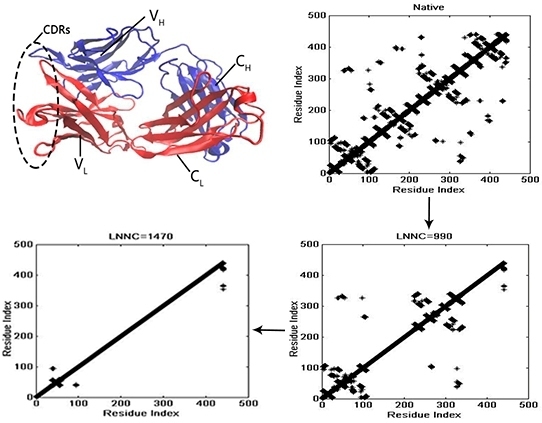
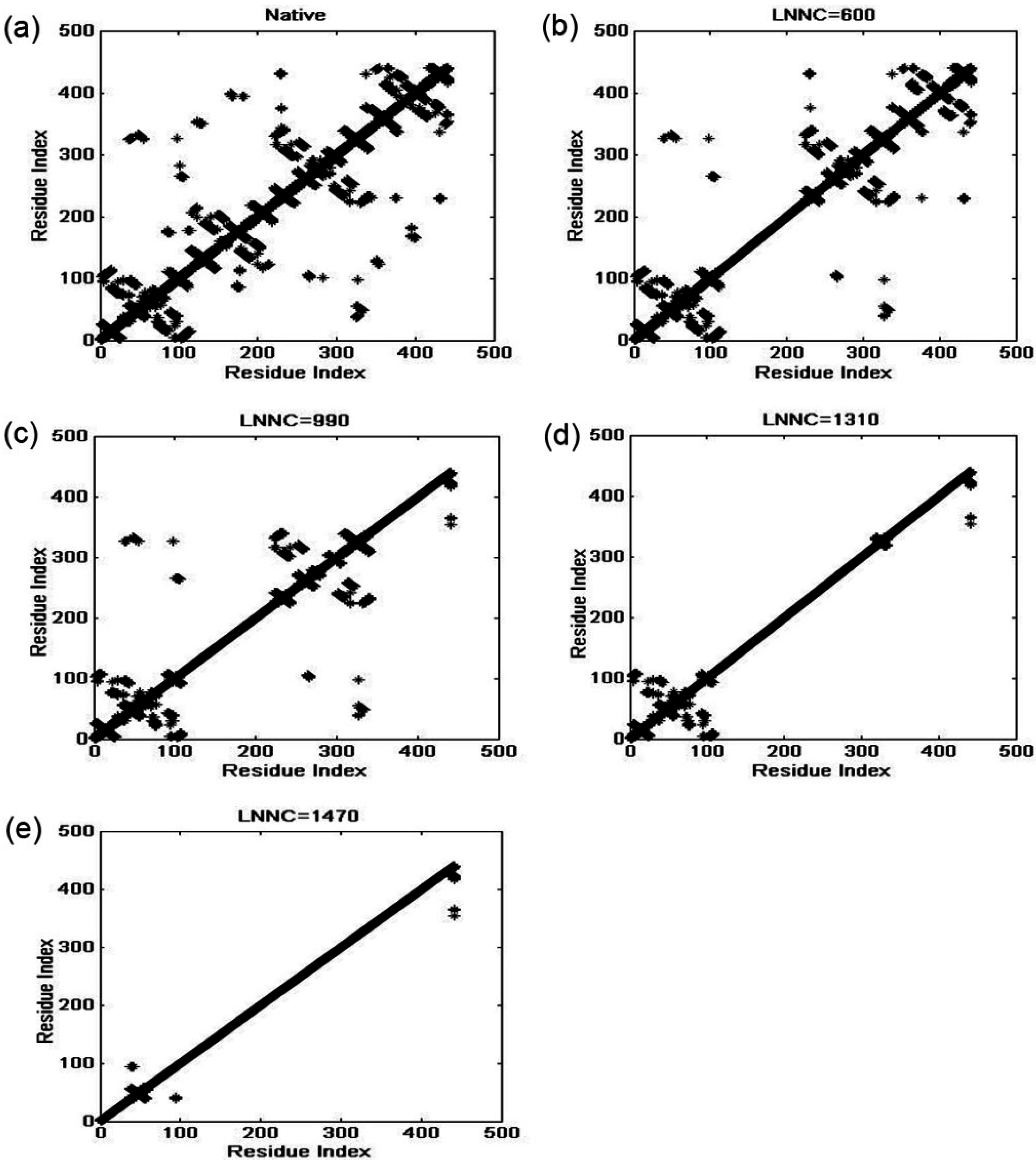
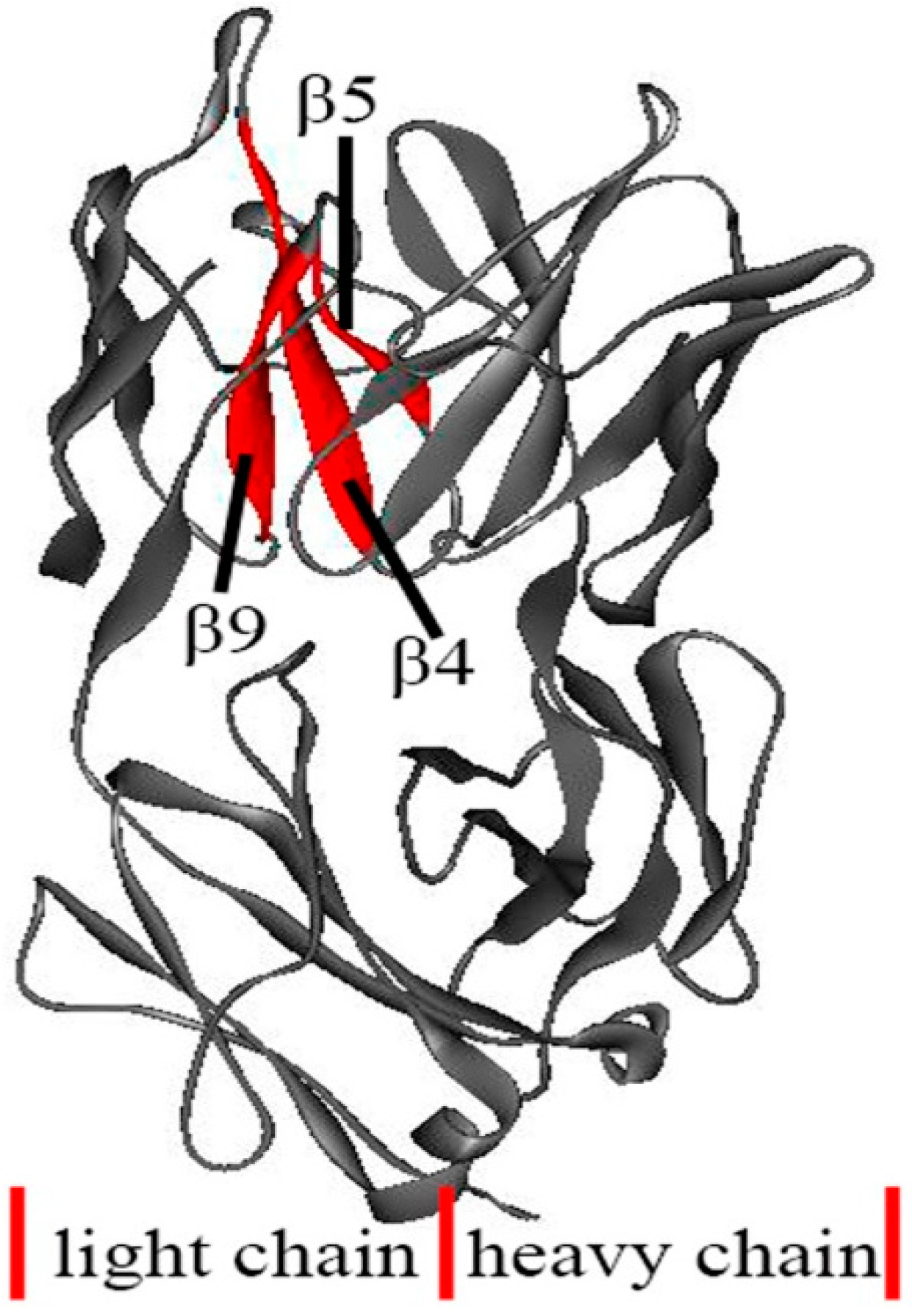
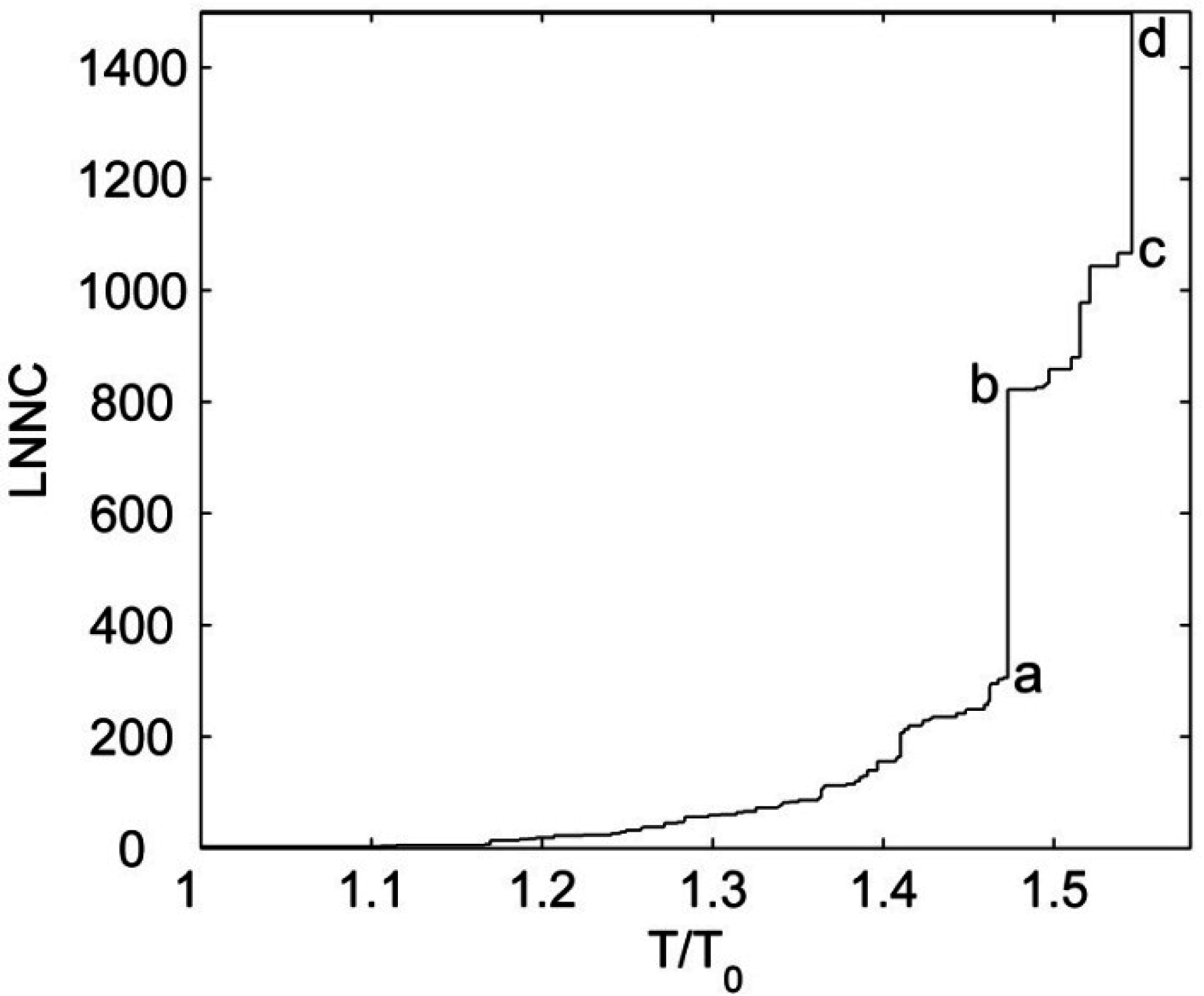
3.4. The Unfolding Process for the Fab Fragment of McPC603



3. Materials and Methods
3.1. The Basic Principle of Gaussian Network Model
3.2. The Iterative Unfolding Method
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sawyer, L.A. Antibodies for the prevention and treatment of viral diseases. Antivir. Res. 2000, 47, 57–77. [Google Scholar] [CrossRef]
- Kuroda, D.; Shirai, H.; Jacobson, M.P.; Nakamura, H. Computer-aided antibody design. Protein Eng. Des. Sel. 2012, 25, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Rouet, R.; Lowe, D.; Christ, D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014, 588, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Baker, D. Prediction of protein-folding mechanisms from free-energy landscapes derived from native structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11305–11310. [Google Scholar] [CrossRef] [PubMed]
- Clementi, C.; Jennings, P.A.; Onuchic, J.N. How native-state topology affects the folding of dihydrofolate reductase and interleukin-1β. Proc. Natl. Acad. Sci. USA 2000, 97, 5871–5876. [Google Scholar] [CrossRef] [PubMed]
- Koga, N.; Takada, S. Roles of native topology and chain-length scaling in protein folding: A simulation study with a Gō-like model. J. Mol. Biol. 2001, 313, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 1997, 36, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; Day, J.; Greenwood, A.; McPherson, A. The three-dimensional structure of an intact monoclonal antibody for canine lymphoma. Nature 1992, 360, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Larson, S.B.; Skaletsky, E.; McPherson, A. Comparision of the conformations of two intact monoclonal antibodies with hinges. Immunol. Rev. 1998, 163, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic structure of an intact IgG1 monoclonal antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Satow, Y.; Cohen, G.H.; Padlan, E.A.; Davies, D.R. Phosphocholine binding immunoglobulin Fab McPC603. An X-ray diffraction study at 2.7 Å. J. Mol. Biol. 1986, 190, 593–604. [Google Scholar] [CrossRef]
- Haliloglu, T.; Bahar, I.; Erman, B. Gaussian dynamics of folded protein. Phys. Rev. Lett. 1997, 79, 3090–3093. [Google Scholar] [CrossRef]
- Yang, L.W.; Liu, X.; Jursa, C.J.; Holliman, M.; Rader, A.J.; Karimi, H.A.; Bahar, I. iGNM: A database of protein functional motions based on Gaussian network model. Bioinformatics 2005, 21, 2978–2987. [Google Scholar] [CrossRef] [PubMed]
- Tirion, M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996, 77, 1905. [Google Scholar] [CrossRef] [PubMed]
- Romo, T.D.; Grossfield, A. Validating and improving elastic network models with molecular dynamics simulations. Proteins 2011, 79, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ma, J. The role of shape in determining molecular motions. Biophys. J. 2005, 89, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Su, J.G.; Jiao, X.; Sun, T.G.; Li, C.H.; Chen, W.Z.; Wang, C.X. Analysis of domain movements in glutamine-binding protein with simple models. Biophys. J. 2007, 92, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Cheng, M.H.; Lee, J.Y.; Kaya, C.; Zhang, S. Structure-encoded global motions and their role in mediating protein-substrate interactions. Biophys. J. 2015, 109, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tekpinar, M. Large-scale evaluation of dynamically important residues in proteins predicted by the perturbation analysis of a coarse-grained elastic model. BMC Struct. Biol. 2009, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Sorensen, D.C.; Phillips, G.N., Jr. Automatic domain decomposition of proteins by a Gaussian network model. Proteins 2004, 57, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Ma, J. New advances in normal mode analysis of supermolecular complexes and applications to structural refinement. Curr. Protein Pept. Sci. 2004, 5, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Su, J.G.; Li, C.H.; Hao, R.; Chen, W.Z.; Wang, C.X. Protein unfolding behavior studied by elastic network model. Biophys. J. 2008, 94, 4586–4596. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Granek, R. Cooperativity in thermal and force-induced protein unfolding: Integration of crack propagation and network elasticity models. Phys. Rev. Lett. 2013, 110, 138101. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Granek, R. Protein unfolding from free-energy calculations: Integration of the Gaussian network model with bond binding energies. Phys. Rev. E 2015, 91, 022708. [Google Scholar] [CrossRef] [PubMed]
- Arkun, Y.; Erman, B. Prediction of optimal folding routes of proteins that satisfy the principle of lowest entropy loss: Dynamic contact maps and optimal control. PLoS ONE 2010, 5, 13275. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Melton, J.S.; Sorensen, D.C.; Philips, G.N. Dynamics of proteins in crystals: Comparison of experiment with simple models. Biophys. J. 2002, 83, 723–732. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Demirel, M.C.; Erman, B. Vibrational dynamics of proteins: Significance of slow and fast modes in relation to function and stability. Phys. Rev. Lett. 1998, 80, 2733–2736. [Google Scholar] [CrossRef]
- Freund, C.; Gehrig, P.; Holak, T.A.; Plückthun, A. Comparison of the amide proton exchange behavior of the rapidly formed folding intermediate and the native state of an antibody scFv fragment. FEBS Lett. 1997, 407, 42–46. [Google Scholar] [CrossRef]
- Freund, C.; Honegger, A.; Hunziker, P.; Holak, T.A.; Plückthun, A. Folding nuclei of the scFv fragment of an antibody. Biochemistry 1996, 35, 8457–8464. [Google Scholar] [CrossRef] [PubMed]
- Roccatano, D.; Daidone, I.; Ceruso, M.; Bossa, C.; Nola, A.D. Selective excitation of native fluctuations during thermal unfolding simulations: horse heart cytochrome c as a case study. Biophys J. 2003, 84, 1876–1883. [Google Scholar] [CrossRef]
- Röthlisberger, D.; Honegger, A.; Plückthun, A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J. Mol. Biol. 2005, 347, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Welfle, K.; Misselwitz, R.; Hausdorf, G.; Höhne, W.; Welfle, H. Conformation, pH-induced conformational changes, and thermal unfolding of anti-p24 (HIV-1) monoclonal antibody CB4-1 and its Fab and Fc fragments. Biochim. Biophys. Acta 1999, 1431, 120–131. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical thermodynamics of random networks. Proc. Roy. Soc. Lond. A 1976, 351, 351–380. [Google Scholar] [CrossRef]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in protein using a single parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef]
- Sulkowska, J.I.; Kloczkowski, A.; Sen, T.Z.; Cieplak, M.; Jernigan, R.L. Predicting the order in which contacts are broken during single molecule protein stretching experiments. Proteins 2008, 71, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Kloczkowski, A.; Mark, J.E. Chain dimensions and fluctuations in random elastomeric networks. 1. Phantom Gaussian networks in the undeformed state. Macromolecules 1989, 22, 1423–1432. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, J.-G.; Zhang, X.; Han, X.-M.; Zhao, S.-X.; Li, C.-H. The Intrinsic Dynamics and Unfolding Process of an Antibody Fab Fragment Revealed by Elastic Network Model. Int. J. Mol. Sci. 2015, 16, 29720-29731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226197
Su J-G, Zhang X, Han X-M, Zhao S-X, Li C-H. The Intrinsic Dynamics and Unfolding Process of an Antibody Fab Fragment Revealed by Elastic Network Model. International Journal of Molecular Sciences. 2015; 16(12):29720-29731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226197
Chicago/Turabian StyleSu, Ji-Guo, Xiao Zhang, Xiao-Ming Han, Shu-Xin Zhao, and Chun-Hua Li. 2015. "The Intrinsic Dynamics and Unfolding Process of an Antibody Fab Fragment Revealed by Elastic Network Model" International Journal of Molecular Sciences 16, no. 12: 29720-29731. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226197