
Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine


Abstract
:
1. Introduction

2. Methods
3. Results

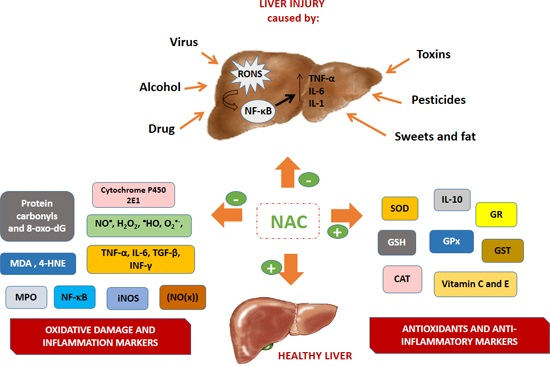{kind=link}
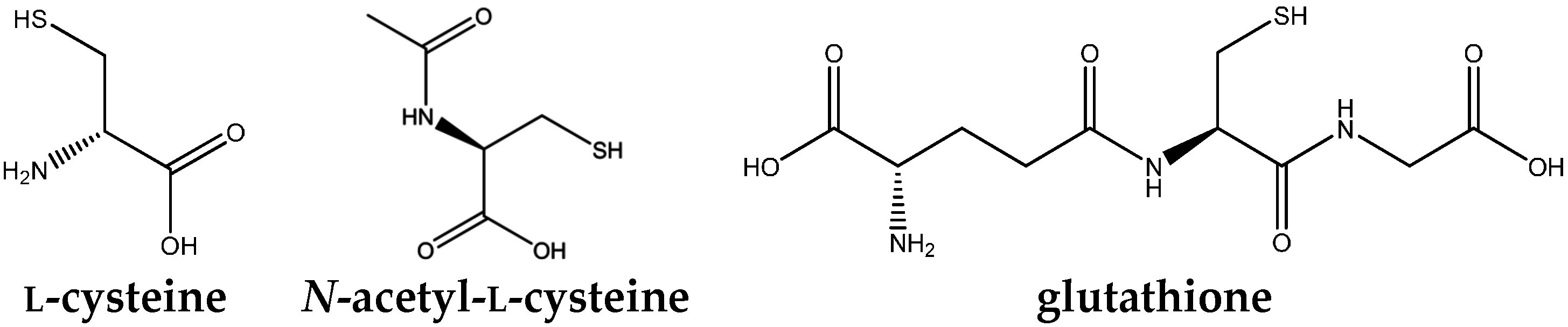{kind=link}
{kind=link}
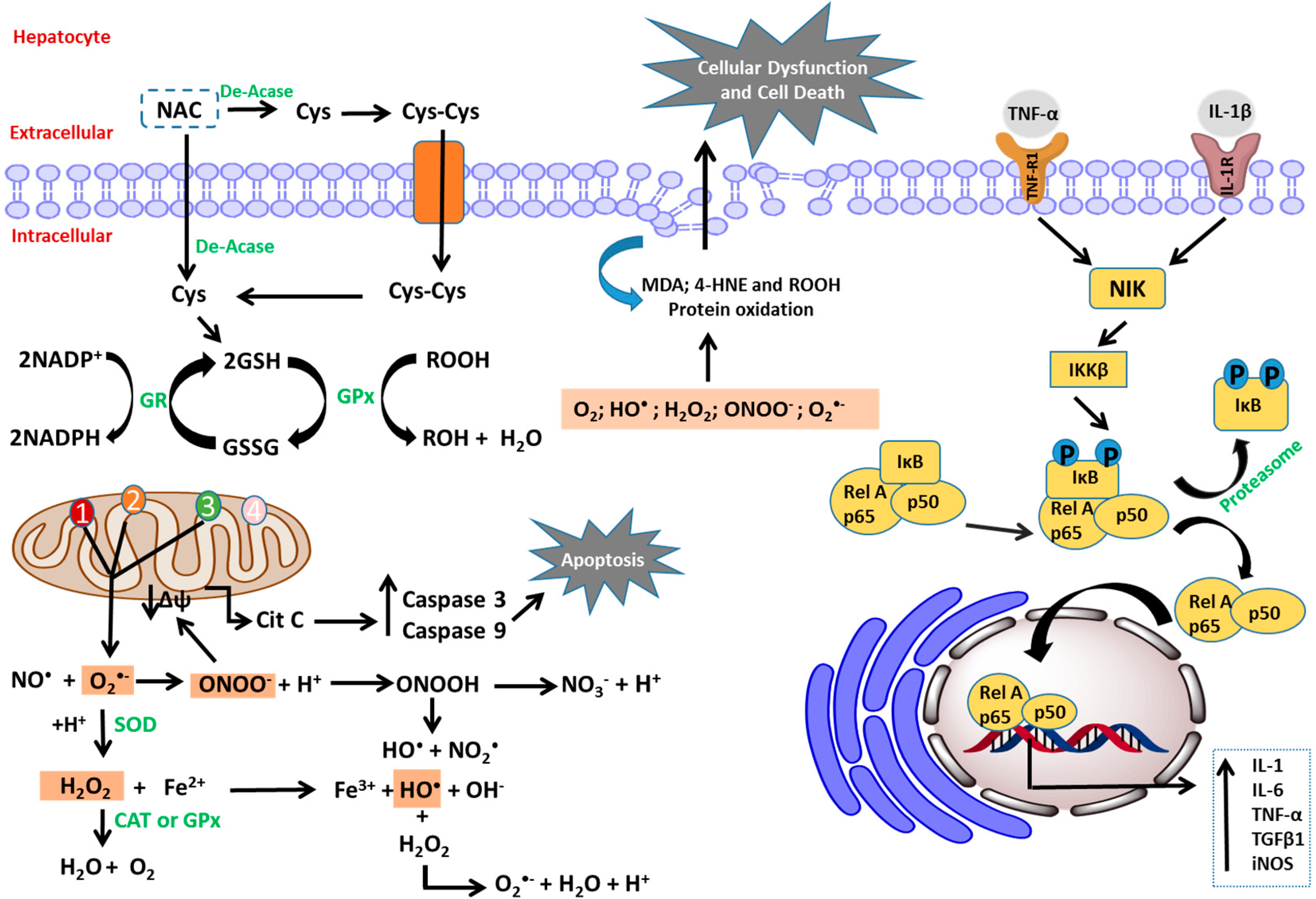{kind=link}
{kind=link}
{kind=link}
| Synthesis of RONS or Damage by Them, Forming ROMs | Increase of AO Defense | Cytokines and Interleukins Synthesis and Levels |
|---|---|---|
| Levels of: O2·−, H2O2, HO·, HOCl, NO·, ONOO−, ONOOH, NO2·−, ROS; NO(x) | Antioxidant enzymes: GPx, GR, GST, SOD, CAT | Levels of: TNF-α, IL-1β, INF-γ, IL-6, IL-10, IL-1, IL-5, IL-12, IL-16, IL-2, IL-4, IL-17, TGF-β |
| Levels of enzymes (activity/expression) related to RONS: iNOS, XO, COX, LPO, MPO; cytochrome P450 2E1, PGE2 | Levels of non-enzymatic defenses: GSH, GSH/GSSG | |
| Levels of producing enzymes (activity/expression) related to pro-oxidants: NF-kB, IkB-α | P-SH, Vit E, Vit C, Zn, Se; Levels of antioxidant capacity (TAS, TAC) | |
| Levels of oxidative damage caused by RONS: formation of AGE, 8-OXOdG, LP, MDA, 4-HNE, TBARS, GSSG, protein carbonyls | Levels of producing enzymes (activity/expression) of antioxidants: Nrf2 |
4. Discussion
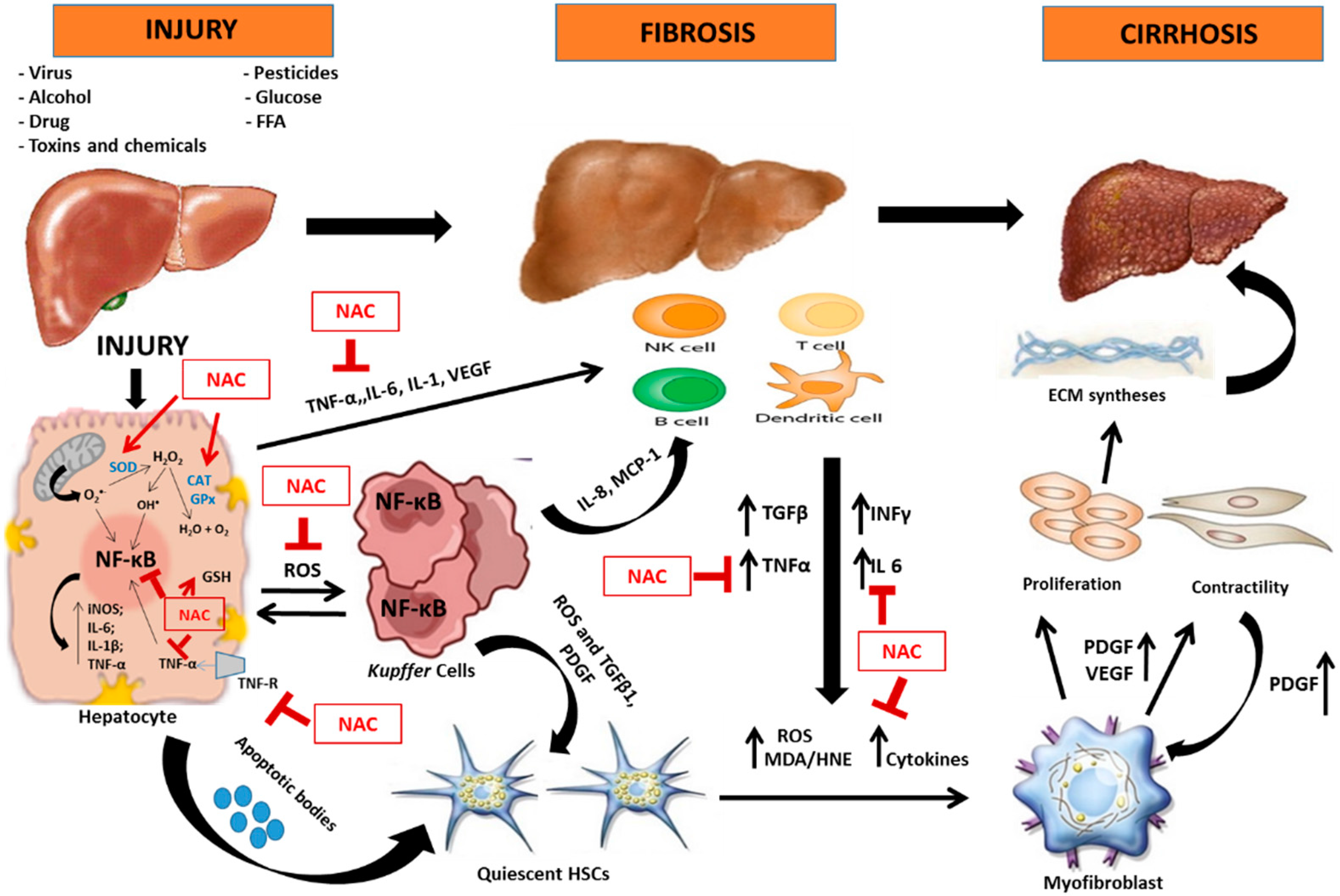
4.1. Oxidative Stress and Inflammation in Hepatic Lesion
4.2. N-Acetylcysteine and Its Antioxidant and Anti-Inflammatory Properties


 inhibition;
inhibition;  stimulation; ↑ increase.
inhibition; stimulation; ↑ increase.
stimulation; ↑ increase.
inhibition; stimulation; ↑ increase.
4.3. Hepatic Diseases and Possibilities of Therapeutic Effect of N-Acetylcysteine
4.3.1. Chronic Viral Hepatitis
Hepatitis C Virus
Hepatitis B Virus
4.3.2. Alcohol
| Type of Damage | Type of Study | Admin. Route | Dose; Time of Admin | RONS Synthesis or Damage | AO Defense | Cytokines and Interleukins Synthesis and Levels | Ref. |
|---|---|---|---|---|---|---|---|
| DISEASES | |||||||
| NASH | In vivo (rats) | Gavage | 2 g/kg/d, 65 d | ↓ LOOH and MDA tissue levels; ↓ Cytochrome P450 2E1 tissue expression | ↑ GSH tissue levels | ↓ TNF-α tissue, mRNA expression IL-1β # mRNA expression | [54] |
| In vivo (rats) | Oral (diet) | 20 mg/kg/d, 6 wk | MDA # tissue levels | ↓ GSH plasm levels | - | [140] | |
| In vivo (rats) | Oral (diet) | 500 mg/kg/d, 4 wk | MDA # tissue levels | GSH # tissue levels | - | [141] | |
| Fibrosis | In vivo (rats) | i.p. | 50 mg/kg/d, 6 wk | - | GSH # tissue levels | ↓ TNF-α and IL-6 tissue levels | [142] |
| In vivo (rats) | i.m. | 50 μmol/kg/d, 2 wk | ↓ TBARS tissue levels; ↓ protein carbonyl tissue levels | ↑ GSH and P-SH tissue levels | - | [143] | |
| Cirrhosis | In vivo (rats) | i.p. | 1 g/kg | - | ↑ GSH tissue levels | - | [144] |
| Diabetes mellitus | In vivo (rats) | i.p. | 1.5 g/kg/d, 4 wk after induction | - | ↑ TAC plasm levels ↑ SOD tissue activity | ↓ TNF-α and IL-6 serum levels | [145] |
| In vivo (rats) | i.p. | 25 a mg/kg/d; 75 b mg/kg/d, 30 d | MDA # tissue levels | ↑ GSH tissue levels; GPx # and SOD # tissue activities | - | [16] | |
| Obesity | In vivo (rats) | Oral (water) | 2 mg/L/d, 30 d | ↓ lipid hydroperoxide tissue levels | ↑ Antioxidant capacity; ↑ GSH tissue levels; ↑ GSH/GSSG tissue levels; ↑ SOD tissue activity; CAT # tissue activity; ↑ GPx tissue activity | - | [146] |
| Ischemia-reperfusion | In vivo (mice) | i.p. | 300 mg/kg/d, 2 h before ischemia | ↓ MDA tissue levels | ↑ GSH tissue levels | - | [147] |
| In vivo (rats) | i.p. | 1 g/kg/d, every second day over a 10 day period | ↓ Protein carbonyl tissue levels; ↓ Protein carbonyl/GSH tissue levels | ↑ GSH tissue levels | - | [148] | |
| In vivo (rats) | i.p. | 500 mg/kg, 20 min before induction | ↓ MDA tissue levels | ↓ GPx tissue activity | - | [149] | |
| In vivo (rats) | i.p. | 150 mg/kg, 15 min before ischemia | ↓ MDA tissue levels; ↓ protein oxidation tissue; ↓ MPO tissue activity | ↑ GSH tissue levels | - | [150] | |
| In vivo (mice) | i.v. | 150 mg/kg, 6 a h, 12 b h and 24 c h after IR | ↓ NF-κB b ↓ ROS a,b,c | - | ↓ IL-6 a,b,c mRNA, TNF-α a,b | [151] | |
| Hepatocellular carcinogenesis | In vivo (rats) | i.p. | 100 mg/kg/d, 3 months before induction | ↓ ROS | - | - | [152] |
| In vivo (mice) | Oral (water) | 4 mg/mL, starting from 2 months of age | ↓ 4-HNE, MDA and 8-OXO-dG tissue levels | - | - | [153] | |
| Cholestasis | In vivo (rats) | Oral (0.5% carboxymethyl cellulose) | 300 mg/kg/d 28 d after bile duct ligation | ↓ MDA tissue levels | ↑ GSH tissue levels; CAT # tissue activity | ↓ TGF-β and IL-6 tissue expression; IL-10 # tissue expression | [154] |
| Obstructive jaundice | In vivo (rats) | s.c. | 100 mg/kg/d, 5 d after induction | ↓ MDA tissue levels; ↓ iNOS tissue expression | - | - | [78] |
| Acute hepatic failure | In vivo (rats) | s.c. | 20 mg/kg, 3 and 6 h after CCl4 | TBARS # tissue levels; ↓ protein carbonyls tissue levels | - | - | [155] |
| In vivo (mice) | i.p. | 1.2 g/kg a, immediately before induction and 1.2 g/kg b injected 1 h after induction | - | ↑ GSH a tissue levels | ↓ IL-5 a, IL-10 a, IL-12 a, IL-17 a and IFN-γ a tissue levels | [156] | |
| Schistosomiasis | In vivo (mice) | Oral (water) | 300 mg/kg, 5 d a wk/4 wk | - | ↓ GSH tissue levels; GST #, GR #, GPx # and SOD # tissue activities | - | [157] |
| Lipopolysaccharide (LPS) | In vivo (rats) | i.v | 150 mg/kg/h (0.3 mL/h) at 60 min and 12.5 mg/kg/h throughout the experiment (0.3 mL/h) | - | - | ↓ TNF-α, IL-6 and IL-10 plasma levels | [158] |
| ALCOHOL | |||||||
| Ethanol | In vivo (mice) | i.p. | 75 a, 150 b or 300 c mg/kg/d, at 30 min before ethanol; 75 d, 150 e or 300 f mg/kg/d, at 4 h after ethanol | ↓ TBARS a,b,c tissue levels | ↑ GSH a,b,c tissue levels | ↓ TNF-α a,b,c tissue mRNA expression | [139] |
| In vivo (rats) | Gavage | 1.2 g/kg/d, 45 d | ↓ MDA and HNE tissue adducts; CYP2E1 # expression | ORAC #; GSH # tissue levels | ↓ TNF-α tissue mRNA expression | [159] | |
| In vivo (rats) | Gavage | 1.2 g/kg/d, for 130 d | - | - | IL-1β #, IL-2 #, IL-4 #, IL-6 #, TNF-α # levels | [160] | |
| In vivo (rats) | Oral (ethanol solution) | 2 g/L with ethanol, 15 d after 30 d of ethanol a, and 2 g/L without ethanol for 15 d after 30 d of ethanol b | - | ↑ GSH/GSSG a,b tissue levels; ↑ GR a,b tissue activity | - | [119] | |
| In vivo (mice) | i.p. | 300 mg/kg/d, 30 d | - | ↑ P-SH tissue levels; ↑ GPx tissue activity | - | [137] | |
| In vivo (rats) | Gavage | 1.7 g/kg/d, 150 d | Protein carbonyls # mitochondrial levels; TBARS # mitochondrial levels; mtDNA # damage | ↑ GSH tissue levels | - | [138] | |
| DRUGS | |||||||
| Acetaminophen (APAP) | In vitro (hepatocyte) | - | 20 mM (dissolved in 10X PBS, pH 7.4) 1 a h before or 2 b h after APAP administration | - | ↑ GSH a,b levels | - | [161] |
| In vivo (rats) | i.p. | 100 mg/kg/d, 5 d | - | - | ↓ TNF-α and IL-6 tissue levels | [162] | |
| In vivo (rats) | i.p. | 2.4 mM/kg/(2 mL dose), 30 min before induction | ↓ MDA tissue and serum levels | ↑ GSH, GSSG, GSH/GSSG tissue and serum levels; ↑ GR, GST tissue and serum activities | - | [163] | |
| In vivo (mice) | i.p. | 1.25 mmol/kg after 1 h after induction | MDA #, ↓ 4-HNE and protein carbonyl tissue levels | ↑ GSH and GSSG tissue levels | - | [164] | |
| In vitro (hepatocyte) | - | 2.0 mM, 30 min after incubation with APAP | - | ↑ GSH levels, P-SH levels | - | [165] | |
| In vitro (hepatocyte) | - | 5.0 mM, after 24 a and 48 b h of APAP exposure | - | ↑ GSH a levels | - | [166] | |
| In vivo (mice) | i.p. | 400 mg/kg, 2 h after induction | - | ↑ GSH tissue levels | - | [167] | |
| In vitro (human hepatocyte) | - | 250 μM, before 12 a or 24 b h | ↓ TBARS a,b levels; ↓ ROS a,b levels | GSH a and GSH/GSSG #,a; ↑ GSH/GSSG b ↑ GR a,b activity | - | [168] | |
| Rifampicin | In vivo (rats) | i.p. | 100 mg/kg/d, 3 wk | MDA # levels; Cytochrome P450 2E1 # tissue levels | SOD # and CAT # tissue activities; GPx # activity GST # activity GR # activity | - | [169] |
| Azathioprine | In vivo (rats) | i.p. | 100 mg/kg/d, 7 d before induction | ↓ MDA tissue levels | ↑ GSH tissue levels | - | [170] |
| Cocaine | In vitro (hepatocyte) | - | 0.5 mM, 24 h before incubation with cocaine and 24 h after incubation | ↓ Peroxide levels | ↑ GSH levels ↑ CAT and GPx RNAm levels | - | [84] |
| N-methyl-methyldopamine | In vitro (hepatocyte) | - | 0.1 a and 1 b mM, 15 min before induction | - | GSH # levels | - | [171] |
| Cyclosporine A | In vivo (rats) | i.m. | 150 mg/kg/d, 11 d starting 1 d before induction | ↓ MDA tissue levels; ↓ NO· tissue levels | ↑ SOD tissue activity | - | [172] |
| Isoniazid | In vivo (rats) | i.p. | 100 mg/kg/d, 3 wk | CYP2E1 # levels | SOD # and CAT # tissue activities; P-SH # levels; GPx # and GR # tissue activities | - | [169] |
| Statins | In vitro (rats) | - | 200 μM | ↓ TBARS levels | - | - | [173] |
| Methotrexate | In vivo (rats) | i.p. | 50 mg/kg/d, 7 d | MDA # tissue levels | ↑ SOD tissue activity; GSH # tissue levels; CAT # tissue activity; TAC # | - | [174] |
| Carbamazepine | In vivo (rats) | Gavage | 50 a, 100 b and 200 c mg/kg/d for 45 d | ↓ TBARS c tissue levels | ↑ SOD c and CAT c tissue activities; ↑ GSH c tissue levels | - | [175] |
| PESTICIDES | |||||||
| Malathion | In vivo (rats) | Oral (water) | 2 g/L, 28 d | ↓ MDA tissue levels; ↓ MPO tissue activity | ↑ GSH tissue levels; ↑ SOD, CAT, GPx and GST tissue activities | ↓ IL-1β, IL-6, INF-γ, mRNA levels | [176] |
| In vitro (hepatocyte) | 200 μM, 30 min before exposure | ↓ ROS | - | - | [177] | ||
| Paraquat | In vivo (rats) | i.p. | 200 mg/kg 2 h before induction | ↓ MDA tissue levels; ↓ iNOS tissue levels; ↓ NO2− tissue levels | ↑ GSH tissue levels ↓ SOD tissue levels ↑ GR tissue levels ↓ GPx tissue levels | ↓ CYP2E1 tissue levels; ↓ TNF-α tissue levels ↓ IL-1β tissue levels | [178] |
| Dichlorodiphenyltrichloroethane (DDT) | In vitro (hepatocyte) | - | 100 mg/mL, 1 h before of exposure | ROS # | - | - | [179] |
| Carbosulfan | In vivo (rats) | Oral (water) | 2 g/L for 30 d | ↓ MDA tissue levels | ↑ GSH tissue levels | ↓ IFN-γ mRNA expression | [180] |
| IONIZING RADIATION | |||||||
| X-Rays | In vivo (mice) | i.p. | 50 a mg/kg/d, 100 b mg/kg/d or 200 c mg/kg/d, 1 h before or after exposure to X-ray irradiation | ↓ MDA a,b,c tissue levels; ↓ DNA a,b,c,* tissue damage | ↑ GSH a,b,c tissue levels 1h before; ↑ GSH b,c tissue levels after exposure; ↑ SOD a,b,c,* tissue activity | - | [181] |
| γ-Rays | In vivo (rats) | i.p | 1 g/kg/d, 7 d before exposure to γ-ray irradiation | ↓ MDA tissue levels; ↓ DNA tissue damage; (NO(x)) # | ↑ GSH tissue levels; ↑ GSH-Px tissue activity; ↑ SOD tissue activity | - | [182] |
| OTHERS | |||||||
| Mercury toxicity | In vivo (rats) | i.p. | 0.6 g/kg/d, 3 d after induction | ↓ MDA tissue levels | ↑ SOD and CAT tissue activities; ↑ GSH tissue levels | - | [183] |
| Carbon tetrachloride | In vivo (rats) | Gavage | 150 mg/kg/d, for 3 months | ↓ TBARS plasm and tissue levels; ↓ HP plasm and tissue levels | ↑ SOD and CAT tissue activities; ↑ GPx tissue activity; ↑ GSH plasm levels; ↑ Vitamin C and vitamin E plasm levels | - | [184] |
| In vivo (rats) | i.p. | 25 a mg/kg/d and 50 b mg/kg/d, 12 wk | ↓ LP a,b tissue levels | ↑ GPx a,b tissue activity ↑ GSH a,b tissue activity ↑ CAT a,b tissue activity | ↓ CYP2E1 a,b tissue activity | [185] | |
| In vivo (rats) | i.p. | 50 a; 100 b; 200 c mg/kg/d, 4 wk | ↓ MDA b,c tissue levels | ↑ SOD a,b,c tissue activity; ↑ GSH a,b,c tissue levels | [186] | ||
| Cadmium (Cd) | In vitro (rats) | - | 5 mM, 2 h pre-, simultaneous or 2 h post-treatment | - | ↑ GR levels ↑ CAT levels GPx # levels | - | [187] |
| In vitro (rats) | - | 1 mM a and 2 b mM, 1.5 or 24 h, added simultaneously with the Cd | ↓ ROS | - | - | [188] | |
| Glycochenodeoxycholic acid (GCDCA) | In vitro (rats) HepG2 cells | - | 0.5 mM, co-administered with GCDCA | ↓ O2·− production; ↓ NO levels | - | - | [189] |
| Methanol | In vivo (rats) | i.p. | 150 mg/kg, two doses, after 12 h and 24 h | ↓ MDA tissue levels | ↑ GSH tissue levels ↑ GSSG tissue levels ↑ GPx tissue activity | - | [190] |
| Fluoride | In vitro (rats) culture | - | 1 mmol/L, 60 a min before and 60 b min simultaneously with fluoride | ↓ MDA a tissue levels | ↑ GPx a tissue levels ↑ GR a tissue levels | - | [191] |
| iNOS | In vivo (rats) | s.c. | 100 mg/kg, 5 d | ↓ MDA tissue levels; ↓ iNOS tissue expression | - | - | [78] |
| Dimethylnitrosamine | In vivo (rats) | Gavage | 50 mg/kg/d, 7 d | ↓ MDA tissue levels | ↑ SOD tissue activity; ↑ Vitamin C tissue levels; ↑ Vitamin E tissue levels; ↑ GPx tissue activity; ↑ GSH tissue levels; ↑ CAT tissue activity; ↑ GST tissue activity | - | [192] |
| Arsenic | In vivo (rats) | i.p. | 10 mg/kg/d, for 3 wk | TBARS tissue levels # | CAT tissue activity # GSH tissue levels # | - | [193] |
| Mercuric chloride | In vitro (mice) | - | 10 a–500 b μM | ↓ MDA b levels | - | - | [194] |
| Polychlorinated biphenyls | In vivo (rats) | Oral (diet) | 1% (10g/kg diet) one wk before induction | CYP1A1 # activity | ↓ GSSG/GSH; GST # tissue activity | - | [145] |
| Isoflurane anaesthesia | Human | i.v. | 12.5 mg/kg/h throughout the operation (laparoscopic surgery) | ↑ MDA plasm levels | ↑ GST plasm levels; ↑ GSH plasm levels | - | [195] |
| Cardiopulmonary bypass | In vivo (rats) | - | 250 mg/kg, at 0.5, 1, 2, 3, and 24 h | ↓ MDA tissue levels ↓ MPO tissue levels ↓ iNOS tissue levels | ↑ GSSG tissue levels ↑ GSH tissue levels ↑ GPx tissue activity | - | [196] |
| Acrylamide (ACR) | In vivo (rats) | Gavage | 250 mg/kg/d, 21 d | ↓ MDA tissue levels | ↑ GSH tissue levels; ↑ GST tissue activity | [197] | |
4.3.3. Non-Alcoholic Steatohepatitis
4.3.4. Lipopolysaccharides
4.3.5. Intoxication
Drug-Induced Liver Injury
Pesticides
Toxins and Miscellaneous Chemicals
4.3.6. Ischemia/Reperfusion
4.3.7. Hepatocelullar Carcinoma
5. Concluding Remarks
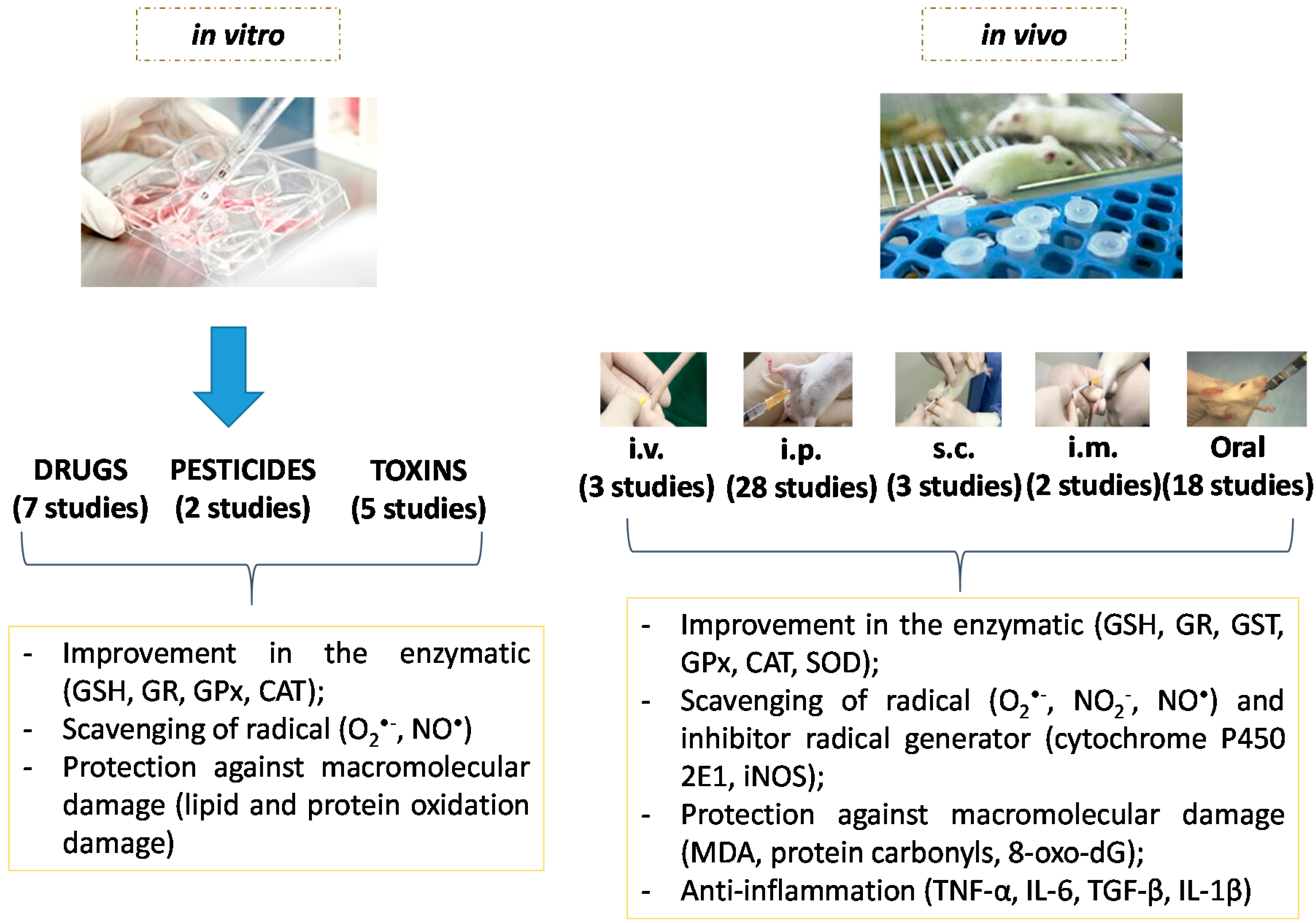
5.1. Type of Study
5.2. Doses, Route of Administration and Study Time

5.3. Biomarkers
6. Challenges and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Chatterjee, R.; Mitra, A. An overview of effective therapies and recent advances in biomarkers for chronic liver diseases and associated liver cancer. Int. Immunopharmacol. 2015, 24, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P. Liver auto-immunology: The paradox of autoimmunity in a tolerogenic organ. J. Autoimmun. 2013, 46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Raschzok, N.; Sallmon, H.; Pratschke, J.; Sauer, I.M. MicroRNAs in liver tissue engineering—New promises for failing organs. Adv. Drug Deliv. Rev. 2015, 88, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.H.; Young, K.K. Clinical aspects of hepatic disease. Anaesth. Intensive Care Med. 2015, 16, 11–13. [Google Scholar] [CrossRef]
- Mehal, W.Z. The Gordian Knot of dysbiosis, obesity and NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Huang, C.; Meng, X.M.; Li, J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie 2015, 116, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Duval, F.; Moreno-Cuevas, J.E.; Gonzalez-Garza, M.T.; Rodriguez-Montalvo, C.; Cruz-Vega, D.E. Protective mechanisms of medicinal plants targeting hepatic stellate cell activation and extracellular matrix deposition in liver fibrosis. Chin. Med. 2014, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Diesen, D.L.; Kuo, P.C. Nitric oxide and redox regulation in the liver: Part II. Redox biology in pathologic hepatocytes and implications for intervention. J. Surg. Res. 2011, 167, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Valentim, I.B.; de Araujo, O.R.; Ataide Tda, R.; Goulart, M.O. Development of nonalcoholic hepatopathy: Contributions of oxidative stress and advanced glycation end products. Int. J. Mol. Sci. 2013, 14, 19846–19866. [Google Scholar] [CrossRef] [PubMed]
- Mormone, E.; George, J.; Nieto, N. Molecular pathogenesis of hepatic fibrosis and current therapeutic approaches. Chem. Biol. Interact. 2011, 193, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Musacco-Sebio, R.; Saporito-Magrina, C.; Semprine, J.; Torti, H.; Ferrarotti, N.; Castro-Parodi, M.; Damiano, A.; Boveris, A.; Repetto, M.G. Rat liver antioxidant response to iron and copper overloads. J. Inorg. Biochem. 2014, 137, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Johansson, E.; Yang, Y.; Miller, M.L.; Shen, D.; Orlicky, D.J.; Shertzer, H.G.; Vasiliou, V.; Nebert, D.W.; Dalton, T.P. Oral N-acetylcysteine rescues lethality of hepatocyte-specific Gclc-knockout mice, providing a model for hepatic cirrhosis. J. Hepatol. 2010, 53, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dong, H.; Thompson, D.C.; Shertzer, H.G.; Nebert, D.W.; Vasiliou, V. Glutathione defense mechanism in liver injury: Insights from animal models. Food Chem. Toxicol. 2013, 60, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Okanoue, T.; Mitsuyoshi, H. Non-alcoholic steatohepatitis. 3. Oxidative stress and NASH. Nihon Naika Gakkai Zasshi 2006, 95, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, G.; Roehrs, M.; Bairros, A.; Moro, A.; Charao, M.; Araujo, F.; Valentini, J.; Arbo, M.; Brucker, N.; Moresco, R.; et al. N-acetylcysteine on oxidative damage in diabetic rats. Drug Chem. Toxicol. 2011, 34, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-acetylcysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr. 2005, 9, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Moura, F.A.; de Andrade, K.Q.; Dos Santos, J.C.; Araujo, O.R.; Goulart, M.O. Antioxidant therapy for treatment of inflammatory bowel disease: Does it work? Redox Biol. 2015, 6, 617–639. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Wu, Y.; Guo, W.; Zhang, Y.; Zhai, W. Protective effects of N-acetylcysteine on the liver of brain-dead Ba-Ma mini pig. Transplant. Proc. 2010, 42, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.M.; Kremer, M.; Sanderlin, E.J.; Wheeler, M.D.; Hines, I.N. Emerging Roles for Lipids in the Hepatic Innate Immune Response. Hum. Nutr. Food. Sci. 2013, 1, 1–9. [Google Scholar]
- Videla, L.A.; Rodrigo, R.; Orellana, M.; Fernandez, V.; Tapia, G.; Quiñones, L.; Varela, N.; Contreras, J.; Lazarte, R.; Csendes, A.; et al. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 2004, 106, 261–268. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Vascotto, C.; Tiribelli, C. Alterations in the redox state and liver damage: Hints from the EASL Basic School of Hepatology. J. Hepatol. 2013, 58, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Center, S.A. Metabolic, antioxidant, nutraceutical, probiotic, and herbal therapies relating to the management of hepatobiliary disorders. Vet. Clin. N. Am. Small Anim. Pract. 2004, 34, 67–172. [Google Scholar] [CrossRef]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. The relative importance of HO· and ONOO− in mediating the toxicity of O·. Free Radic. Biol. Med. 1999, 26, 777–778. [Google Scholar] [PubMed]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.C.; Cerchiaro, G.; Honório, K.M. Relações patofisiológicas entre estresse oxidativo e arteriosclerose. Quim. Nova 2011, 34, 300–305. [Google Scholar] [CrossRef]
- Giavarotti, K.A.S. Estudo do estresse oxidativo hepático induzido por lindano em um modelo de hipertireoidismo experimental. Ph.D. Thesis, Universidade de São Paulo, São Paulo, Brazil, 9 February 2001. [Google Scholar]
- Possamai, F.P. Estudo do Estresse Oxidativo em Órgãos de Ratos Wistar Adultos Induzidos à Intoxicação por Malation. Master’s Thesis, Universidade do Extremo Sul Catarinense, Santa Catarina, Brazil, 2005. [Google Scholar]
- Marí, M.; Colell, A.; Morales, A.; von Montfort, C.; Garcia-Ruiz, C.; Fernández-Checa, J.C. Redox control of liver function in health and disease. Antioxid. Redox Signal. 2010, 12, 1295–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, S.M.L.; Goulart, M.O.F.; Moura, J.B.F.; Manfredini, V.; Benfato, M.S.; Kubota, L.T. Espécies reativas de oxigênio e de nitrogênio, antioxidantes e marcadores de dano oxidativo em sangue humano: Principais métodos analíticos para sua determinação. Quím. Nova 2007, 30, 1323–1338. [Google Scholar] [CrossRef]
- Edwards, L.; Wanless, I.R. Mechanisms of liver involvement in systemic disease. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; de Araujo, O.R.; Valentim, I.B.; de Andrade, K.Q.; Moura, F.A.; Smaniotto, S.; dos Santos, J.M.; Gasparotto, J.; Gelain, D.P.; Goulart, M.O. Choline and Cystine deficient diets in animal models with hepatocellular injury: Evaluation of oxidative stress and expression of RAGE, TNF-α, and IL-1β. Oxid. Med. Cell. Longev. 2015, 2015, 121925. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kanai, T. Role of toll-like receptors in immune activation and tolerance in the liver. Front. Immunol. 2014, 5, 221. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J. Oxygen homeostas is, thiol equilibrium and redox regulation of signalling transcription factors in the alveolar epithelium. Cell Signal. 2002, 14, 799–810. [Google Scholar] [CrossRef]
- Bhogal, R.H.; Curbishley, S.M.; Weston, C.J.; Adams, D.H.; Afford, S.C. Reactive oxygen species mediate human hepatocyte injury during hypoxia/reoxygenation. Liver Transpl. 2010, 16, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World J. Gastroenterol. 2014, 20, 2515–2532. [Google Scholar] [CrossRef] [PubMed]
- Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.M.; de Maria, S.; Carteni, M.; Nardone, G. Gut—Liver axis: The impact of gut microbiota on non alcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, J.; Zhang, J.; Dai, C.; Liu, X.; Wang, J.; Gao, Z.; Guo, H.; Wang, R.; Lu, S.; et al. S100A4 promotes liver fibrosis via activation of hepatic stellate cells. J. Hepatol. 2015, 62, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Giraudi, P.J.; Becerra, V.J.; Marin, V.; Chavez-Tapia, N.C.; Tiribelli, C.; Rosso, N. The importance of the interaction between hepatocyte and hepatic stellate cells in fibrogenesis induced by fatty accumulation. Exp. Mol. Pathol. 2015, 98, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Pelz, S.; Stock, P.; Bruckner, S.; Christ, B. A methionine-choline-deficient diet elicits NASH in the immunodeficient mouse featuring a model for hepatic cell transplantation. Exp. Cell. Res. 2012, 318, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Naftaly, M.; Friedman, S.L. Current status of novel antifibrotic therapies in patients with chronic liver disease. Ther. Adv. Gastroenterol. 2011, 4, 391–417. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Conrad, C.K.; Bottiglieri, T.; Herzenberg, L.A.; Moss, R.B.; Herzenberg, L.A. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 4628–4633. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Fernandez, C.; Rodriguez, A.; Ribera, J.M.; de la Fuente, M. The glutathione precursor N-acetylcysteine improves immune function in postmenopausal women. Free Radic. Biol. Med. 2008, 45, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Baumgardner, J.N.; Shankar, K.; Hennings, L.; Albano, E.; Badger, T.M.; Ronis, M.J.J. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J. Nutr. 2008, 138, 1872–1879. [Google Scholar] [PubMed]
- Garaiová, I.; Muchová, J.; Šustrová, M.; Blažíček3, P.; Sivoňová, M.; Kvasnička, P.; Pueschel, S.; Ďuračková, Z. The relationship b etween antioxidant systems and some markers of oxidative stress in persons with down syndrome. Biol. Bratisl. 2004, 59, 787–794. [Google Scholar]
- Atkinson, M.C. The Use of N-acetylcysteine in Intensive Care. Lit. Rev. 2002, 4, 21–27. [Google Scholar]
- Aitio, M.L. N-acetylcysteine—Passe-partout or much ado about nothing? Br. J. Clin. Pharmacol. 2005, 61, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Noszál, B.; Visky, D.; Kraszni, M. Population, acid-base, and redox properties of N-acetylcysteine conformers. J. Med. Chem. 2000, 43, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Sadowska, A.M.; Manuel, Y.K.B.; de Backer, W.A. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: Discordant in vitro and in vivo dose-effects: A review. Pulm Pharmacol. Ther. 2007, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Akca, T.; Canbaz, H.; Tataroglu, C.; Caglikulekci, M.; Tamer, L.; Colak, T.; Kanik, A.; Bilgin, O.; Aydin, S. The effect of N-acetylcysteine on pulmonary lipid peroxidation and tissue damage. J. Surg. Res. 2005, 129, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Kasperczyk, S.; Dobrakowski, M.; Kasperczyk, A.; Machnik, G.; Birkner, E. Effect of N-acetylcysteine administration on the expression and activities of antioxidant enzymes and the malondialdehyde level in the blood of lead-exposed workers. Environ. Toxicol. Pharmacol. 2014, 37, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.A.; Oliveira, C.S.; Mesquita, M.; Pedroso, T.F.; Costa, L.M.; Fiuza Tda, L.; Pereira, M.E. Zinc and N-acetylcysteine modify mercury distribution and promote increase in hepatic metallothionein levels. J. Trace Elem. Med. Biol. 2015, 32, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Lawrence, A.; Wardman, P.; Burkitt, M.J. Kinetics of superoxide scavenging by glutathione: An evaluation of its role in the removal of mitochondrial superoxide. Biochem Soc. Trans. 2003, 31, 1337–1339. [Google Scholar] [CrossRef] [PubMed]
- Sarnstrand, B.; Jansson, A.H.; Matuseviciene, G.; Scheynius, A.; Pierrou, S.; Bergstrand, H. N,N′-Diacetyl-l-cystine-the disulfide dimer of N-acetylcysteine—Is a potent modulator of contact sensitivity/delayed type hypersensitivity reactions in rodents. J. Pharmacol. Exp. Ther. 1999, 288, 1174–1184. [Google Scholar] [PubMed]
- Dodd, S.; Dean, O.; Copolov, D.L.; Malhi, G.S.; Berk, M. N-acetylcysteine for antioxidant therapy: Pharmacology and clinical utility. Expert Opin. Biol. Ther. 2008, 8, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chai, F.Y.; Yan, H.; Guo, Y.; Harding, J.J. Effects of N-acetylcysteine and glutathione ethyl ester drops on streptozotocin-induced diabetic cataract in rats. Mol. Vis. 2008, 14, 862–870. [Google Scholar] [PubMed]
- Griffith, O.W.; Meister, A. Glutathione: Interorgan translocation, turnover, and metabolism. Proc. Natl. Acad. Sci. USA 1979, 76, 5606–5610. [Google Scholar] [CrossRef] [PubMed]
- Lasram, M.M.; Dhouib, I.B.; Annabi, A.; El Fazaa, S.; Gharbi, N. A review on the possible molecular mechanism of action of N-acetylcysteine against insulin resistance and type-2 diabetes development. Clin. Biochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, L.; Gazzaniga, A. Toxicological, pharmacokinetic and metabolic studies on acetylcysteine. Eur. J. Respir. Dis. Suppl. 1980, 111, 45–51. [Google Scholar] [PubMed]
- Sheffner, A.L.; Medler, E.M.; Bailey, K.R.; Gallo, D.G.; Mueller, A.J.; Sarett, H.P. Metabolic studies with acetylcysteine. Biochem. Pharmacol. 1966, 15, 1523–1535. [Google Scholar] [CrossRef]
- Borgström, L.; Kågedal, B.; Paulsen, O. Pharmacokinetics of N-acetylcysteine in man. Eur. J. Clin. Pharmacol. 1986, 31, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.C.; Zaretsky, M.D.; Dubs, J.G.; Roederer, M.; Anderson, M.; Green, A.; Mitra, D.; Watanabe, N.; Nakamura, H.; Tjioe, I.; et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur. J. Clin. Investig. 2000, 30, 915–929. [Google Scholar] [CrossRef]
- Cetinkaya, A.; Bulbuloglu, E.; Kurutas, E.B.; Ciralik, H.; Kantarceken, B.; Buyukbese, M.A. Beneficial effects of N-acetylcysteine on acetic acid-induced colitis in rats. Tohoku J. Exp. Med. 2005, 206, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, L.G.; Mate, J.; Gisbert, J.P.; Perez-Calle, J.L.; Marin-Jimenez, I.; Arriaza, E.; Olleros, T.; Delgado, M.; Castillejo, M.S.; Prieto-Merino, D.; et al. N-acetyl-l-cysteine combined with mesalamine in the treatment of ulcerative colitis: Randomized, placebo-controlled pilot study. World J. Gastroenterol. 2008, 14, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.L.; Anglade, M.W.; Baker, E.L.; White, C.M.; Kluger, J.; Coleman, C.I. Use of N-acetylcysteine to reduce post-cardiothoracic surgery complications: A meta-analysis. Eur. J. Cardiothorac. Surg. 2009, 35, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Eck, H.P.; Mihm, S. HIV-induced cysteine deficiency and T-cell dysfunction—A rationale for treatment with N-acetylcysteine. Immunol. Today 1992, 13, 211–214. [Google Scholar] [CrossRef]
- Berk, M.; Malhi, G.S.; Gray, L.J.; Dean, O.M. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 2013, 34, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Çağlıkülekci, M.; Pata, C.; Apa, D.D.; Dirlik, M.; Tamer, L.; Yaylak, F.; Kanik, A.; Aydin, S. The effect of N-acetylcysteine (NAC) on liver and renal tissue inducible nitric oxide synthase (iNOS) and tissue lipid peroxidation in obstructive jaundice stimulated by lipopolysaccharide (LPS). Pharmacol. Res. 2004, 49, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Miyashita, H.; Sakamoto, I.; Kitagawa, M.; Tanaka, H.; Yasuda, H.; Karin, M.; Kikugawa, K. Evidence that reactive oxygen species do not mediate NF-κB activation. EMBO J. 2003, 22, 3356–3366. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, L.; Yi, D.; Ding, B.; Yang, Z.; Li, J.; Chen, X.; Qiu, Y.; Wu, G. N-acetylcysteine reduces inflammation in the small intestine by regulating redox, EGF and TLR4 signaling. Amino Acids 2013, 45, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Yu, K.H.; Kim do, K.; Kim, S.; Sapkota, K.; Kim, S.J.; Kim, C.S.; Chun, H.S. Protective effects of N-acetylcysteine against monosodium glutamate-induced astrocytic cell death. Food Chem. Toxicol. 2014, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; Brunelli, R.; Bracci-Laudiero, L.; Greco, G.; Gustafsson, A.C.; Krasnowska, E.K.; Lundeberg, J.; Lundeberg, T.; Pittaluga, E.; Romano, M.C.; et al. Differentiation of normal and cancer cells induced by sulfhydryl reduction: Biochemical and molecular mechanisms. Cell Death Differ. 2005, 12, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.T.; Xue, J.Y.; Sun, F.F.; Wong, P.Y. Reactive oxygen species participate in peroxynitrite-induced apoptosis in HL-60 cells. Biochem. Biophys. Res. Commun. 1997, 230, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, A.; Díez-Fernándeza, C.; Alvarezb, A.M.; Andrésa, D.; Cascales, M. Effect of N-acetylcysteine and deferoxamine on endogenous antioxidant defense system gene expression in a rat hepatocyte model of cocaine cytotoxicity. Biochim. Biophys. Acta 2000, 1496, 183–195. [Google Scholar] [CrossRef]
- Mahapatra, S.K.; Bhattacharjee, S.; Chakraborty, S.P.; Majumdar, S.; Roy, S. Alteration of immune functions and Th1/Th2 cytokine balance in nicotine-induced murine macrophages: Immunomodulatory role of eugenol and N-acetylcysteine. Int. Immunopharmacol. 2011, 11, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Hafiz, A.M.A.E.; Wakeel, L.M.E.; Hady, H.M.E.; Mourad, A.E.R. High dose N-acetyl cysteine improves inflammatory response and outcome in patients with COPD exacerbations. Egypt. J. Chest Dis. Tuberc. 2013, 62, 51–57. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell. Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Afford, S.C.; Humphreys, E.H.; Reid, D.T.; Russell, C.L.; Banz, V.M.; Oo, Y.; Vo, T.; Jenne, C.; Adams, D.H.; Eksteen, B. Vascular cell adhesion molecule 1 expression by biliary epithelium promotes persistence of inflammation by inhibiting effector T-cell apoptosis. Hepatology 2014, 59, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Csontos, C.; Rezman, B.; Foldi, V.; Bogar, L.; Drenkovics, L.; Roth, E.; Weber, G.; Lantos, J. Effect of N-acetylcysteine treatment on oxidative stress and inflammation after severe burn. Burns 2012, 38, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.L.; Galley, H.F.; Webster, N.R. The effect of N-acetylcysteine on nuclear factor-κB activation, interleukin-6, interleukin-8, and intercellular adhesion molecule-1 expression in patients with sepsis. Crit. Care Med. 2003, 31, 2574–2578. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Chang, E.J.; Kim, H.M.; Lee, S.B.; Kim, H.D.; Su Kim, G.; Kim, H.H. Antioxidant α-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-κB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-κB ligand and tumor necrosis factor-α. Free Radic. Biol. Med. 2006, 40, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Pajonk, F.; Riess, K.; Sommer, A.; McBride, W.H. N-acetyl-l-cysteine inhibits 26S proteasome function: Implications for effects on NF-κB activation. Free Radic. Biol. Med. 2002, 32, 536–543. [Google Scholar] [CrossRef]
- Oka, S.; Kamata, H.; Kamata, K.; Yagisawa, H.; Hirata, H. N-acetylcysteine suppresses TNF-induced NF-κB activation through inhibition of IκB kinases. FEBS Lett. 2000, 472, 196–202. [Google Scholar] [CrossRef]
- Ingaramo, P.I.; Ronco, M.T.; Frances, D.E.; Monti, J.A.; Pisani, G.B.; Ceballos, M.P.; Galleano, M.; Carrillo, M.C.; Carnovale, C.E. Tumor necrosis factor α pathways develops liver apoptosis in type 1 diabetes mellitus. Mol. Immunol. 2011, 48, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Ronco, M.T.; Alvarez Mde, L.; Monti, J.A.; Carrillo, M.C.; Pisani, G.B.; Lugano, M.C.; Carnovale, C.E. Role of nitric oxide increase on induced programmed cell death during early stages of rat liver regeneration. Biochim. Biophys. Acta 2004, 1690, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, S.W.; Klassen, S.S. Nitric oxide differentially regulates the gene expression of caspase genes but not some autophagic genes. Nitric Oxide 2007, 16, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Mohd Hanafiah, K.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P.; Parkin, D.M.; Munoz, N.; Ferlay, J. Cancer and infection: Estimates of the attributable fraction in 1990. Cancer Epidemiol. Biomark. Prev. 1997, 6, 387–400. [Google Scholar]
- Poli, G. Pathogenesis of liver fibrosis: Role of oxidative stress. Mol. Asp. Med. 2000, 21, 49–98. [Google Scholar] [CrossRef]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.; Dias, J.F.; Dalmarco, E.M.; Frode, T.S.; Pedrosa, R.C.; Wilhelm Filho, D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol. Hepatol. 2012, 35, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Ko, W.S.; Guo, C.H.; Yeh, M.S.; Lin, L.Y.; Hsu, G.S.; Chen, P.C.; Luo, M.C.; Lin, C.Y. Blood micronutrient, oxidative stress, and viral load in patients with chronic hepatitis C. World J. Gastroenterol. 2005, 11, 4697–4702. [Google Scholar] [PubMed]
- Miyanishi, K.; Hoki, T.; Tanaka, S.; Kato, J. Prevention of hepatocellular carcinoma: Focusing on antioxidant therapy. World J. Hepatol. 2015, 7, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-κB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005, 13, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.; Goulart, M.F.O. Lipoic acid: Its antioxidant and anti-inflammatory role and clinical applications. Curr. Top. Med. Chem. 2015, 15, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Shedlofsky, S.I. Role of iron in the natural history and clinical course of hepatitis C disease. Hepatogastroenterology 1998, 45, 349–355. [Google Scholar] [PubMed]
- Metwally, M.A.; Zein, C.O.; Zein, N.N. Clinical significance of hepatic iron deposition and serum iron values in patients with chronic hepatitis C infection. Am. J. Gastroenterol. 2004, 99, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Matos, B.; Santiago, N.; Reymunde, A.; Matta, J.L. Hepatitis C patients in puerto rico have an altered iron balance. Biol. Trace Elem. Res. 2001, 84, 239–245. [Google Scholar] [CrossRef]
- Shan, Y.; Lambrecht, R.W.; Bonkovsky, H.L. Association of hepatitis C virus infection with serum iron status: Analysis of data from the third National Health and Nutrition Examination Survey. Clin. Infect. Dis. 2005, 40, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Bulatova, I.A.; Tret'iakova, I.I.; Shchekotov, V.V.; Shchekotova, A.P.; Ulitina, P.V.; Krivtsov, A.V.; Nenasheva, O.I. Catalase gene rs1001179 polymorphism and oxidative stress in patients with chronic hepatitis C and ulcerative colitis. Ter. Arkh. 2015, 87, 49–53. [Google Scholar] [PubMed]
- Bitetto, D.; Bortolotti, N.; Falleti, E.; Vescovo, S.; Fabris, C.; Fattovich, G.; Cussigh, A.; Cmet, S.; Fornasiere, E.; Ceriani, E.; et al. Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy. Hepatology 2013, 57, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Masri, O.A.; Chalhoub, J.M.; Sharara, A.I. Role of vitamins in gastrointestinal diseases. World J. Gastroenterol. 2015, 21, 5191–5209. [Google Scholar] [CrossRef] [PubMed]
- Khadem Ansari, M.H.; Omrani, M.D.; Kheradmand, F. Oxidative stress response in patients infected by diverse hepatitis C virus genotypes. Hepat. Mon. 2015, 15, e22069. [Google Scholar] [CrossRef] [PubMed]
- Gabr, S.A.; Alghadir, A.H. Prediction of fibrosis in hepatitis C patients: Assessment using hydroxyproline and oxidative stress biomarkers. Virusdisease 2014, 25, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bunchorntavakul, C.; Wootthananont, T.; Atsawarungruangkit, A. Effects of vitamin E on chronic hepatitis C genotype 3: A randomized, double-blind, placebo-controlled study. J. Med. Assoc. Thail. 2014, 97, 31–40. [Google Scholar]
- Vonghia, L.; Michielsen, P.; Francque, S. Immunological mechanisms in the pathophysiology of non-alcoholic steatohepatitis. Int. J. Mol. Sci. 2013, 14, 19867–19890. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- McMahon, B.J. Natural history of chronic hepatitis B. Clin. Liver Dis. 2010, 14, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Seiva, F.R.F.; Amauchi, J.F.; Rocha, K.K.R.; Souza, G.A.; Ebaid, G.X.; Burneiko, R.M.; Novelli, E.L. Effects of N-acetylcysteine on alcohol abstinence and alcohol-induced adverse effects in rats. Alcohol 2009, 43, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.; Wu, L.; Lin, Q.; Lau, Q.C.; Zhao, X.; Li, J.; Chen, P.; Chen, L.; Tang, H.; Huang, C.; Wei, Y.Q. Proteomic analysis of cellular protein alterations using a hepatitis B virus-producing cellular model. Proteomics 2008, 8, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Hajjou, M.; Norel, R.; Carver, R.; Marion, P.; Cullen, J.; Rogler, L.E.; Rogler, C.E. cDNA microarray analysis of HBV transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by HBV. J. Med. Virol. 2005, 77, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Seo, H.W.; Jung, G. Reactive oxygen species promote heat shock protein 90-mediated HBV capsid assembly. Biochem. Biophys. Res. Commun. 2015, 457, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Acar, A.; Gorenek, L.; Aydin, A.; Eyigun, C.P.; Eken, A.; Sayal, A.; Pahsa, A. Investigation of oxidative stress and antioxidant defense in patients with hepatitis B virus infection and the effect of interferon-α plus lamivudine combination therapy on oxidative stress. Mikrobiyol. Bulteni 2009, 43, 411–423. [Google Scholar]
- Tasdelen Fisgin, N.; Aydin, B.K.; Sarikaya, H.; Tanyel, E.; Esen, S.; Sunbul, M.; Leblebicioglu, H. Oxidative stress and antioxidant defense in patients with chronic hepatitis B. Clin. Lab. 2012, 58, 273–280. [Google Scholar] [PubMed]
- Dikici, I.; Mehmetoglu, I.; Dikici, N.; Bitirgen, M.; Kurban, S. Investigation of oxidative stress and some antioxidants in patients with acute and chronic viral hepatitis B and the effect of interferon-α treatment. Clin. Biochem. 2005, 38, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Duygu, F.; Karsen, H.; Aksoy, N.; Taskin, A. Relationship of oxidative stress in hepatitis B infection activity with HBV DNA and fibrosis. Ann. Lab. Med. 2012, 32, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.S.; Dasarathy, S.; McCullough, A.J.; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010, 51, 307–328. [Google Scholar] [PubMed]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef]
- MacSween, R.N.; Burt, A.D. Histologic spectrum of alcoholic liver disease. Semin. Liver Dis. 1986, 6, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Wands, J.R.; Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Forrest, E.; Reed, E. Alcohol and the liver. Medicine 2011, 39, 532–535. [Google Scholar] [CrossRef]
- Qu, B.G.; Wang, H.; Jia, Y.G.; Su, J.L.; Wang, Z.D.; Wang, Y.F.; Han, X.H.; Liu, Y.X.; Pan, J.D.; Ren, G.Y. Changes in tumor necrosis factor-α, heat shock protein 70, malondialdehyde, and superoxide dismutase in patients with different severities of alcoholic fatty liver disease: A prospective observational study. Medicine 2015, 94, e643. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Compalati, A.D.; Voci, A.; Vecchione, G.; Ragazzoni, M.; Gallo, G.; Borro, P.; Sumberaz, A.; Testino, G.; Vergani, L. Altered oxidative stress/antioxidant status in blood of alcoholic subjects is associated with alcoholic liver disease. Drug Alcohol Depend. 2014, 143, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Kattimani, S.; Sridhar, M.G. Oxidative stress during alcohol withdrawal and its relationship with withdrawal severity. Indian J. Psychol. Med. 2015, 37, 175–180. [Google Scholar] [PubMed]
- Pivetta, L.A.; Pereira, R.P.; Farinon, M.; de Bem, A.F.; Perottoni, J.; Soares, J.C.; Duarte, M.M.; Zeni, G.; Rocha, J.B.; Farina, M. Ethanol inhibits δ-aminolevulinate dehydratase and glutathione peroxidase activities in mice liver: Protective effects of ebselen and N-acetylcysteine. Environ. Toxicol. Pharmacol. 2006, 21, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.A.; Bell, M.; Ejiofor, S.; Zurcher, G.; Petersen, D.R.; Ronis, M.J. N-acetylcysteine inhibits the up-regulation of mitochondrial biogenesis genes in livers from rats fed ethanol chronically. Alcohol. Clin. Exp. Res. 2014, 38, 2896–2906. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Wang, J.P.; Wang, H.; Chen, Y.H.; Zhao, L.; Wang, L.S.; Wei, W.; Xu, D.X. A dual effect of N-acetylcysteine on acute ethanol-induced liver damage in mice. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2006, 34, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Thong-Ngam, D.; Samuhasaneeto, S.; Kulaputana, O.; Klaikeaw, N. N-acetylcysteine attenuates oxidative stress and liver pathology in rats with non-alcoholic steatohepatitis. World J. Gastroenterol. 2007, 13, 5127–5132. [Google Scholar] [CrossRef] [PubMed]
- Samuhasaneeto, S.; Thong-Ngam, D.; Kulaputana, O.; Patumraj, S.; Klaikeaw, N. Effects of N-acetylcysteine on oxidative stress in rats with non-alcoholic steatohepatitis. J. Med. Assoc. Thail. 2007, 90, 788–797. [Google Scholar]
- Demiroren, K.; Dogan, Y.; Kocamaz, H.; Ozercan, I.H.; Ilhan, S.; Ustundag, B.; Bahcecioglu, I.H. Protective effects of l-carnitine, N-acetylcysteine and genistein in an experimental model of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Vendemiale, G.; Grattagliano, I.; Caruso, M.L.; Serviddio, G.; Valentini, A.M.; Pirrelli, M.; Altomare, E. Increased oxidative stress in dimethylnitrosamine-induced liver fibrosis in the rat: Effect of N-acetylcysteine and interferon-α. Toxicol. Appl. Pharmacol. 2001, 175, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Tsuyuki, S.; Yamauchi, A.; Nakamura, H.; Nakamura, Y.; Kinoshita, K.; Gomi, T.; Kawai, Y.; Hirose, T.; Furuke, K.; Ikai, I.; et al. N-acetylcysteine improves cytotoxic activity of cirrhotic rat liver-associated mononuclear cells. Int. Immunol. 1998, 10, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Liu, Y.; Liu, H.; Yu, H.; Wang, H.; Xia, Z. Effects of N-acetylcysteine on nicotinamide dinucleotide phosphate oxidase activation and antioxidant status in heart, lung, liver and kidney in streptozotocin-induced diabetic rats. Yonsei Med. J. 2012, 53, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Diniz, Y.S.; Rocha, K.K.; Souza, G.A.; Galhardi, C.M.; Ebaid, G.M.; Rodrigues, H.G.; Novelli Filho, J.L.; Cicogna, A.C.; Novelli, E.L. Effects of N-acetylcysteine on sucrose-rich diet-induced hyperglycaemia, dyslipidemia and oxidative stress in rats. Eur. J. Pharmacol. 2006, 543, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Pu, L.Y.; Lu, L.; Wang, X.H.; Zhang, F.; Rao, J.H. N-acetylcysteine attenuates reactive-oxygen-species-mediated endoplasmic reticulum stress during liver ischemia-reperfusion injury. World J. Gastroenterol. 2014, 20, 15289–15298. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, V.; Vargas, R.; Castillo, V.; Cadiz, N.; Bastias, D.; Roman, S.; Tapia, G.; Videla, L.A. Reestablishment of ischemia-reperfusion liver injury by N-acetylcysteine administration prior to a preconditioning iron protocol. Sci. World J. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Inal-Erden, M. Pentoxifylline and N-acetylcysteine in hepatic ischemia/reperfusion injury. Clin. Chim. Acta 1998, 275, 127–135. [Google Scholar] [CrossRef]
- Sener, G.; Tosun, O.; Şehirli, A.Ö.; Kaçmaz, A.; Arbak, S.; Ersoy, Y.; Ayanoğlu-Dülger, G. Melatonin and N-acetylcysteine have beneficial effects during hepatic ischemia and reperfusion. Life Sci. 2003, 72, 2707–2718. [Google Scholar] [CrossRef]
- Wang, C.; Chen, K.; Xia, Y.; Dai, W.; Wang, F.; Shen, M.; Cheng, P.; Wang, J.; Lu, J.; Zhang, Y.; et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS ONE 2014, 9, e108855. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, X.B.; Yu, J.J.; Hua, F.; Hu, Z.W. Antioxidant N-acetylcysteine attenuates hepatocarcinogenesis by inhibiting ROS/ER stress in TLR2 deficient mouse. PLoS ONE 2013, 8, e74130. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kuo, C.F.; Sir, D.; Wang, L.; Govindarajan, S.; Petrovic, L.M.; Ou, J.H. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell. Death Differ. 2015, 22, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Moreno, M.; Favari, L.; Muriel, P. Antifibrotic and antioxidant effects of N-acetylcysteine in an experimental cholestatic model. Eur. J. Gastroenterol. Hepatol. 2012, 24, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; Reinke, A.; Andrades, M.; Martins, M.R.; Rocha, J.; Menna-Barreto, S.; Quevedo, J.; Moreira, J.C.F.; Dal-Pizzol, F. Protective effect of N-acetylcysteine and deferoxamine on carbon tetrachloride-induced acute hepatic failure in rats. Crit. Care Med. 2004, 32, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Bemeur, C.; Vaquero, J.; Desjardins, P.; Butterworth, R.F. N-acetylcysteine attenuates cerebral complications of non-acetaminophen-induced acute liver failure in mice: Antioxidant and anti-inflammatory mechanisms. Metab. Brain Dis. 2010, 25, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Seif el-Din, S.H.; Al-Hroob, A.M.; Ebeid, F.A. Schistosoma mansoni: N-acetylcysteine downregulates oxidative stress and enhances the antischistosomal activity of artemether in mice. Exp. Parasitol. 2011, 128, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Hsu, B.G.; Lee, R.P.; Yang, F.L.; Harn, H.J.; Chen, H.I. Post-treatment with N-acetylcysteine ameliorates endotoxin shock-induced organ damage in conscious rats. Life Sci. 2006, 79, 2010–2016. [Google Scholar] [CrossRef] [PubMed]
- Ronis, M.J.; Butura, A.; Sampey, B.P.; Shankar, K.; Prior, R.L.; Korourian, S.; Albano, E.; Ingelman-Sundberg, M.; Petersen, D.R.; Badger, T.M. Effects of N-acetylcysteine on ethanol-induced hepatotoxicity in rats fed via total enteral nutrition. Free Radic. Biol. Med. 2005, 39, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Setshedi, M.; Longato, L.; Petersen, D.R.; Ronis, M.; Chen, W.C.; Wands, J.R.; de la Monte, S.M. Limited therapeutic effect of N-acetylcysteine on hepatic insulin resistance in an experimental model of alcohol-induced steatohepatitis. Alcohol. Clin. Exp. Res. 2011, 35, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Bajt, M.L.; Knight, T.R.; Lemasters, J.J.; Jaeschke, H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: Protection by N-acetyl cysteine. Toxicol. Sci. 2004, 80, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Taslipinar, M.Y.; Aydin, I.; Kaldirim, U.; Aydin, F.N.; Agilli, M.; Eyi, Y.E.; Tuncer, S.K.; Altayli, E.; Ucar, F.; Macit, E.; et al. Hyperbaric oxygen treatment and N-acetylcysteine ameliorate acetaminophen-induced liver injury in a rat model. Hum. Exp. Toxicol. 2013, 32, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.; Lau-Cam, C.A. Comparison of the protective actions of N-acetylcysteine, hypotaurine and taurine against acetaminophen-induced hepatotoxicity in the rat. J. Biomed. Sci. 2010, 17 (Suppl. S1), 1–11. [Google Scholar] [CrossRef] [PubMed]
- Terneus, M.V.; Brown, J.M.; Carpenter, A.B.; Valentovic, M.A. Comparison of S-adenosyl-l-methionine (SAMe) and N-acetylcysteine (NAC) protective effects on hepatic damage when administered after acetaminophen overdose. Toxicology 2008, 244, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Rafeiro, E.; Barr, S.G.; Harrison, J.J.; Racz, W.J. Effects of N-acetylcysteine and dithiothreitol on glutathione and protein thiol replenishment during acetaminophen-induced toxicity in isolated mouse hepatocytes. Toxicology 1994, 93, 209–224. [Google Scholar] [CrossRef]
- Manov, I.; Hirsh, M.; Iancu, T.C. Acetaminophen hepatotoxicity and mechanisms of its protection by N-acetylcysteine: A study of Hep3B cells. Exp. Toxicol. Pathol. 2002, 53, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Al-Mustafa, Z.H.; Al-Ali, A.K.; Qaw, F.S.; Abdul-Cader, Z. Cimetidine enhances the hepatoprotective action of N-acetylcysteine in mice treated with toxic doses of paracetamol. Toxicology 1997, 121, 223–228. [Google Scholar] [CrossRef]
- Tobwala, S.; Khayyat, A.; Fan, W.; Ercal, N. Comparative evaluation of N-acetylcysteine and N-acetylcysteineamide in acetaminophen-induced hepatotoxicity in human hepatoma HepaRG cells. Exp. Biol. Med. 2015, 240, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.V.; Attri, S.; Vaiphei, K.; Pal, R.; Attri, A.; Singh, K. Role of N-acetylcysteine in rifampicin-induced hepatic injury of young rats. World J. Gastroenterol. 2006, 12, 287–291. [Google Scholar] [PubMed]
- Raza, M.; Ahmad, M.; Gado, A.; Al-Shabanah, O.A. A comparison of hepatoprotective activities of aminoguanidine and N-acetylcysteine in rat against the toxic damage induced by azathioprine. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2003, 134, 451–456. [Google Scholar] [CrossRef]
- Carvalho, M.; Remiao, F.; Milhazes, N.; Borges, F.; Fernandes, E.; Carvalho, F.; Bastos, M.L. The toxicity of N-methyl-α-methyldopamine to freshly isolated rat hepatocytes is prevented by ascorbic acid and N-acetylcysteine. Toxicology 2004, 200, 193–203. [Google Scholar] [PubMed]
- Kaya, H.; Koc, A.; Sogut, S.; Duru, M.; Yilmaz, H.R.; Uz, E.; Durgut, R. The protective effect of N-acetylcysteine against cyclosporine A-induced hepatotoxicity in rats. J. Appl. Toxicol. 2008, 28, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Abdoli, N.; Azarmi, Y.; Eghbal, M.A. Protective Effects of N-acetylcysteine against the Statins Cytotoxicity in Freshly Isolated Rat Hepatocytes. Adv. Pharm. Bull. 2014, 4, 249–254. [Google Scholar] [PubMed]
- Akbulut, S.; Elbe, H.; Eris, C.; Dogan, Z.; Toprak, G.; Otan, E.; Erdemli, E.; Turkoz, Y. Cytoprotective effects of amifostine, ascorbic acid and N-acetylcysteine against methotrexate-induced hepatotoxicity in rats. World J. Gastroenterol. 2014, 20, 10158–10165. [Google Scholar] [CrossRef] [PubMed]
- Maheswari, E.; Saraswathy, G.R.; Santhranii, T. Hepatoprotective and antioxidant activity of N-acetyl cysteine in carbamazepine-administered rats. Indian J. Pharmacol. 2014, 46, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Lasram, M.M.; Lamine, A.J.; Dhouib, I.B.; Bouzid, K.; Annabi, A.; Belhadjhmida, N.; Ahmed, M.B.; El Fazaa, S.; Abdelmoula, J.; Gharbi, N. Antioxidant and anti-inflammatory effects of N-acetylcysteine against malathion-induced liver damages and immunotoxicity in rats. Life Sci. 2014, 107, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Mostafalou, S.; Abdollahi, M.; Eghbal, M.A.; Saeedi Kouzehkonani, N. Protective effect of NAC against malathion-induced oxidative stress in freshly isolated rat hepatocytes. Adv. Pharm. Bull. 2012, 2, 79–88. [Google Scholar] [PubMed]
- Ahmad, I.; Shukla, S.; Kumar, A.; Singh, B.K.; Kumar, V.; Chauhan, A.K.; Singh, D.; Pandey, H.P.; Singh, C. Biochemical and molecular mechanisms of N-acetyl cysteine and silymarin-mediated protection against maneb- and paraquat-induced hepatotoxicity in rats. Chem. Biol. Interact. 2013, 201, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Van Tonder, J.J.; Gulumian, M.; Cromarty, A.D.; Steenkamp, V. In vitro effect of N-acetylcysteine on hepatocyte injury caused by dichlorodiphenyltrichloroethane and its metabolites. Hum. Exp. Toxicol. 2014, 33, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Dhouib, I.E.-B.; Annabi, A.; Lasram, M.M.; Gharbi, N.; El-Fazaa, S. Anti-inflammatory Effects of N-acetylcysteine against Carbosulfan-induced Hepatic Impairment in Male Rats. Recent Adv. Biol. Med. 2015, 1, 29–40. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.H.; Hafez, H.F.; Fahmy, N.M.; Hanafi, N. Protective effect of N-acetylcysteine against radiation induced DNA damage and hepatic toxicity in rats. Biochem. Pharmacol. 2008, 75, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Joshi, D.; Mittal, D.K.; Shukla, S.; Srivastav, A.K.; Srivastav, S.K. N-acetyl cysteine and selenium protects mercuric chloride-induced oxidative stress and antioxidant defense system in liver and kidney of rats: A histopathological approach. J. Trace Elem. Med. Biol. 2014, 28, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Kamalakkannan, N.; Rukkumani, R.; Aruna, K.; Varma, P.; Viswanathan, P.; Padmanabhan Menon, V. Protective effect of N-acetyl cysteine in carbon tetrachloride-induced hepatotoxicity in rats. Iran. J. Pharmacol. Ther. 2005, 4, 118–123. [Google Scholar]
- Nissar, A.U.; Farrukh, M.R.; Kaiser, P.J.; Rafiq, R.A.; Afnan, Q.; Bhushan, S.; Adil, H.S.; Subhash, B.C.; Tasduq, S.A. Effect of N-acetyl cysteine (NAC), an organosulfur compound from Allium plants, on experimentally induced hepatic prefibrogenic events in Wistar rat. Phytomedicine 2013, 20, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lou, Q.; Wang, F.; Li, E.; Sun, J.; Fang, H.; Xi, J.; Ju, L. N-acetylcysteine protects against liver injure induced by carbon tetrachloride via activation of the Nrf2/HO-1 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 8655–8662. [Google Scholar] [PubMed]
- Odewumi, C.O.; Badisa, V.L.; Le, U.T.; Latinwo, L.M.; Ikediobi, C.O.; Badisa, R.B.; Darling-Reed, S.F. Protective effects of N-acetylcysteine against cadmium-induced damage in cultured rat normal liver cells. Int. J. Mol. Med. 2011, 27, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, H.; Liu, X.; Liu, Z. N-acetylcysteine protects against cadmium-induced oxidative stress in rat hepatocytes. J. Vet. Sci. 2014, 15, 485. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rubio, S.; Linares, C.I.; Bello, R.I.; Gonzalez, R.; Ferrin, G.; Hidalgo, A.B.; Munoz-Gomariz, E.; Rodriguez, B.A.; Barrera, P.; Ranchal, I.; et al. Calcium-dependent nitric oxide production is involved in the cytoprotective properties of N-acetylcysteine in glycochenodeoxycholic acid-induced cell death in hepatocytes. Toxicol. Appl. Pharmacol. 2010, 242, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Skrzydlewska, E.; Farbiszewski, R. Protective effect of N-acetylcysteine on reduced glutathione, reduced glutathione-related enzymes and lipid peroxidation in methanol intoxication. Drug Alcohol. Depend. 1999, 57, 61–67. [Google Scholar] [CrossRef]
- Pawlowska-Goral, K.; Kurzeja, E.; Stec, M. N-acetylcysteine protects against fluoride-induced oxidative damage in primary rat hepatocytes. Toxicol. Vitro 2013, 27, 2279–2282. [Google Scholar] [CrossRef] [PubMed]
- Sathish, P.; Paramasivan, V.; Palani, V.; Sivanesan, K. N-acetylcysteine attenuates dimethylnitrosamine induced oxidative stress in rats. Eur. J. Pharmacol. 2011, 654, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Modi, M.; Kaul, R.K.; Kannan, G.M.; Flora, S.J. Co-administration of zinc and N-acetylcysteine prevents arsenic-induced tissue oxidative stress in male rats. J. Trace Elem. Med. Biol. 2006, 20, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Brandao, R.; Nogueira, C.W. Inhibition of hepatic delta-aminolevulinate dehydratase activity induced by mercuric chloride is potentiated by N-acetylcysteine in vitro. Food Chem. Toxicol. 2011, 49, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Beyaz, S.G.; Yelken, B.; Kanbak, G. The effects of N-acetylcysteine on hepatic function during isoflurane anaesthesia for laparoscopic surgery patients. Indian J. Anaesth. 2011, 55, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yin, R.; Zhu, J.; Feng, X.; Wang, C.; Sheng, Y.; Dong, G.; Li, D.; Jing, H. Protective effects of melatonin and N-acetylcysteine on hepatic injury in a rat cardiopulmonary bypass model. J. Surg. Res. 2007, 142, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Altinoz, E.; Turkoz, Y.; Vardi, N. The protective effect of N-acetylcysteine against acrylamide toxicity in liver and small and large intestine tissues. Bratisl. Lek. Listy 2015, 116, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Lindor, K.D. Nonalcoholic fatty liver disease. Ann. Epidemiol. 2007, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros, I.C.; de Lima, J.G. Is nonalcoholic fatty liver disease an endogenous alcoholic fatty liver disease?—A mechanistic hypothesis. Med. Hypotheses 2015, 85, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Al Rifai, M.; Silverman, M.G.; Nasir, K.; Budoff, M.J.; Blankstein, R.; Szklo, M.; Katz, R.; Blumenthal, R.S.; Blaha, M.J. The association of nonalcoholic fatty liver disease, obesity, and metabolic syndrome, with systemic inflammation and subclinical atherosclerosis: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2015, 239, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Ozenirler, S.; Erkan, G.; Konca Degertekin, C.; Ercin, U.; Cengiz, M.; Bilgihan, A.; Yilmaz, G.; Akyol, G. The relationship between advanced oxidation protein products (AOPP) and biochemical and histopathological findings in patients with nonalcoholic steatohepatitis. J. Dig. Dis. 2014, 15, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Lin, D.; Yuan, Q.; Zhu, X.; Tang, X. Protein adducts generated from products of lipid oxidation: Focus on HNE and one. Drug Metabol. Rev. 2006, 38, 651–675. [Google Scholar] [CrossRef] [PubMed]
- Farhangi, M.A.; Alipour, B.; Jafarvand, E.; Khoshbaten, M. Oral coenzyme Q10 supplementation in patients with nonalcoholic fatty liver disease: Effects on serum vaspin, chemerin, pentraxin 3, insulin resistance and oxidative stress. Arch. Med. Res. 2014, 45, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Horoz, M.; Bolukbas, C.; Bolukbas, F.F.; Sabuncu, T.; Aslan, M.; Sarifakiogullari, S.; Gunaydin, N.; Erel, O. Measurement of the total antioxidant response using a novel automated method in subjects with nonalcoholic steatohepatitis. BMC Gastroenterol. 2005, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar] [PubMed]
- Athyros, V.G.; Tziomalos, K.; Katsiki, N.; Doumas, M.; Karagiannis, A.; Mikhailidis, D.P. Cardiovascular risk across the histological spectrum and the clinical manifestations of non-alcoholic fatty liver disease: An update. World J. Gastroenterol. 2015, 21, 6820–6834. [Google Scholar] [PubMed]
- Linhart, K.B.; Glassen, K.; Peccerella, T.; Waldherr, R.; Linhart, H.; Bartsch, H.; Seitz, H.K. The generation of carcinogenic etheno-DNA adducts in the liver of patients with nonalcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 117–123. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Liu, Y.; Liu, H.; Yu, H.; Wang, H.; Xia, Z. Effects of N-acetylcysteine on nicotinamide dinucleotide phosphate oxidase activation and antioxidant status in heart, lung, liver and kidney in streptozotocin-induced diabetic rats. Yonsei Med. J. 2012, 53, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, S.H.; Song, E.H.; Park, Y.M.; Lim, J.Y.; Kim, D.J.; Choi, K.H.; Park, S.I.; Gao, B.; Kim, W.H. A critical role of STAT1 in streptozotocin-induced diabetic liver injury in mice: Controlled by ATF3. Cell Signal. 2009, 21, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.P.; Jena, G.B. Ulcerative colitis-induced hepatic damage in mice: Studies on inflammation, fibrosis, oxidative DNA damage and GST-P expression. Chem. Biol. Interact. 2013, 201, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.P.; Jena, G.B. Role of α-lipoic acid in dextran sulfate sodium-induced ulcerative colitis in mice: Studies on inflammation, oxidative stress, DNA damage and fibrosis. Food Chem. Toxicol. 2013, 59, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, C.; Tiso, A.; del Prete, A.; Cariello, R.; Tuccillo, C.; Cotticelli, G.; del Vecchio Blanco, C.; Loguercio, C. Gut microbiota and probiotics in chronic liver diseases. Dig. Liver Dis. 2011, 43, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Thaiss, C.A.; Licona-Limon, P.; Flavell, R.A. Role of the intestinal microbiome in liver disease. J. Autoimmun. 2013, 46, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Koppe, S.W. Obesity and the liver: Nonalcoholic fatty liver disease. Transl. Res. 2014, 164, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Sourianarayanane, A.; Garg, G.; Smith, T.H.; Butt, M.I.; McCullough, A.J.; Shen, B. Risk factors of non-alcoholic fatty liver disease in patients with inflammatory bowel disease. J. Crohns Colitis 2013, 7, e279–e285. [Google Scholar] [CrossRef] [PubMed]
- Carter-Kent, C.; Zein, N.N.; Feldstein, A.E. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: Implications for treatment. Am. J. Gastroenterol. 2008, 103, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-induced liver injury: Interactions between drug properties and host factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Au, J.S.; Pockros, P.J. Drug-induced liver injury from antiepileptic drugs. Clin. Liver Dis. 2013, 17, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Kaplowitz, N. Mechanisms of drug-induced liver injury. Clin. Liver Dis. 2013, 17, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E.S.; Bergmann, O.M.; Björnsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Sgro, C.; Clinard, F.; Ouazir, K.; Chanay, H.; Allard, C.; Guilleminet, C.; Lenoir, C.; Lemoine, A.; Hillon, P. Incidence of drug-induced hepatic injuries: A French population-based study. Hepatology 2002, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Amirana, S.; Babby, J. A Review of Drug-induced Liver Injury. J. Nurs. Pract. 2015, 11, 270–271. [Google Scholar] [CrossRef]
- Tan, C.Y.; Saw, T.Y.; Fong, C.W.; Ho, H.K. Comparative hepatoprotective effects of tocotrienol analogs against drug-induced liver injury. Redox Biol. 2015, 4, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Baveco, J.M.; Roos, A.M.D. Assessing the Impact of Pesticides on Lumbricid Populations: An Individual Based Modelling Approach. J. Appl. Ecol. 1996, 33, 1451–1468. [Google Scholar] [CrossRef]
- Ojha, A.; Yaduvanshi, S.K.; Pant, S.C.; Lomash, V.; Srivastava, N. Evaluation of DNA damage and cytotoxicity induced by three commonly used organophosphate pesticides individually and in mixture, in rat tissues. Environ. Toxicol. 2013, 28, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Morita, S. Hepatotoxicity of trichlorfon and dichlorvos in isolated rat hepatocytes. Toxicology 1992, 76, 69–77. [Google Scholar] [CrossRef]
- Numan, I.T.; Hassan, M.Q.; Stohs, S.J. Protective effects of antioxidants against endrin-induced lipid peroxidation, glutathione depletion, and lethality in rats. Arch. Environ. Contam. Toxicol. 1990, 19, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.Q.; Numan, I.T.; Al-Nasiri, N.; Stohs, S.J. Endrin-induced histopathological changes and lipid peroxidation in livers and kidneys of rats, mice, guinea pigs and hamsters. Toxicol. Pathol. 1991, 19, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Dalton, S.R.; Lee, S.M.; King, R.N.; Nanji, A.A.; Kharbanda, K.K.; Casey, C.A.; McVicker, B.L. Carbon tetrachloride-induced liver damage in asialoglycoprotein receptor-deficient mice. Biochem. Pharmacol. 2009, 77, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Muriel, P. Role of free radicals in liver diseases. Hepatol. Int. 2009, 3, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Domenicali, M.; Vendemiale, G.; Serviddio, G.; Grattagliano, I.; Pertosa, A.M.; Nardo, B.; Principe, A.; Viola, A.; Trevisani, F.; Altomare, E.; et al. Oxidative injury in rat fatty liver exposed to ischemia-reperfusion is modulated by nutritional status. Dig. Liver Dis. 2005, 37, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Jave, E.E.; Escalante-Tattersfield, T.; Ortega-Salgado, J.A.; Pina, E.; Geller, D.A. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J. Surg. Res. 2008, 147, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Serracino-Inglott, F.; Habib, N.A.; Mathie, R.T. Hepatic ischemia-reperfusion injury. Am. J. Surg. 2001, 181, 160–166. [Google Scholar] [CrossRef]
- Fukuda, K.; Asoh, S.; Ishikawa, M.; Yamamoto, Y.; Ohsawa, I.; Ohta, S. Inhalation of hydrogen gas suppresses hepatic injury caused by ischemia/reperfusion through reducing oxidative stress. Biochem. Biophys. Res. Commun. 2007, 361, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Kireev, R.; Bitoun, S.; Cuesta, S.; Tejerina, A.; Ibarrola, C.; Moreno, E.; Vara, E.; Tresguerres, J.A. Melatonin treatment protects liver of Zucker rats after ischemia/reperfusion by diminishing oxidative stress and apoptosis. Eur. J. Pharmacol. 2013, 701, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Glantzounis, G.K.; Yang, W.; Koti, R.S.; Mikhailidis, D.P.; Seifalian, A.M.; Davidson, B.R. The role of thiols in liver ischemia-reperfusion injury. Curr. Pharm. Des. 2006, 12, 2891–2901. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, L.; Chen, L.; Cai, G.H.; Ren, Q.Y.; Chen, J.Z.; Shi, H.J.; Xie, Y.H. As2O3 induces apoptosis in human hepatocellular carcinoma HepG2 cells through a ROS-mediated mitochondrial pathway and activation of caspases. Int. J. Clin. Exp. Med. 2015, 8, 2190–2196. [Google Scholar] [PubMed]
- Borro, P.; Leone, S.; Testino, G. Liver disease and hepatocellular carcinoma in alcoholics: The role of anticraving therapy. Curr. Drug Targets 2015, 16, 13. [Google Scholar]
- Miura, K.; Taura, K.; Kodama, Y.; Schnabl, B.; Brenner, D.A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008, 48, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Nakajima, T.; Katagishi, T.; Okada, Y.; Jo, M.; Kagawa, K.; Okanoue, T.; Itoh, Y.; Yoshikawa, T. Oxidative stress may enhance the malignant potential of human hepatocellular carcinoma by telomerase activation. Liver Int. 2009, 29, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2'-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health Part C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 120–139. [Google Scholar] [CrossRef] [PubMed]
- Roessner, A.; Kuester, D.; Malfertheiner, P.; Schneider-Stock, R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol. Res. Pract. 2008, 204, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Hamilton, S.J.; Angel, D.; Fung, K.; Franklin, J.; Parnes, L.S.; Lewis, D.; Venkatesan, V.; Winquist, E. Cisplatin otoprotection using transtympanic l-N-acetylcysteine: A pilot randomized study in head and neck cancer patients. Laryngoscope 2014, 124, E87–E94. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Lee, M.Y.; Wang, W.S.; Yen, C.C.; Chao, T.C.; Hsiao, L.T.; Yang, M.H.; Chen, P.M.; Lin, K.P.; Chiou, T.J. N-acetylcysteine has neuroprotective effects against oxaliplatin-based adjuvant chemotherapy in colon cancer patients: Preliminary data. Support. Care Cancer 2006, 14, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of substances to laboratory animals: Routes of administration and factors to consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [Google Scholar] [PubMed]
- Lukas, G.; Brindle, S.D.; Greengard, P. The route of absorption of intraperitoneally administered compounds. J. Pharmacol. Exp. Ther. 1971, 178, 562–564. [Google Scholar] [PubMed]
- Abu-Hijleh, M.F.; Habbal, O.A.; Moqattash, S.T. The role of the diaphragm in lymphatic absorption from the peritoneal cavity. J. Anat. 1995, 186 Pt 3, 453–467. [Google Scholar] [PubMed]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Kadiiska, M.B.; Peddada, S.; Herbert, R.A.; Basu, S.; Hensley, K.; Jones, D.P.; Hatch, G.E.; Mason, R.P. Biomarkers of oxidative stress study VI. Endogenous plasma antioxidants fail as useful biomarkers of endotoxin-induced oxidative stress. Free Radic. Biol. Med. 2015, 81, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Albers, R.; Bosco, N.; Bourdet-Sicard, R.; Haller, D.; Holgate, S.T.; Jonsson, L.S.; Latulippe, M.E.; Marcos, A.; et al. A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br. J. Nutr. 2013, 109 (Suppl. S1), S1–S34. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Andrade, K.Q.; Moura, F.A.; Dos Santos, J.M.; De Araújo, O.R.P.; De Farias Santos, J.C.; Goulart, M.O.F. Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine. Int. J. Mol. Sci. 2015, 16, 30269-30308. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226225
De Andrade KQ, Moura FA, Dos Santos JM, De Araújo ORP, De Farias Santos JC, Goulart MOF. Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine. International Journal of Molecular Sciences. 2015; 16(12):30269-30308. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226225
Chicago/Turabian StyleDe Andrade, Kívia Queiroz, Fabiana Andréa Moura, John Marques Dos Santos, Orlando Roberto Pimentel De Araújo, Juliana Célia De Farias Santos, and Marília Oliveira Fonseca Goulart. 2015. "Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine" International Journal of Molecular Sciences 16, no. 12: 30269-30308. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms161226225