
PPARs Link Early Life Nutritional Insults to Later Programmed Hypertension and Metabolic Syndrome



Abstract
:1. Introduction
2. The Impact of Renal Programming in Programmed Hypertension and Metabolic Syndrome
3. Peroxisome Proliferator-Activated Receptors (PPARs) and the Kidney in Hypertension and Metabolic Syndrome
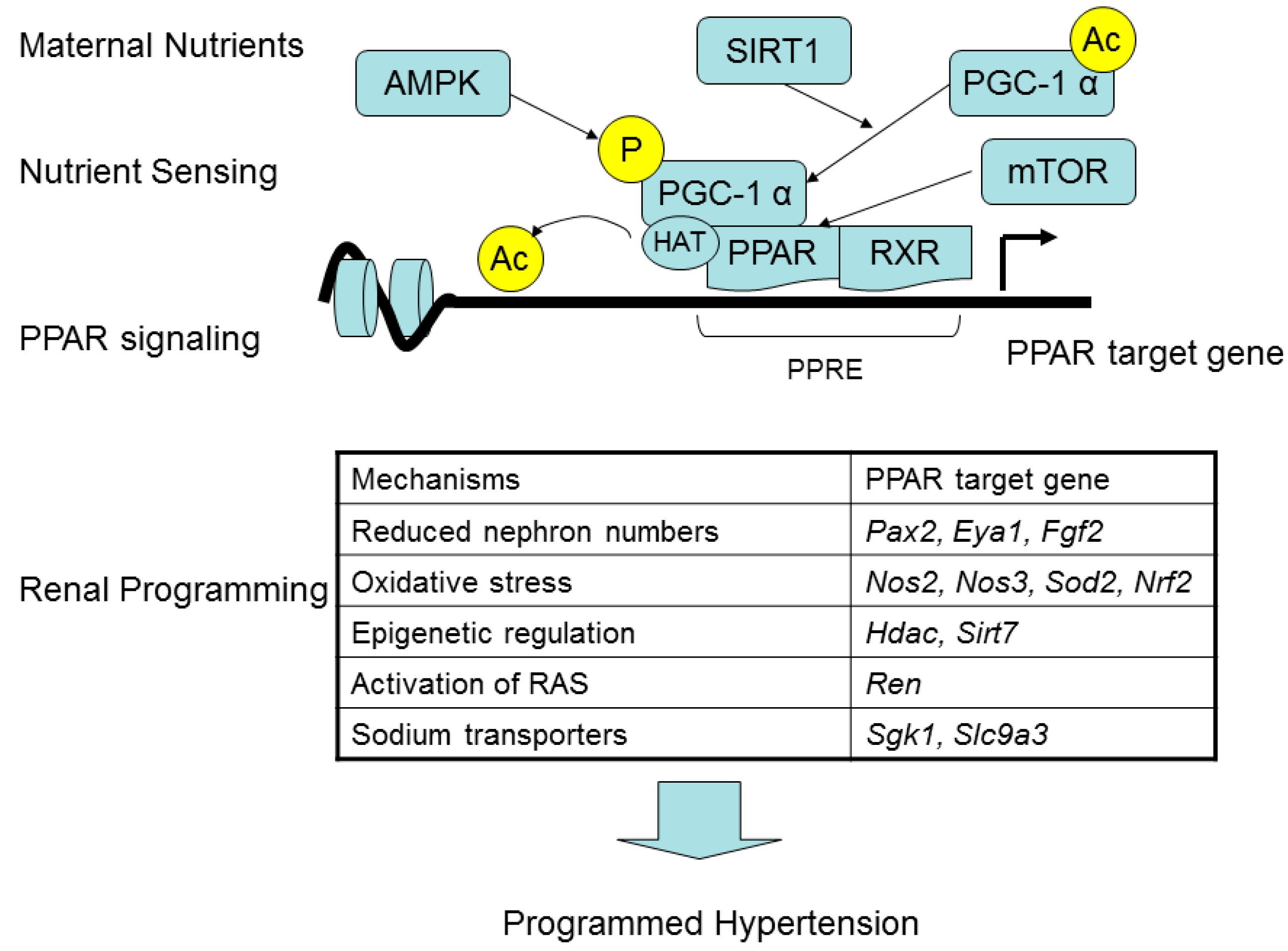
4. PPARs Link Maternal Nutritional Insults to Programmed Hypertension and Metabolic Syndrome

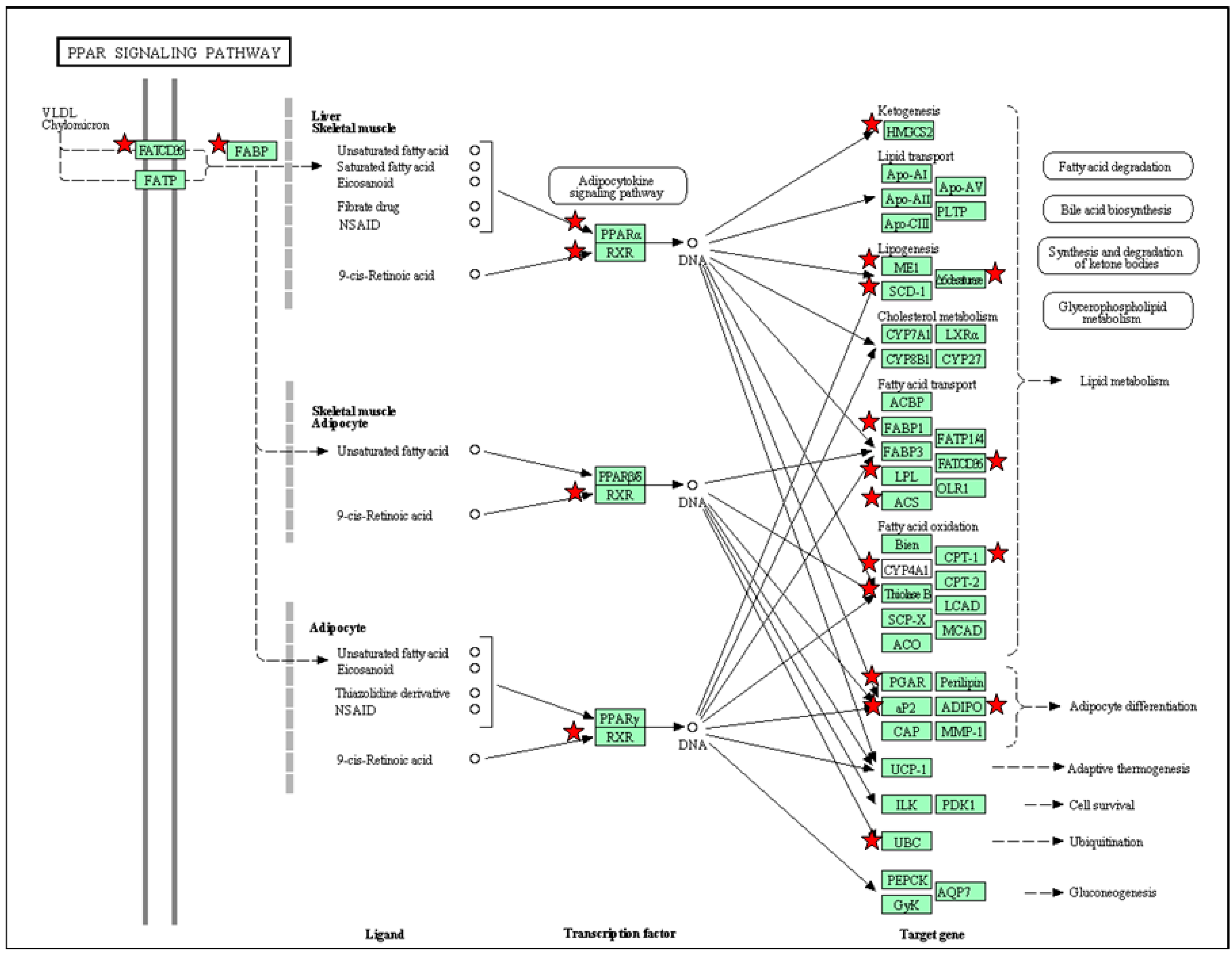
5. PPAR Signaling Pathway in Response to Maternal High-Fructose Intake

{kind=link}
{kind=link}
| Organ | Count | Gene Symbol | p-Value | Benjamini |
|---|---|---|---|---|
| Liver | 9 | Adipoq, Ehhadh, Fabp3, Fabp4, Pparg, Cd36, Scd1, Scl27a5 and Sorbs1 | 5.1 × 10−2 | 4.0 × 10−1 |
| Heart | 14 | Hmgcs2, Acsl1, Angptl4, Aqp7, Cpt1a, Cpt1b, Ctp2, Dbi, Fabp4, Olr1, Rxrg, Acaa1, Cd36 and Ubc | 1.1 × 10−2 | 2.1 × 10−1 |
| Kidney | 19 | Hmgcs2, Acsl3, Adipoq, Angptl4, Cpt1b, Cyp4a8, Cyp4a1, Fabp1, Fabp4, Fabp7, Fads2, Lpl, Me1, Ppara, Rxrg, Acaa1, Cd36, Ubc and Scd | 6.7 × 10−4 | 4.0 × 10−2 |

6. Targeting on PPARs to Prevent Programmed Hypertension and Metabolic Syndrome
| Programming Model [Reference] | Strain | PPAR Isoform | Treatment | Reprograming Effects |
|---|---|---|---|---|
| Prenatal dexamethasone exposure [49] | Wistar | PPARα PPARγ | Diet high in ω-3 fatty acids from three weeks to six months of age | Prevented hypertension and hyperleptinemia at six months of age |
| Low protein diet [50] | Wistar | PPARγ | Losartan between two and four weeks of age | Prevented hypertension at 12 weeks of age |
| Low protein diet [51] | Wistar | PPARγ | Rosiglitazone from three to six months of age | Prevented hypertension at six months of age |
| 50% caloric restriction [52] | Sprague-Dawley | PPARγ | Losartan between two and four weeks of age | Prevented hypertension at 12 weeks of age |
| High fat diet [53] | Sprague-Dawley | PPARγ | Conjugated linoleic acid during pregnancy and lactation | Failed to confer antihypertensive effect at 130 days of age |
| Genetic hypertension [54] | SHR | PPARα | Clofibrate between nine and 12 weeks of age | Prevented hypertension at 12 weeks of age |
| Genetic hypertension plus high-fat diet [55] | SHR | PPARα | Fenofibrate between 8 and 20 weeks of age | Prevented hypertension at 20 weeks of age |
| Genetic hypertension [56] | SHR | PPARα PPARγ | Wy14643 or rosiglitazone between five and 13 weeks of age | Prevented hypertension at 13 weeks of age |
| Genetic hypertension [26] | SHR | PPARβ/δ | GW0742 between 12 and 17 weeks of age | Prevented hypertension at 17 weeks of age |
| Genetic hypertension [57] | SHR | PPARγ | Pioglitazone between five and seven weeks of age | Prevented hypertension at seven weeks of age |
| Genetic hypertension [58] | SHR | PPARγ | Magnolol between four and seven weeks of age | Prevented hypertension at seven weeks of age |
| Genetic hypertension plus high-fat diet [59] | SHR | PPARγ | Telmisartan between eight and 17 weeks of age | Prevented hypertension and renal injury at 17 weeks of age |
| Genetic hypertension [60] | SHRSP | PPARα PPARγ | Fenofibrate, clofibrate, or rosiglitazone between five and 10 weeks of age | Failed to confer antihypertensive effect at 14 weeks of age |
| Genetic hypertension [61] | FHH | PPARγ | Pioglitazone from two weeks before birth to four weeks of age | Failed to confer antihypertensive effect at 28 weeks of age |
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ojeda, N.B.; Grigore, D.; Alexander, B.T. Developmental programming of hypertension: Insight from animal models of nutritional manipulation. Hypertension 2008, 52, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, P.; Wang, E. Fetal programming and metabolic syndrome. Annu. Rev. Physiol. 2012, 74, 107–130. [Google Scholar] [CrossRef] [PubMed]
- McMullen, S.; Langley-Evans, S.C.; Gambling, L.; Lang, C.; Swali, A.; McArdle, H.J. A common cause for a common phenotype: The gatekeeper hypothesis in fetal programming. Med. Hypotheses 2012, 78, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Azhar, S. Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease. Future Cardiol. 2010, 6, 657–691. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013, 549627. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Zheng, F.; Guan, Y. PPARs and the kidney in metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2008, 294, F1032–F1047. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.C.; Xiao, L.; Nuyt, A.M. Mechanisms of developmental programming of the metabolic syndrome and related disorders. World J. Diabetes 2010, 1, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal l-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Leu, S.; Wu, K.L.; Chan, J.Y. High salt exacerbates programmed hypertension in maternal fructose-fed male offspring. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Yu, H.R.; Chen, C.C.; Tiao, M.M.; Hsu, C.N.; Lin, Y.J.; Kuo, K.C.; Huang, L.T. Maternal melatonin therapy rescues prenatal dexamethasone and postnatal high-fat diet induced programmed hypertension in male rat offspring. Front. Physiol. 2015, 6, 377. [Google Scholar] [CrossRef]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Huang, L.T. Long-term effects of maternal citrulline supplementation on renal transcriptome prevention of nitric oxide depletion-related programmed hypertension: The impact of gene-nutrient interactions. Int. J. Mol. Sci. 2014, 15, 23255–23268. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chen, C.C.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Melatonin attenuates prenatal dexamethasone-induced blood pressure increase in a rat model. J. Am. Soc. Hypertens. 2014, 8, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.H.; Kuo, H.C.; Lin, I.C.; Chien, S.J.; Huang, L.T.; Tain, Y.L. Melatonin prevents neonatal dexamethasone induced programmed hypertension: Histone deacetylaseinhibition. J. Steroid Biochem. Mol. Biol. 2014, 144, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Meher, A.; Sundrani, D.; Joshi, S. Maternal nutrition influences angiogenesis in the placenta through peroxisome proliferator activated receptors: A novel hypothesis. Mol. Reprod. Dev. 2015, 82, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Rees, W.D.; McNeil, C.J.; Maloney, C.A. The roles of PPARs in the fetal origins of metabolic health and disease. PPAR Res. 2008, 2008, 459030. [Google Scholar] [CrossRef] [PubMed]
- Zana-Taieb, E.; Pham, H.; Franco-Montoya, M.L.; Jacques, S.; Letourneur, F.; Baud, O.; Jarreau, P.H.; Vaiman, D. Impaired alveolarization and intra-uterine growth restriction in rats: A postnatal genome-wide analysis. J. Pathol. 2015, 235, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Diep, Q.N.; Amiri, F.; Touyz, R.M.; Cohn, J.S.; Endemann, D.; Neves, M.F.; Schiffrin, E.L. PPARα activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension 2002, 40, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Usuda, D.; Kanda, T. Peroxisome proliferator-activated receptors for hypertension. World J. Cardiol. 2014, 6, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Zarzuelo, M.J.; Jiménez, R.; Galindo, P.; Sánchez, M.; Nieto, A.; Romero, M.; Quintela, A.M.; López-Sepúlveda, R.; Gómez-Guzmán, M.; Bailón, E.; et al. Antihypertensive effects of peroxisome proliferator-activated receptor-β activation in spontaneously hypertensive rats. Hypertension 2011, 58, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.; Chang, L.; Zhang, J.; Chen, Y.E. The role of peroxisome proliferator-activated receptor γ in blood pressure regulation. Curr. Hypertens. Rep. 2009, 11, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.Z.; Ivashchenko, C.Y.; Whitesall, S.E.; D’Alecy, L.G.; Duquaine, D.C.; Brosius, F.C., III; Gonzalez, F.J.; Vinson, C.; Pierre, M.A.; Milstone, D.S.; et al. Hypotension, lipodystrophy, and insulin resistance in generalized PPARγ-deficient mice rescued from embryonic lethality. J. Clin. Investig. 2007, 117, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Todorov, V.T.; Desch, M.; Schmitt-Nilson, N.; Todorova, A.; Kurtz, A. Peroxisome proliferator-activated receptor-γ is involved in the control of renin gene expression. Hypertension 2007, 50, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Park, J.; Choi, J.H. Revisiting PPARγ as a target for the treatment of metabolic disorders. BMB Rep. 2014, 47, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [PubMed]
- Sugden, M.C.; Caton, P.W.; Holness, M.J. PPAR control: It’s SIRTainly as easy as PGC. J. Endocrinol. 2010, 204, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brûlé, S.; Turcotte, V.; Côté, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Lemay, D.G.; Hwang, D.H. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. J. Lipid Res. 2006, 47, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, S.; Strokin, M.; Sergeeva, M.; Reiser, G. Peroxisome proliferator-activated receptor (PPAR)β/δ, a possible nexus of PPARα- and PPARγ-dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem. Int. 2013, 63, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Racasan, S.; Braam, B.; Koomans, H.A.; Joles, J.A. Programming blood pressure in adult SHR by shifting perinatal balance of NO and reactive oxygen species toward NO: The inverted Barker phenomenon. Am. J. Physiol. Ren. Physiol. 2005, 288, F626–F636. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Restoration of asymmetric dimethylarginine-nitric oxide balance to prevent the development of hypertension. Int. J. Mol. Sci. 2014, 15, 11773–11782. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Galli, A. PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO. PPAR Res. 2012, 2012, 641087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Shao, Z.; Alibin, C.P.; Acosta, C.; Anderson, H.D. Liganded peroxisome proliferator-activated receptors (PPARs) preserve nuclear histone deacetylase 5 levels in endothelin-treated Sprague-Dawley rat cardiac myocytes. PLoS ONE 2014, 9, e115258. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.; Agapiou, D.J.; Chen, X.M.; Stevens, V.; Pollock, C.A. The role of Sgk-1 in the upregulation of transport proteins by PPAR-γ agonists in human proximal tubule cells. Nephrol. Dial. Transplant. 2009, 24, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Lang, F. New insights into the role of serum- and glucocorticoid-inducible kinase SGK1 in the regulation of renal function and blood pressure. Curr. Opin. Nephrol. Hypertens. 2005, 14, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Rizkalla, S.W. Health implications of fructose consumption: A review of recent data. Nutr. Metab. 2010, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y. Maternal fructose-intake-induced renal programming in adult male offspring. J. Nutr. Biochem. 2015, 26, 642–650. [Google Scholar] [CrossRef] [PubMed]
- NIH DAVID Bioinformatics Resources 6.7. Available online: http://david.abcc.ncifcrf.gov/ (accessed on 1 December 2015).
- Koeners, M.P.; Wesseling, S.; Ulu, A.; Sepúlveda, R.L.; Morisseau, C.; Braam, B.; Hammock, B.D.; Joles, J.A. Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E691–E698. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Saitoh, S.; Shimamoto, K.; Miura, T. Fatty acid-binding protein 4 (FABP4): Pathophysiological insights and potent clinical biomarker of metabolic and cardiovascular diseases. Clin. Med. Insights Cardiol. 2015, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.H.; Xie, Z.; Shapiro, J.I.; Silverstein, R.L. CD36 and Na/K-ATPase-α1 form a proinflammatory signaling loop in kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Wyrwoll, C.S.; Mark, P.J.; Mori, T.A.; Puddey, I.B.; Waddell, B.J. Prevention of programmed hyperleptinemia and hypertension by postnatal dietary ω-3 fatty acids. Endocrinology 2006, 147, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.C.; Langley-Evans, S.C. Antihypertensive treatment in early postnatal life modulates prenatal dietary influences upon blood pressure in the rat. Clin. Sci. 2000, 98, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.S.; D’Oliveira Silva, G.; Aguila, M.B.; de Carvalho, J.J.; Mandarim-De-Lacerda, C.A. Effects of rosiglitazone (a peroxysome proliferator-activated receptor γ agonist) on the blood pressure and aortic structure in metabolically programmed (perinatal low protein) rats. Hypertens. Res. 2008, 31, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lee, C.T.; Huang, L.T.; Tain, Y.L. Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Vickers, M.H.; Segovia, S.A.; Zhang, X.D.; Reynolds, C.M. A maternal high fat diet programmes endothelial function and cardiovascular status in adult male offspring independent of body weight, which is reversed by maternal conjugated linoleic acid (CLA) supplementation. PLoS ONE 2015, 10, e0115994. [Google Scholar] [CrossRef] [PubMed]
- Yousefipour, Z.; Newaz, M. PPARα ligand clofibrate ameliorates blood pressure and vascular reactivity in spontaneously hypertensive rats. Acta Pharmacol. Sin. 2014, 35, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.W.; Lim, J.H.; Kim, M.Y.; Shin, S.J.; Chung, S.; Choi, B.S.; Kim, H.W.; Kim, Y.S.; Park, C.W.; Chang, Y.S. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPARα-FoxO3a-PGC-1α pathway. Nephrol. Dial. Transplant. 2012, 27, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, R.; de Champlain, J.; Wilson, T.W. Beneficial and deleterious effects of rosiglitazone on hypertension development in spontaneously hypertensive rats. Am. J. Hypertens. 2004, 17, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Dovinová, I.; Barancik, M.; Majzunova, M.; Zorad, S.; Gajdosechová, L.; Gresová, L.; Cacanyiova, S.; Kristek, F.; Balis, P.; Chan, J.Y. Effects of PPAR γ agonist pioglitazone on redox-sensitive cellular signaling in young spontaneously hypertensive rats. PPAR Res. 2013, 2013, 541871. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xing, W.; He, J.; Fu, F.; Zhang, W.; Su, F.; Liu, F.; Ji, L.; Gao, F.; Su, H.; et al. Magnolol administration in normotensive young spontaneously hypertensive rats postpones the development of hypertension: Role of increased PPARγ, reduced TRB3 and resultant alleviative vascular insulin resistance. PLoS ONE 2015, 10, e0120366. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Imig, J.D. Telmisartan provides better renal protection than valsartan in a rat model of metabolic syndrome. Am. J. Hypertens. 2011, 24, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gelosa, P.; Banfi, C.; Gianella, A.; Brioschi, M.; Pignieri, A.; Nobili, E.; Castiglioni, L.; Cimino, M.; Tremoli, E.; Sironi, L. Peroxisome proliferator-activated receptor α agonism prevents renal damage and the oxidative stress and inflammatory processes affecting the brains of stroke-prone rats. J. Pharmacol. Exp. Ther. 2010, 335, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Wesseling, S.; Sánchez, M.; Braam, B.; Joles, J.A. Perinatal Inhibition of NF-κB has long-term antihypertensive and renoprotective effects in fawn-hooded hypertensive rats. Am. J. Hypertens. 2015. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Hsu, C.-N.; Chan, J.Y.H. PPARs Link Early Life Nutritional Insults to Later Programmed Hypertension and Metabolic Syndrome. Int. J. Mol. Sci. 2016, 17, 20. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010020
Tain Y-L, Hsu C-N, Chan JYH. PPARs Link Early Life Nutritional Insults to Later Programmed Hypertension and Metabolic Syndrome. International Journal of Molecular Sciences. 2016; 17(1):20. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010020
Chicago/Turabian StyleTain, You-Lin, Chien-Ning Hsu, and Julie Y. H. Chan. 2016. "PPARs Link Early Life Nutritional Insults to Later Programmed Hypertension and Metabolic Syndrome" International Journal of Molecular Sciences 17, no. 1: 20. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010020